




A Novel Paclitaxel Derivative for Triple-Negative Breast Cancer Chemotherapy


Abstract
1. Introduction
2. Results
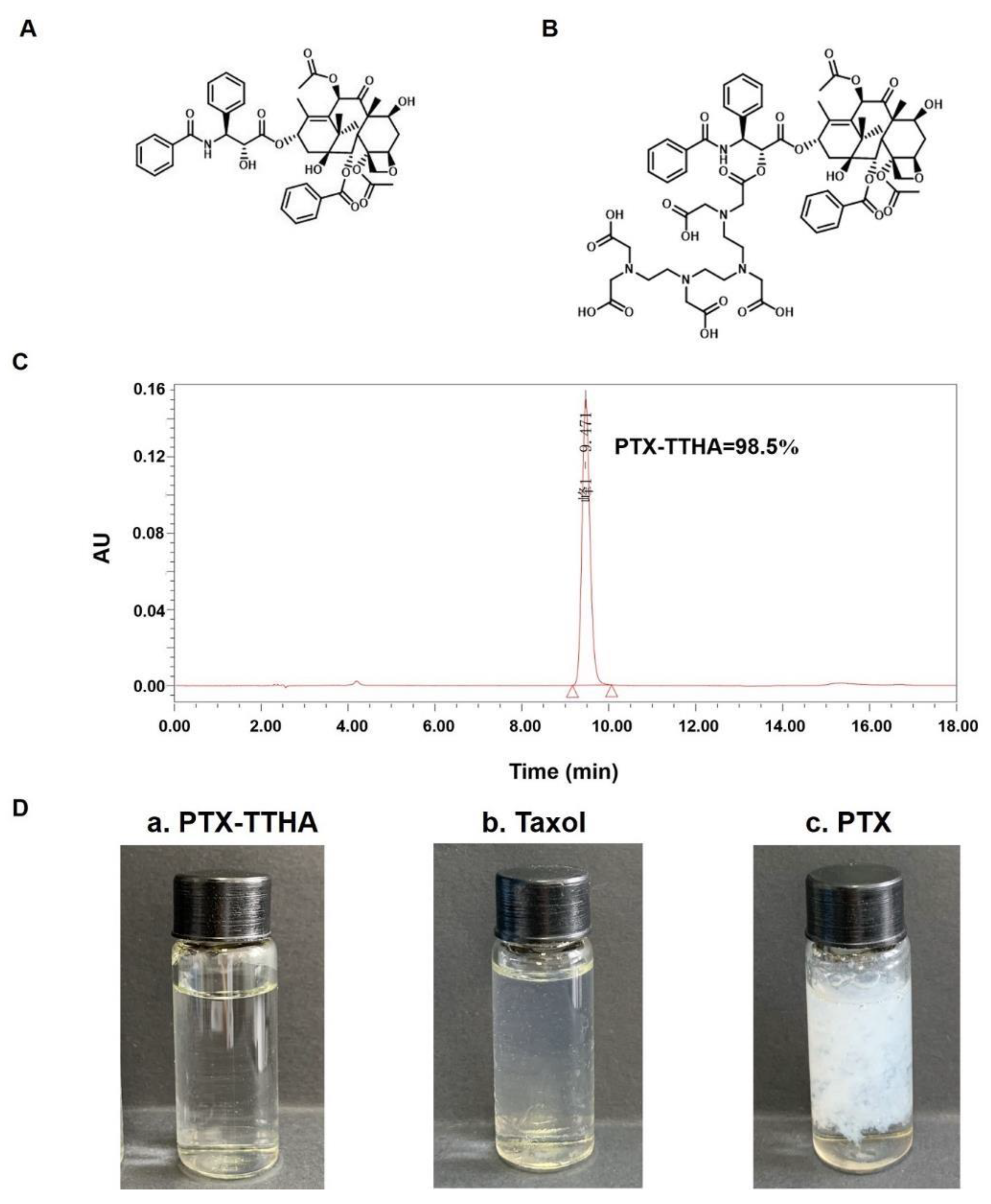
2.1. Preparation and Purity of PTX-TTHA
2.2. Improved Solubility of PTX-TTHA
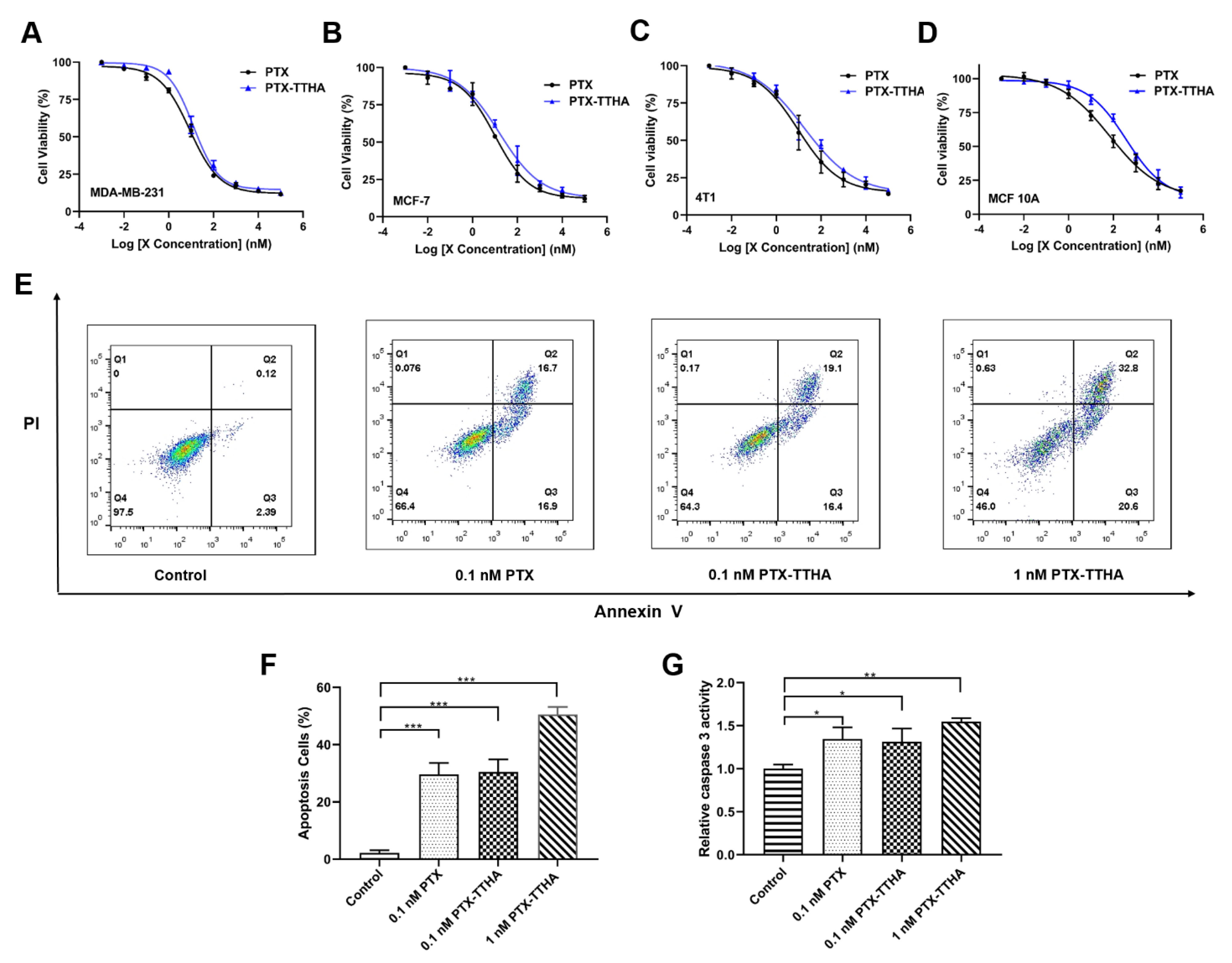
2.3. PTX-TTHA Inhibited the Proliferation of Breast Cancer Cells
2.4. PTX-TTHA Induced Cell Apoptosis in the MDA-MB-231 TNBC Cell Line
2.5. PTX-TTHA Induced Morphological Changes of Apoptosis in the MDA-MB-231 TNBC Cell Line
2.6. PTX-TTHA Stabilized Microtubules and Arrested the Cell Cycle in the G2/M Phase in MDA-MB-231 Cells
2.7. PTX-TTHA Reduced Tumor Size in the MDA-MB-231 Xenograft Breast Tumor Model
2.8. PTX-TTHA Induced Apoptosis in the MDA-MB-231 Xenograft Breast Tumor Model
2.9. PTX-TTHA Inhibited Ki-67 Expression in Tumor Tissues
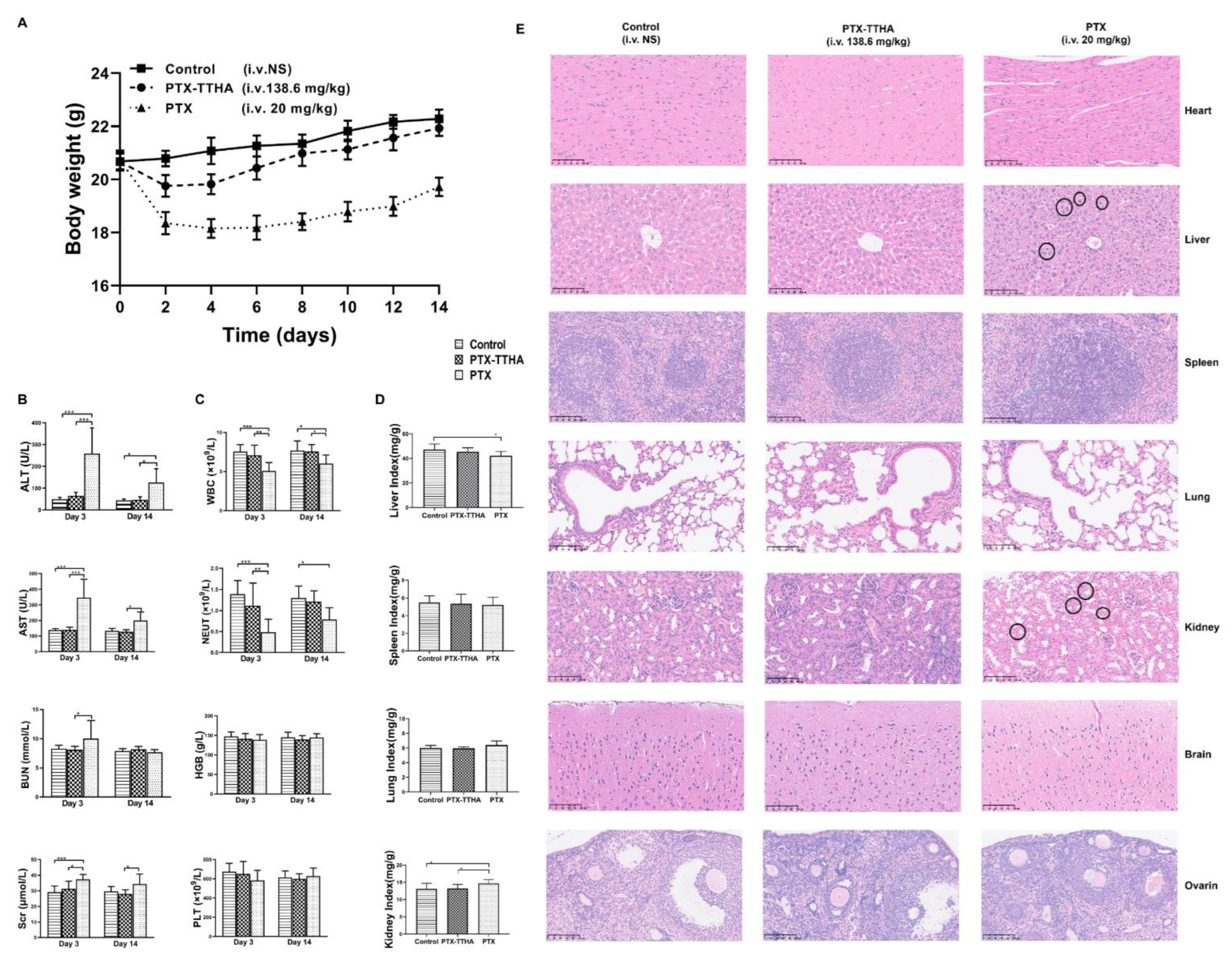
2.10. PTX-TTHA Reduced In Vivo Toxicity Compared with PTX
3. Discussion
4. Materials and Methods
4.1. Preparation of PTX-TTHA
4.2. Solubility Experiment
4.3. Cell Culture
4.4. MTT Assay [73]
4.5. Annexin V-FITC/PI and Flow Cytometry Assay
4.6. Caspase 3 Activity Assay
4.7. Transmission Electron Microscopy [76]
4.8. Immunofluorescence Staining
4.9. Cell Cycle Analysis
4.10. Animals
4.11. In Vivo Antitumor Assay
4.12. Western Blot Analysis [80]
4.13. Hematoxylin and Eosin (H&E) Staining [81]
4.14. Ki-67 Immunohistochemical Staining
4.15. TdT-Mediated dUTP Nick End Labeling Assay (TUNEL) Assay
4.16. Acute-Toxicity Experiment
4.17. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Fisusi, F.A.; Akala, E.O. Drug combinations in breast cancer therapy. Pharm. Nanotechnol. 