
Target Identification of 22-(4-Pyridinecarbonyl) Jorunnamycin A, a Tetrahydroisoquinoline Derivative from the Sponge Xestospongia sp., in Mediating Non-Small-Cell Lung Cancer Cell Apoptosis

 ,
, 

 and
and
Abstract

1. Introduction
2. Results
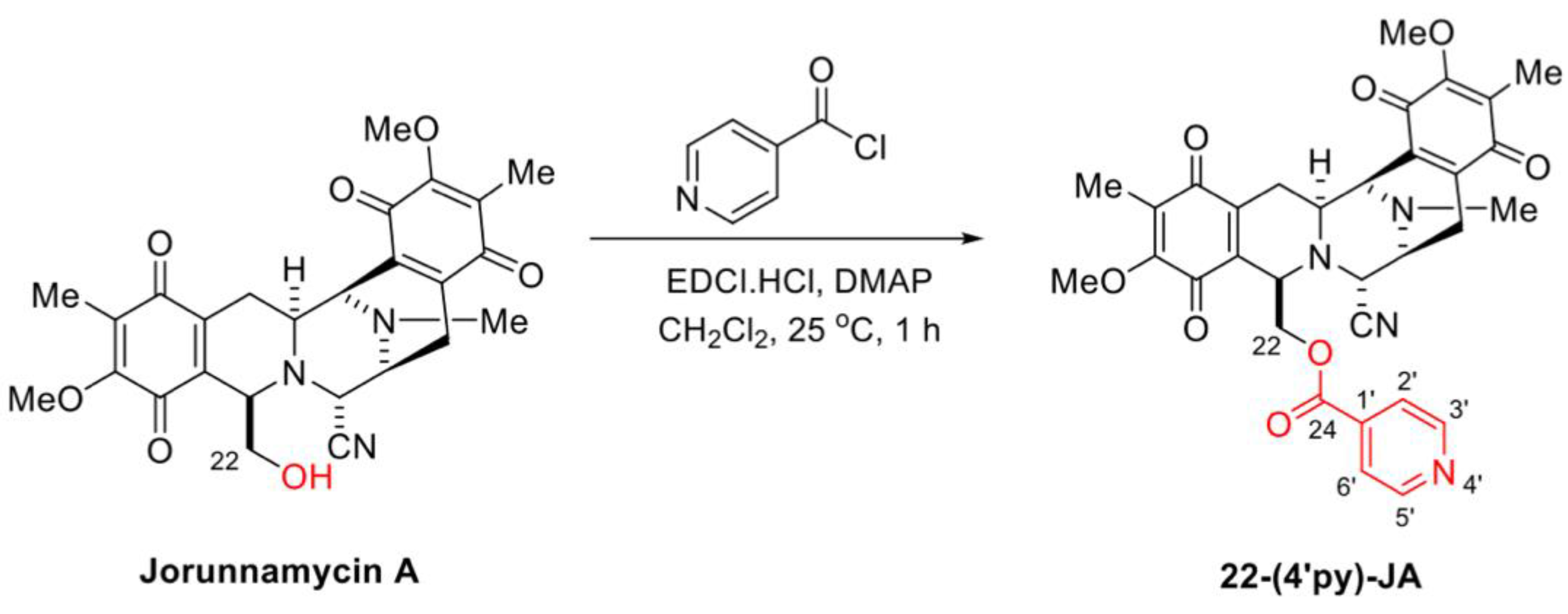
2.1. Semisynthesis of 22-(4′py)-JA
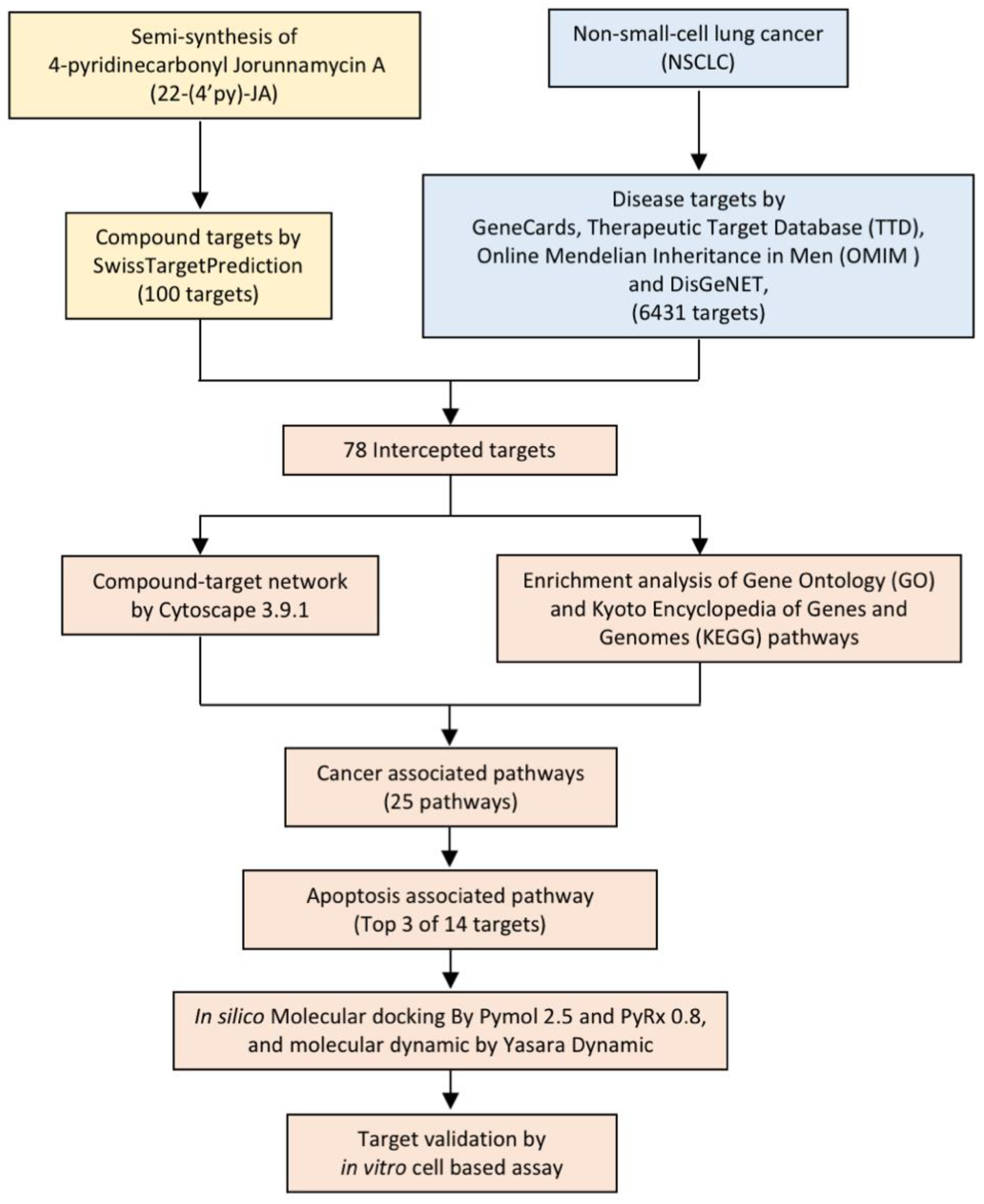
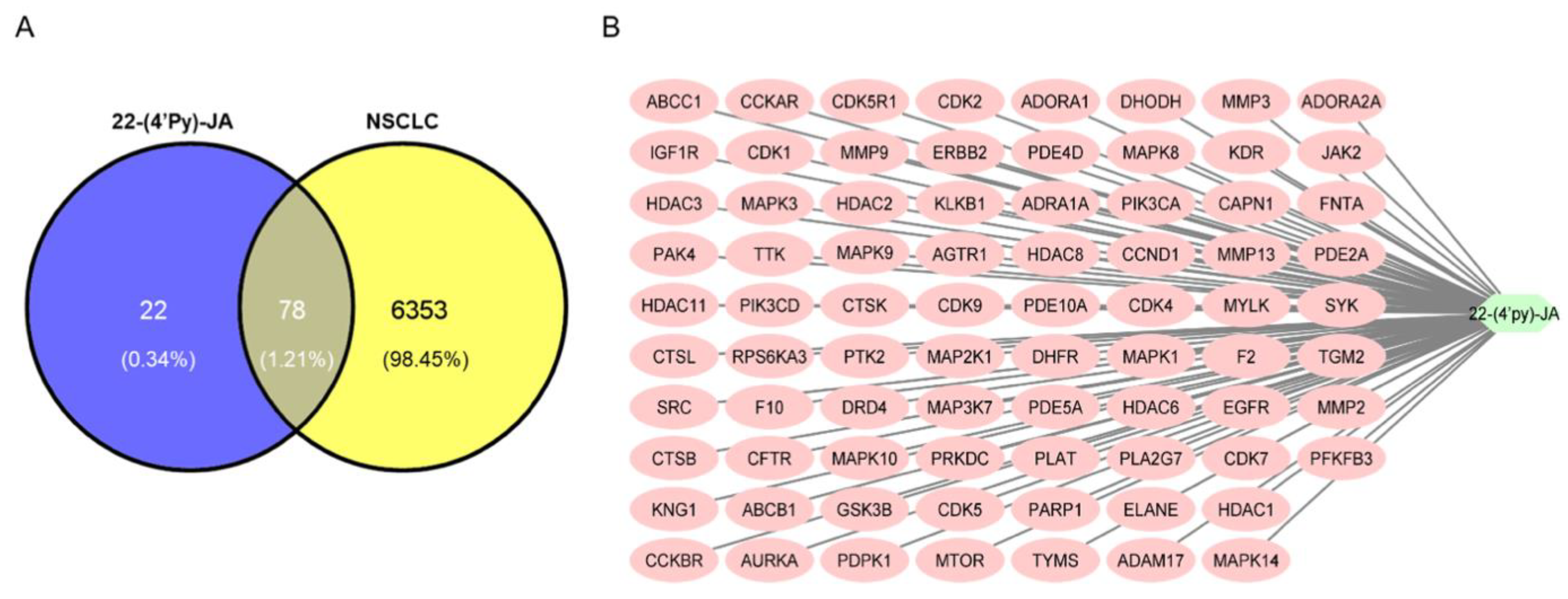
2.2. Target Identification of 22-(4′py)-JA against NSCLC
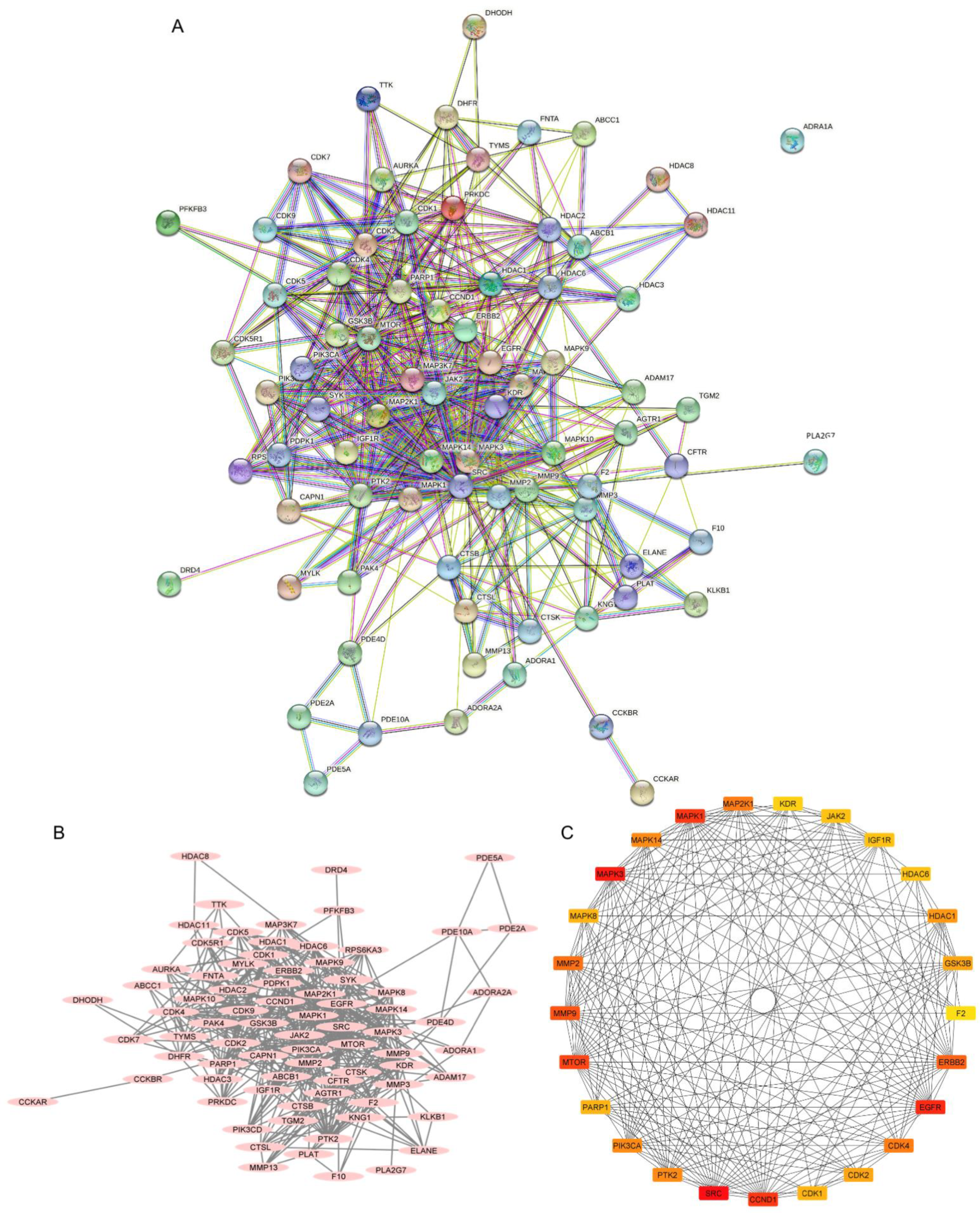
2.3. Investigation of the Protein–Protein Interaction (PPI) Network
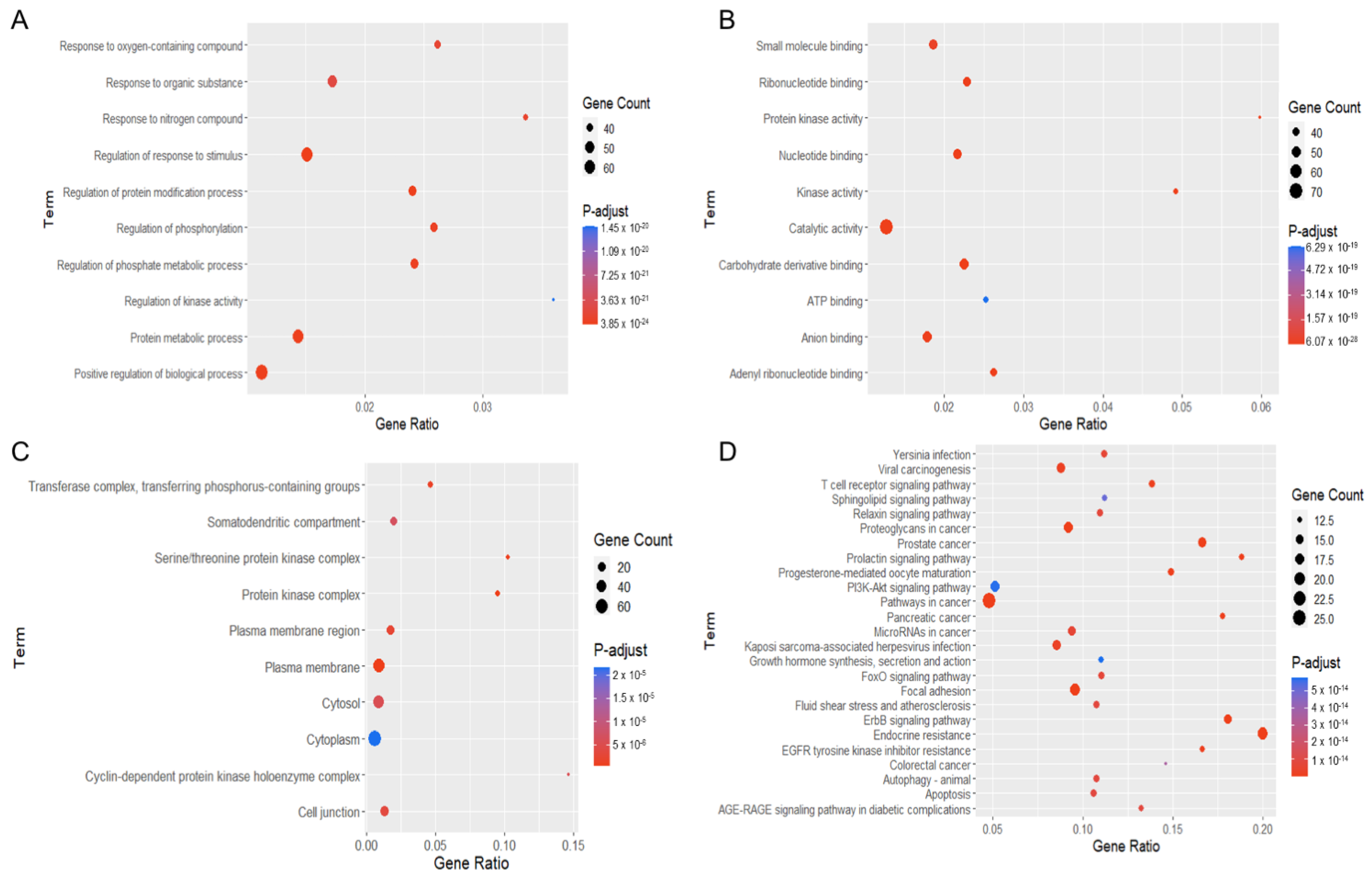
2.4. Enrichment Analysis of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways
2.5. Potential Targets and Signaling Pathway of 22-(4′py)-JA in Regulating Cancer Apoptosis
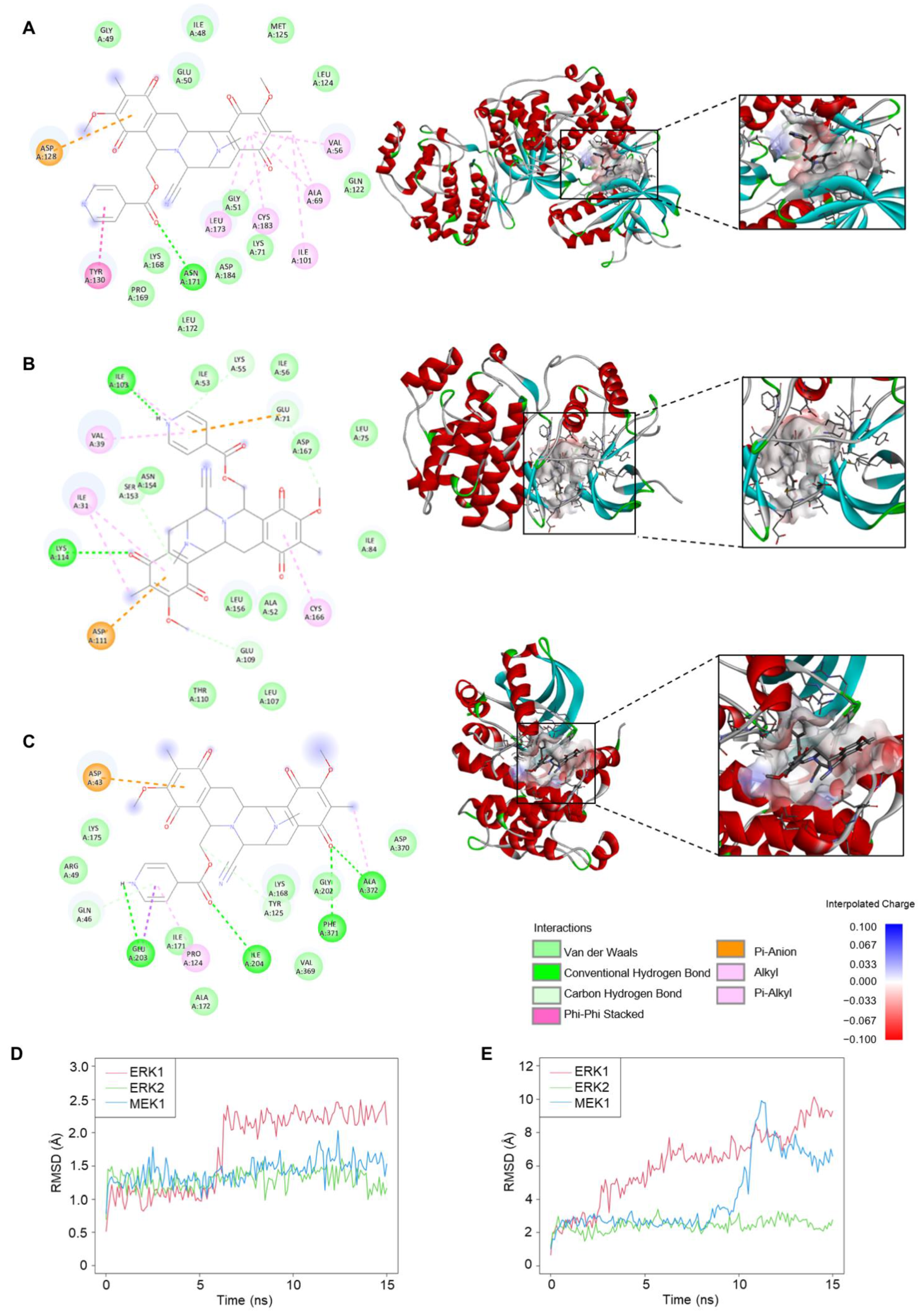
2.6. Molecular Docking and Molecular Dynamic Analysis of 22-(4′py)-JA-Target Interactions
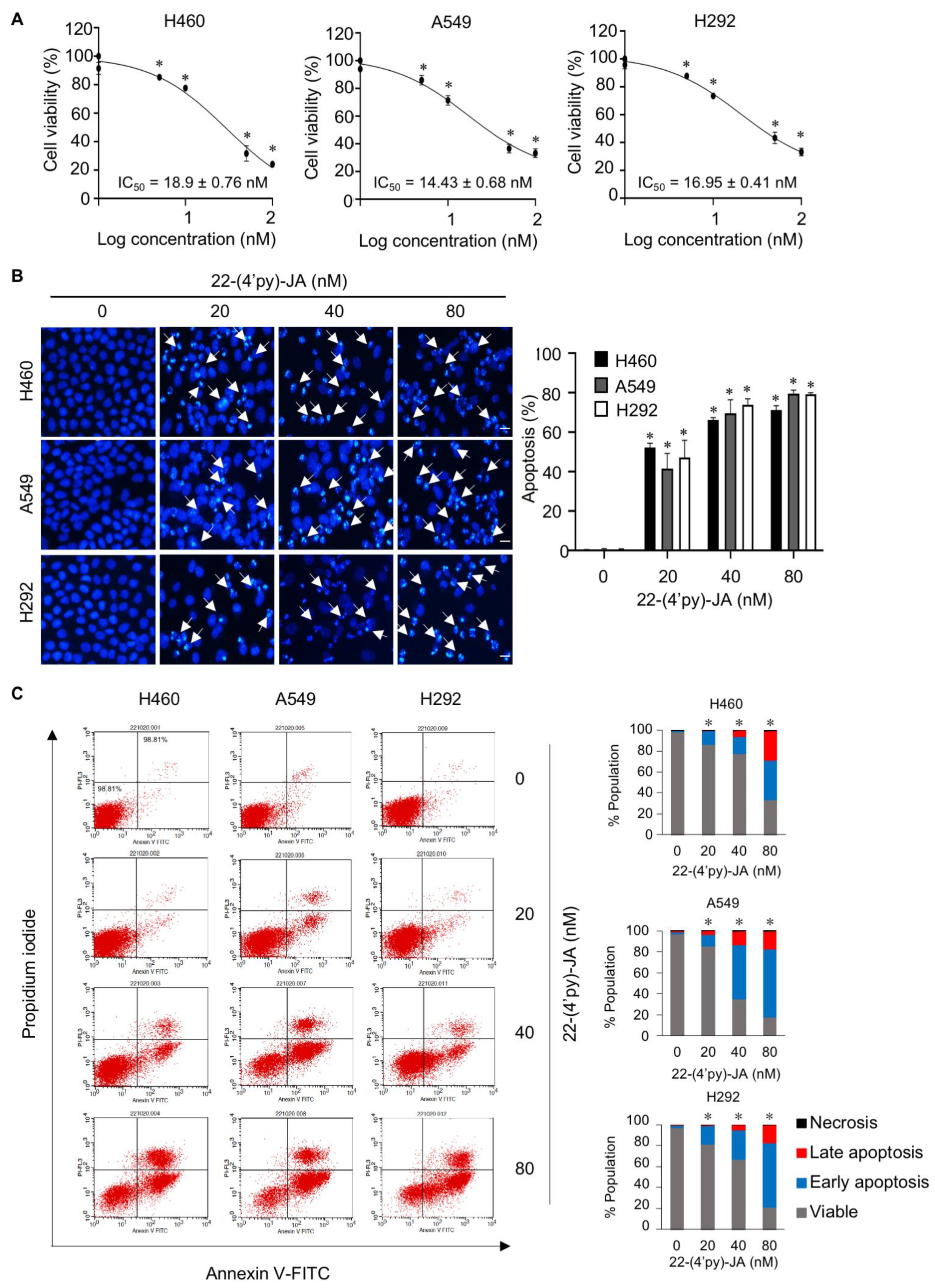
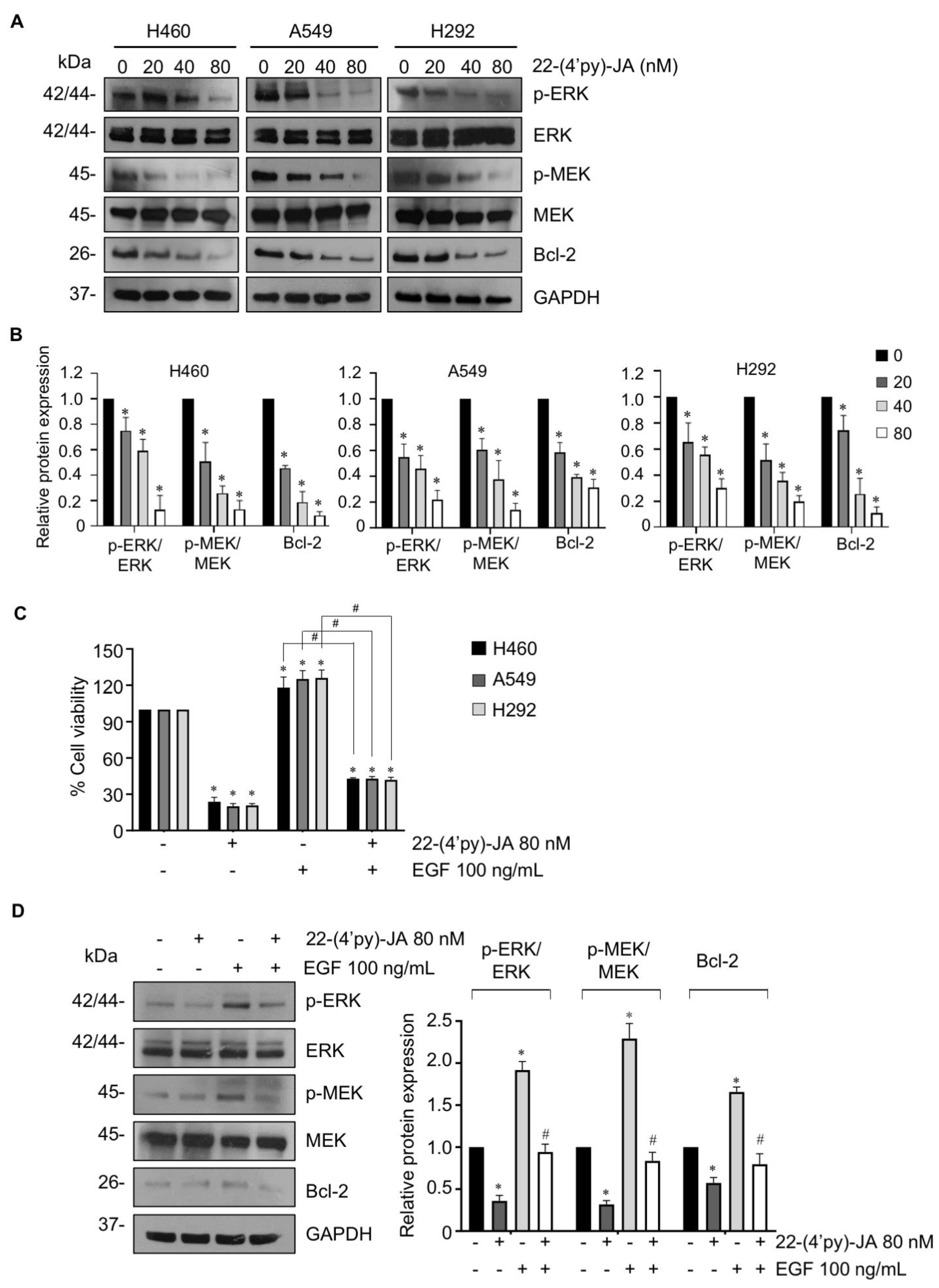
2.7. In Vitro Target Validation of 22-(4′py)-JA in Apoptosis-Inducing Effects
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Semisynthesis of 22-(4′py)-JA from Jorunnamycin A
4.3. Target Identification of 22-(4′py)-JA and NSCLC-Related Genes
4.4. Compound–Target and Protein–Protein Network Construction
4.5. Bioinformatic Analysis of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways
4.6. Molecular Docking and Dynamics
4.7. Cell Culture
4.8. Cytotoxicity Testing
4.9. Apoptosis Assay
4.10. Immunoblotting Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Aluru, J.S.; Barsouk, A. Epidemiology of lung cancer. Contemp. Oncol. 2021, 25, 45–52. [Google Scholar]
- Cersosimo, R.J. Lung cancer: A review. Am. J. Health Syst. Pharm. 2002, 59, 611–642. [Google Scholar] [CrossRef] [PubMed]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Chemotherapy resistance in lung cancer. Adv. Exp. Med. Biol. 2016, 893, 189–209. [Google Scholar]
- Scheff, R.J.; Schneider, B.J. Non-small-cell lung cancer: Treatment of late stage disease: Chemotherapeutics and new frontiers. Semin. Intervent. Radiol. 2013, 30, 191–198. [Google Scholar] [CrossRef]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.E.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, H.J.; Lee, S.H. Therapeutic application of diverse marine-derived natural products in cancer therapy. Anticancer Res. 2019, 39, 5261–5284. [Google Scholar] [CrossRef]
- Suksamai, D.; Racha, S.; Sriratanasak, N.; Chaotham, C.; Aphicho, K.; Lin, A.C.K.; Chansriniyom, C.; Suwanborirux, K.; Chamni, S.; Chanvorachote, P. 5-O-(N-Boc-l-Alanine)-Renieramycin T induces cancer stem cell apoptosis via targeting Akt signaling. Mar. Drugs 2022, 20, 235. [Google Scholar] [CrossRef]
- Saito, N. Chemical research on antitumor isoquinoline marine natural products and related compounds. Chem. Pharm. Bull. 2021, 69, 155–177. [Google Scholar] [CrossRef]
- Fang, Y.; Li, H.; Ji, B.; Cheng, K.; Wu, B.; Li, Z.; Zheng, C.; Hua, H.; Li, D. Renieramycin-type alkaloids from marine-derived organisms: Synthetic chemistry, biological activity and structural modification. Eur. J. Med. Chem. 2021, 210, 113092. [Google Scholar] [CrossRef]
- Hongwiangchan, N.; Sriratanasak, N.; Wichadakul, D.; Aksorn, N.; Chamni, S.; Chanvorachote, P. Hydroquinone 5-O-Cinnamoyl ester of renieramycin M suppresses lung cancer stem cells by targeting Akt and destabilizes c-Myc. Pharmaceuticals 2021, 30, 1112. [Google Scholar] [CrossRef]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for soft tissue sarcoma: Current status and future perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef]
- Barone, A.; Chi, D.C.; Theoret, M.R.; Chen, H.; He, K.; Kufrin, D.; Helms, W.S.; Subramaniam, S.; Zhao, H.; Patel, A.; et al. FDA approval summary: Trabectedin for unresectable or metastatic liposarcoma or leiomyosarcoma following an anthracycline-containing regimen. Clin. Cancer Res. 2017, 23, 7448–7453. [Google Scholar] [CrossRef] [PubMed][Green Version]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Trigo, J.; Subbiah, V.; Besse, B.; Moreno, V.; López, R.; Sala, M.A.; Peters, S.; Ponce, S.; Fernández, C.; Alfaro, V.; et al. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: A single-arm, open-label, phase 2 basket trial. Lancet. Oncol. 2020, 21, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.S.; Shah, A.; Zia-Ur-Rehman; Barker, D. Chemistry of DNA minor groove binding agents. J. Photochem. Photobiol. B Biol. 2012, 115, 105–118. [Google Scholar] [CrossRef]