2019, 7, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Howard, F.M.; Olopade, O.I. Epidemiology of triple-negative breast cancer: A review. Cancer J. 2021, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Yan, H.; Luo, B.; Wu, X.; Guan, F.; Yu, X.; Zhao, L.; Ke, X.; Wu, J.; Yuan, J. Cisplatin induces pyroptosis via activation of meg3/nlrp3/caspase-1/gsdmd pathway in triple-negative breast cancer. Int. J. Biol. Sci. 2021, 17, 2606–2621. [Google Scholar] [CrossRef]
- Weaver, B.A. How taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Abu, S.T.; Samec, M.; Liskova, A.; Kubatka, P.; Busselberg, D. Paclitaxel’s mechanistic and clinical effects on breast cancer. Biomolecules 2019, 9, 798. [Google Scholar]
- Gao, Y.; Nai, J.; Yang, Z.; Zhang, J.; Ma, S.; Zhao, Y.; Li, H.; Li, J.; Yang, Y.; Yang, M.; et al. A novel preparative method for nanoparticle albumin-bound paclitaxel with high drug loading and its evaluation both in vitro and in vivo. PLoS ONE 2021, 16, e250670. [Google Scholar] [CrossRef]
- Bernabeu, E.; Gonzalez, L.; Cagel, M.; Gergic, E.P.; Moretton, M.A.; Chiappetta, D.A. Novel soluplus((r))-tpgs mixed micelles for encapsulation of paclitaxel with enhanced in vitro cytotoxicity on breast and ovarian cancer cell lines. Colloids Surf. B Biointerfaces 2016, 140, 403–411. [Google Scholar] [CrossRef]
- Meng, Z.; Lv, Q.; Lu, J.; Yao, H.; Lv, X.; Jiang, F.; Lu, A.; Zhang, G. Prodrug strategies for paclitaxel. Int. J. Mol. Sci. 2016, 17, 796. [Google Scholar] [CrossRef]
- Bernabeu, E.; Cagel, M.; Lagomarsino, E.; Moretton, M.; Chiappetta, D.A. Paclitaxel: What has been done and the challenges remain ahead. Int. J. Pharm. 2017, 526, 474–495. [Google Scholar] [CrossRef]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor el: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Gradishar, W.J. Albumin-bound paclitaxel: A next-generation taxane. Expert Opin. Pharmacother. 2006, 7, 1041–1053. [Google Scholar] [CrossRef]
- Kim, S.C.; Yoon, H.J.; Lee, J.W.; Yu, J.; Park, E.S.; Chi, S.C. Investigation of the release behavior of dehp from infusion sets by paclitaxel-loaded polymeric micelles. Int. J. Pharm. 2005, 293, 303–310. [Google Scholar] [CrossRef]
- Park, J.H.; Chi, S.C.; Lee, W.S.; Lee, W.M.; Koo, Y.B.; Yong, C.S.; Choi, H.G.; Woo, J.S. Toxicity studies of cremophor-free paclitaxel solid dispersion formulated by a supercritical antisolvent process. Arch. Pharm. Res. 2009, 32, 139–148. [Google Scholar] [CrossRef]
- Nehate, C.; Jain, S.; Saneja, A.; Khare, V.; Alam, N.; Dubey, R.D.; Gupta, P.N. Paclitaxel formulations: Challenges and novel delivery options. Curr. Drug Deliv. 2014, 11, 666–686. [Google Scholar] [CrossRef]
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef]
- Zhang, Z.; Mei, L.; Feng, S.S. Paclitaxel drug delivery systems. Expert Opin. Drug Deliv. 2013, 10, 325–340. [Google Scholar] [CrossRef]
- Marupudi, N.I.; Han, J.E.; Li, K.W.; Renard, V.M.; Tyler, B.M.; Brem, H. Paclitaxel: A review of adverse toxicities and novel delivery strategies. Expert Opin. Drug Saf. 2007, 6, 609–621. [Google Scholar] [CrossRef]
- Liu, K.K.; Zheng, W.W.; Wang, C.C.; Chiu, Y.C.; Cheng, C.L.; Lo, Y.S.; Chen, C.; Chao, J.I. Covalent linkage of nanodiamond-paclitaxel for drug delivery and cancer therapy. Nanotechnology 2010, 21, 315106. [Google Scholar] [CrossRef] [PubMed]
- Cserhati, T.; Forgacs, E.; Hollo, J. Interaction of taxol and other anticancer drugs with alpha-cyclodextrin. J. Pharm. Biomed. Anal. 1995, 13, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, F.; Song, X.; Wu, J.; Yao, W.; Gao, X. Conjugation of paclitaxel to c-6 hexanediamine-modified hyaluronic acid for targeted drug delivery to enhance antitumor efficacy. Carbohydr. Polym. 