- Halim, H.; Chunhacha, P.; Suwanborirux, K.; Chanvorachote, P. Anticancer and antimetastatic activities of renieramycin M, a marine tetrahydroisoquinoline alkaloid, in human non-small cell lung cancer cells. Anticancer Res. 2011, 31, 193–201. [Google Scholar]
- Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P. Renieramycin M sensitizes anoikis resistant H460 lung cancer cells to anoikis. Anticancer Res. 2016, 36, 1665–1671. [Google Scholar]
- Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P. Renieramycin M attenuates cancer stem cell-like phenotypes in H460 lung cancer cells. Anticancer Res. 2017, 37, 615–621. [Google Scholar] [CrossRef]
- Ecoy, G.A.U.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P.; Chaotham, C. Jorunnamycin A from Xestospongia Sp. suppresses epithelial to mesenchymal transition and sensitizes anoikis in human lung cancer cells. J. Nat. Prod. 2019, 82, 1861–1873. [Google Scholar] [CrossRef]
- Sumkhemthong, S.; Chamni, S.; Ecoy, G.U.; Taweecheep, P.; Suwanborirux, K.; Prompetchara, E.; Chanvorachote, P.; Chaotham, C. Jorunnamycin A suppresses stem-like phenotypes and sensitizes cisplatin-induced apoptosis in cancer stem-like cell enriched spheroids of human lung cancer cells. Mar. Drugs 2021, 19, 261. [Google Scholar] [CrossRef] [PubMed]
- Charupant, K.; Daikuhara, N.; Saito, E.; Amnuoypol, S.; Suwanborirux, K.; Owa, T.; Saito, N. Chemistry of renieramycins. Part 8: Synthesis and cytotoxicity evaluation of renieramycin M-jorunnamycin A analogues. Bioorg. Med. Chem. 2009, 17, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Sirimangkalakitti, N.; Chamni, S.; Charupant, K.; Chanvorachote, P.; Mori, N.; Saito, N.; Suwanborirux, K. Chemistry of renieramycins. 15. Synthesis of 22- O -ester derivatives of jorunnamycin A and their cytotoxicity against non-small cell lung cancer cells. J. Nat. Prod. 2016, 79, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 17, 52. [Google Scholar] [CrossRef]
- Alqahtani, F.Y.; Aleanizy, F.S.; El Tahir, E.; Alkahtani, H.M.; AlQuadeib, B.T. Paclitaxel. In Profiles of Drug Substances, Excipients and Related Methodology; Academic Press: Cambridge, MA, USA, 2019; Volume 44, pp. 205–238. [Google Scholar]
- Edwards, V.; Benkendorff, K.; Young, F. An in vitro high-throughput assay for screening reproductive and toxic effects of anticancer compounds. Biotechnol. Appl. Biochem. 2014, 61, 582–592. [Google Scholar] [CrossRef]
- Tomizaki, K.; Usui, K.; Mihara, H. Protein-protein interactions and selection: Array-based techniques for screening disease-associated biomarkers in predictive/early diagnosis. FEBS J. 2010, 277, 1996–2005. [Google Scholar] [CrossRef]
- Earm, K.; Earm, Y.E. Integrative approach in the era of failing drug discovery and development. Integr. Med. Res. 2014, 3, 211–216. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Chandran, U.; Mehendale, N.; Patil, S.; Chaguturu, R.; Patwardhan, B. Network pharmacology. In Innovative Approaches in Drug Discovery, 1st ed.; Patwardhan, B., Chaguturu, R., Eds.; Academic Press: London, UK, 2017; pp. 127–164. [Google Scholar]
- Zhang, G.B.; Li, Q.Y.; Chen, Q.L.; Su, S.B. Network pharmacology: A new approach for chinese herbal medicine research. Evid. Based. Complement. Alternat. Med. 2013, 2013, 621423. [Google Scholar]
- Stein, A.; Thomopoulou, P.H.N.; Frias, C.; Hopff, S.M.; Varela, P.; Wilke, N.; Mariappan, A.; Neudörfl, J.M.; Fedorov, A.Y.; Gopalakrishnan, J.; et al. B-nor-methylene colchicinoid PT-100 selectively induces apoptosis in multidrug-resistant human cancer cells via an intrinsic pathway in a caspase-independent manner. ACS Omega 2022, 7, 2591–2603. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Ren, C.; Wang, J.; Wang, S.; Yang, L.; Han, X.; Chen, Y.; Tong, G.; Yang, G. The crosstalk between sTAT3 and p53/RAS signaling controls cancer cell metastasis and cisplatin resistance via the Slug/MAPK/PI3K/AKT mediated regulation of EMT and autophagy. Oncogenesis 2019, 8, 59. [Google Scholar] [CrossRef]
- Lu, K.H.; Yang, J.S.; Hsieh, Y.H.; Chu, H.J.; Chou, C.H.; Lu, E.W.H.; Lin, C.W.; Yang, S.F. Lipocalin-2 inhibits osteosarcoma cell metastasis by suppressing MET expression via the MEK-ERK pathway. Cancers 2021, 13, 3181. [Google Scholar] [CrossRef] [PubMed]