2018, 181, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Thapa, P.; Li, M.; Bio, M.; Rajaputra, P.; Nkepang, G.; Sun, Y.; Woo, S.; You, Y. Far-red light-activatable prodrug of paclitaxel for the combined effects of photodynamic therapy and site-specific paclitaxel chemotherapy. J. Med. Chem. 2016, 59, 3204–3214. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lu, J.; Liu, J.; Liang, C.; Wang, M.; Wang, L.; Li, D.; Yao, H.; Zhang, Q.; Wen, J.; et al. A water-soluble nucleolin aptamer-paclitaxel conjugate for tumor-specific targeting in ovarian cancer. Nat. Commun. 2017, 8, 1390. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Yoshii, Y.; Matsumoto, H.; Shinada, M.; Takahashi, M.; Igarashi, C.; Hihara, F.; Tachibana, T.; Doi, A.; Higashi, T.; et al. Evaluation of aminopolycarboxylate chelators for whole-body clearance of free 225Ac: A feasibility study to reduce unexpected radiation exposure during targeted alpha therapy. Pharmaceutics 2021, 13, 1706. [Google Scholar] [CrossRef]
- Liu, S.; Edwards, D.S. Bifunctional chelators for therapeutic lanthanide radiopharmaceuticals. Bioconjug. Chem. 2001, 12, 7–34. [Google Scholar] [CrossRef]
- Chong, H.S.; Song, H.A.; Ma, X.; Lim, S.; Sun, X.; Mhaske, S.B. Bile acid-based polyaminocarboxylate conjugates as targeted antitumor agents. Chem. Commun. 2009, 21, 3011–3013. [Google Scholar] [CrossRef]
- Brard, L.; Granai, C.O.; Swamy, N. Iron chelators deferoxamine and diethylenetriamine pentaacetic acid induce apoptosis in ovarian carcinoma. Gynecol. Oncol. 2006, 100, 116–127. [Google Scholar] [CrossRef]
- Kucharzewski, M.; Braziewicz, J.; Majewska, U.; Gozdz, S. Iron concentrations in intestinal cancer tissue and in colon and rectum polyps. Biol. Trace Elem. Res. 2003, 95, 19–28. [Google Scholar] [CrossRef]