- Ausawasamrit, A.; Itthiwarapornkul, N.; Chaotham, C.; Sukrong, S.; Chanvorachote, P. Lupalbigenin from Derris scandens sensitizes detachment-induced cell death in human lung cancer cells. Anticancer Res. 2015, 25, 2827–2834. [Google Scholar]
- Arunmanee, W.; Ecoy, G.A.U.; Khine, H.E.E.; Duangkaew, M.; Prompetchara, E.; Chanvorachote, P.; Chaotham, C. Colicin N mediates apoptosis and suppresses integrin-modulated survival in human lung cancer cells. Molecules 2020, 25, 816. [Google Scholar] [CrossRef]
- Boonjing, S.; Pothongsrisit, S.; Wattanathamsan, O.; Sritularak, B.; Pongrakhananon, V. Correction: Erianthridin induces non-small cell lung cancer cell apoptosis through the suppression of extracellular signal-regulated kinase activity. Planta Med. 2021, 87, 283–293. [Google Scholar] [CrossRef]
- Pénzváltó, Z.; Lánczky, A.; Lénárt, J.; Meggyesházi, N.; Krenács, T.; Szoboszlai, N.; Denkert, C.; Pete, I.; Győrffy, B. MEK1 is associated with carboplatin resistance and is a prognostic biomarker in epithelial ovarian cancer. BMC Cancer 2014, 14, 837. [Google Scholar] [CrossRef]
- Lee, J.; Lim, B.; Pearson, T.; Choi, K.; Fuson, J.A.; Bartholomeusz, C.; Paradiso, L.J.; Myers, T.; Tripathy, D.; Ueno, N.T. Anti-tumor and Anti-metastasis efficacy of E6201, a MEK1 inhibitor, in preclinical models of triple-negative breast cancer. Breast Cancer Res. Treat. 2019, 175, 339–351. [Google Scholar] [CrossRef]
- Sakakibara, K.; Tsujioka, T.; Kida, J.I.; Kurozumi, N.; Nakahara, T.; Suemori, S.I.; Kitanaka, A.; Arao, Y.; Tohyama, K. Binimetinib, a novel MEK1/2 inhibitor, exerts anti-leukemic effects under inactive status of PI3Kinase/Akt pathway. Int. J. Hematol. 2019, 110, 213–227. [Google Scholar] [CrossRef]
- Larif, S.; Ben Salem, C.; Hmouda, H.; Bouraoui, K. In silico screening and study of novel ERK2 inhibitors using 3D QSAR, docking and molecular dynamics. J. Mol. Graph. Model. 2014, 53, 1–12. [Google Scholar] [CrossRef]
- Safran, M.; Dalah, I.; Alexander, J.; Rosen, N.; Iny Stein, T.; Shmoish, M.; Nativ, N.; Bahir, I.; Doniger, T.; Krug, H.; et al. GeneCards version 3: The human gene integrator. Database 2010, 2010, baq020. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Lian, X.; Li, F.; Wang, C.; Zhu, F.; Qiu, Y.; Chen, Y. Therapeutic target database update 2022: Facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022, 50, D1398–D1407. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.Org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef]
- Queralt-Rosinach, N.; Piñero, J.; Bravo, À.; Sanz, F.; Furlong, L.I. DisGeNET-RDF: Harnessing the innovative power of the semantic web to explore the genetic basis of diseases. Bioinformatics 2016, 32, 2236–2238. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html. (accessed on 25 July 2022).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. CytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Rstudio Team. RStudio: Integrated Development for R. Available online: http://www.rstudio.com/ (accessed on 7 August 2022).
- Pongrakhananon, V.; Wattanathamsan, O.; Takeichi, M.; Chetprayoon, P.; Chanvorachote, P. Loss of CAMSAP3 promotes EMT via the modification of microtubule-Akt machinery. J. Cell Sci. 2018, 131, jcs216168. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathways | Targets |
|---|---|
| AGE-RAGE signaling pathway in diabetic complications | MAPK1, MMP2, CCND1, MAPK14, CDK4, MAPK3, PIK3CA, MAPK10, PIK3CD, JAK2, MAPK8, MAPK9, AGTR1 |
| Apoptosis | MAPK1, MAPK3, PIK3CA, CTSK, MAP2K1, PDPK1, CTSL, CTSB, MAPK10, PARP1, PIK3CD, MAPK8, MAPK9, CAPN1 |
| Autophagy–animal | MAPK1, MAPK3, PIK3CA, IGF1R, MAP2K1, PDPK1, CTSL, CTSB, MAPK10, MTOR, MAP3K7, PIK3CD, MAPK8, MAPK9 |
| Colorectal cancer | MAPK1, CCND1, MAPK3, PIK3CA, EGFR, MAP2K1, GSK3B, MAPK10, MTOR, PIK3CD, MAPK8, MAPK9 |
| EGFR tyrosine kinase inhibitor resistance | MAPK1, MAPK3, KDR, PIK3CA, IGF1R, ERBB2, EGFR, MAP2K1, GSK3B, MTOR, SRC, PIK3CD, JAK2 |
| Endocrine resistance | MAPK1, MMP2, CCND1, MAPK14, CDK4, MAPK3, PIK3CA, IGF1R, ERBB2, EGFR, MAP2K1, PTK2, MAPK10, MTOR, MMP9, SRC, PIK3CD, MAPK8, MAPK9 |
| ErbB signaling pathway | MAPK1, MAPK3, PIK3CA, ERBB2, EGFR, MAP2K1, GSK3B, PTK2, MAPK10, MTOR, SRC, PIK3CD, MAPK8, MAPK9, PAK4 |
| Fluid shear stress and atherosclerosis | MMP2, PLAT, MAPK14, KDR, PIK3CA, PTK2, CTSL, MAPK10, MAP3K7, MMP9, SRC, PIK3CD, MAPK8, MAPK9 |
| Focal adhesion | MAPK1, CCND1, MAPK3, KDR, PIK3CA, IGF1R, ERBB2, EGFR, MAP2K1, GSK3B, PTK2, PDPK1, MAPK10, MYLK, SRC, PIK3CD, MAPK8, MAPK9, PAK4 |
| FoxO signaling pathway | MAPK1, CCND1, MAPK14, MAPK3, PIK3CA, CDK2, IGF1R, EGFR, MAP2K1, PDPK1, MAPK10, PIK3CD, MAPK8, MAPK9 |
| Growth hormone synthesis, secretion and action | MAPK1, MAPK14, MAPK3, PIK3CA, MAP2K1, GSK3B, PTK2, MAPK10, MTOR, PIK3CD, JAK2, MAPK8, MAPK9 |
| Kaposi sarcoma-associated herpesvirus infection | MAPK1, CCND1, MAPK14, CDK4, MAPK3, PIK3CA, MAP2K1, GSK3B, MAPK10, MTOR, SRC, SYK, PIK3CD, JAK2, MAPK8, MAPK9 |
| MicroRNAs in cancer | MAPK1, CCND1, MAPK3, PIK3CA, ERBB2, EGFR, MAP2K1, MTOR, MMP9, HDAC1, PIK3CD, ABCC1, HDAC2, PAK4, ABCB1 |
| Pancreatic cancer | MAPK1, CCND1, CDK4, MAPK3, PIK3CA, ERBB2, EGFR, MAP2K1, MAPK10, MTOR, PIK3CD, MAPK8, MAPK9 |
| Pathways in cancer | MAPK1, MMP2, CCND1, CDK4, MAPK3, PIK3CA, KNG1, CDK2, IGF1R, ERBB2, EGFR, MAP2K1, F2, GSK3B, PTK2, MAPK10, MTOR, MMP9, HDAC1, PIK3CD, JAK2, MAPK8, MAPK9, AGTR1, HDAC2 |
| PI3K-Akt signaling pathway | MAPK1, CCND1, CDK4, MAPK3, KDR, PIK3CA, CDK2, IGF1R, ERBB2, EGFR, MAP2K1, GSK3B, PTK2, PDPK1, MTOR, SYK, PIK3CD, JAK2 |
| Progesterone-mediated oocyte maturation | MAPK1, AURKA, MAPK14, MAPK3, PIK3CA, CDK2, IGF1R, MAP2K1, MAPK10, PIK3CD, RPS6KA3, CDK1, MAPK8, MAPK9 |
| Prolactin signaling pathway | MAPK1, CCND1, MAPK14, MAPK3, PIK3CA, MAP2K1, GSK3B, MAPK10, SRC, PIK3CD, JAK2, MAPK8, MAPK9 |
| Prostate cancer | MAPK1, PLAT, CCND1, MAPK3, PIK3CA, CDK2, IGF1R, ERBB2, EGFR, MMP3, MAP2K1, GSK3B, PDPK1, MTOR, MMP9, PIK3CD |
| Proteoglycans in cancer | MAPK1, MMP2, CCND1, MAPK14, MAPK3, KDR, PIK3CA, IGF1R, ERBB2, EGFR, MAP2K1, PTK2, PDPK1, CTSL, MTOR, MMP9, SRC, PIK3CD |
| Relaxin signaling pathway | MAPK1, MMP2, MAPK14, MMP13, MAPK3, PIK3CA, EGFR, MAP2K1, MAPK10, MMP9, SRC, PIK3CD, MAPK8, MAPK9 |
| Sphingolipid signaling pathway | MAPK1, MAPK14, MAPK3, PIK3CA, KNG1, MAP2K1, PDPK1, MAPK10, ADORA1, PIK3CD, MAPK8, ABCC1, MAPK9 |
| T-cell receptor signaling pathway | MAPK1, MAPK14, CDK4, MAPK3, PIK3CA, MAP2K1, GSK3B, PDPK1, MAPK10, MAP3K7, PIK3CD, MAPK8, MAPK9, PAK4 |
| Viral carcinogenesis | MAPK1, CCND1, CDK4, MAPK3, PIK3CA, CDK2, HDAC11, HDAC3, HDAC6, HDAC1, HDAC8, SRC, SYK, PIK3CD, CDK1, HDAC2 |
| Yersinia infection | MAPK1, MAPK14, MAPK3, PIK3CA, MAP2K1, GSK3B, PTK2, MAPK10, MAP3K7, SRC, PIK3CD, RPS6KA3, MAPK8, MAPK9 |
| Targets | PDB | Ligand Efficiency (kcal/mol per Heavy Atom) | Binding Energy (kcal/mol) | Interacting Position of Targets | |||
|---|---|---|---|---|---|---|---|
| H-Bonding | van der Waals | Hydro-Phobic | Electro-Static | ||||
| ERK1 (MAPK3) | 6GES | −0.225 | −9.9 | ASN171 | ILE48, GLY49, GLU50, GLY51, LYS71, GLN122, LEU124, MET125, LYS168, PRO169, ASN171, LEU172 | VAL56 ALA69 ILE101 TYR130 LEU173 CYS183 | ASP128 |
| ERK2 (MAPK1) | 1WZY | −0.2 | −8.8 | LYS55 GLU71 ILE103 GLU109 LYS114 SER153 | ALA52, ILE53, ILE56, LEU75, ILE84, LEU107, THR110, LEU156, ASN154 | ILE31 VAL39 CYS166 | ASP111 |
| MEK1 (MAP2K1) | 7PQV | −0.207 | −9.1 | GLN46 TYR125 GLU203 PHE371 ALA372 | ARG49, LYS168, ILE171, ALA172, LYS175, GLY202, VAL369ASP370 | PRO124 | ASP43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iksen, I.; Sinsook, S.; Wattanathamsan, O.; Buaban, K.; Chamni, S.; Pongrakhananon, V. Target Identification of 22-(4-Pyridinecarbonyl) Jorunnamycin A, a Tetrahydroisoquinoline Derivative from the Sponge Xestospongia sp., in Mediating Non-Small-Cell Lung Cancer Cell Apoptosis. Molecules 2022, 27, 8948. https://doi.org/10.3390/molecules27248948
Iksen I, Sinsook S, Wattanathamsan O, Buaban K, Chamni S, Pongrakhananon V. Target Identification of 22-(4-Pyridinecarbonyl) Jorunnamycin A, a Tetrahydroisoquinoline Derivative from the Sponge Xestospongia sp., in Mediating Non-Small-Cell Lung Cancer Cell Apoptosis. Molecules. 2022; 27(24):8948. https://doi.org/10.3390/molecules27248948
Chicago/Turabian StyleIksen, Iksen, Suwimon Sinsook, Onsurang Wattanathamsan, Koonchira Buaban, Supakarn Chamni, and Varisa Pongrakhananon. 2022. "Target Identification of 22-(4-Pyridinecarbonyl) Jorunnamycin A, a Tetrahydroisoquinoline Derivative from the Sponge Xestospongia sp., in Mediating Non-Small-Cell Lung Cancer Cell Apoptosis" Molecules 27, no. 24: 8948. https://doi.org/10.3390/molecules27248948
APA StyleIksen, I., Sinsook, S., Wattanathamsan, O., Buaban, K., Chamni, S., & Pongrakhananon, V. (2022). Target Identification of 22-(4-Pyridinecarbonyl) Jorunnamycin A, a Tetrahydroisoquinoline Derivative from the Sponge Xestospongia sp., in Mediating Non-Small-Cell Lung Cancer Cell Apoptosis. Molecules, 27(24), 8948. https://doi.org/10.3390/molecules27248948