- Faulk, W.P.; Hsi, B.L.; Stevens, P.J. Transferrin and transferrin receptors in carcinoma of the breast. Lancet 1980, 2, 390–392. [Google Scholar] [CrossRef]
- Ozcan, A.G. Cytoprotective effects of amifostine and cysteamine on cultured normal and tumor cells treated with paclitaxel in terms of mitotic index and 3h-thymidine labeling index. Cancer Chemother. Pharmacol. 2005, 56, 221–229. [Google Scholar] [CrossRef]
- Matsuoka, H.; Furusawa, M.; Tomoda, H.; Seo, Y. Difference in cytotoxicity of paclitaxel against neoplastic and normal cells. Anticancer Res. 1994, 14, 163–167. [Google Scholar]
- Cordes, N.; Plasswilm, L.; Sauer, R. Interaction of paclitaxel (taxol) and irradiation. In-vitro differences between tumor and fibroblastic cells. Strahlenther. Onkol. 1999, 175, 175–181. [Google Scholar] [CrossRef]
- Jiang, S.; Song, M.J.; Shin, E.C.; Lee, M.O.; Kim, S.J.; Park, J.H. Apoptosis in human hepatoma cell lines by chemotherapeutic drugs via fas-dependent and fas-independent pathways. Hepatology 1999, 29, 101–110. [Google Scholar] [CrossRef]
- de Bruin, E.C.; Meersma, D.; de Wilde, J.; den Otter, I.; Schipper, E.M.; Medema, J.P.; Peltenburg, L.T. A serine protease is involved in the initiation of dna damage-induced apoptosis. Cell Death Differ. 2003, 10, 1204–1212. [Google Scholar] [CrossRef]
- Xia, H.; Huang, Y.; Zhang, L.; Luo, L.; Wang, X.; Lu, Q.; Xu, J.; Yang, C.; Jiwa, H.; Liang, S.; et al. Inhibition of macropinocytosis enhances the sensitivity of osteosarcoma cells to benzethonium chloride. Cancers 2023, 15, 961. [Google Scholar] [CrossRef]
- Yin, J.; Cai, Z.; Zhang, L.; Zhang, J.; He, X.; Du, X.; Wang, Q.; Lu, J. A recombined fusion protein ptd-grb2-sh2 inhibits the proliferation of breast cancer cells in vitro. Int. J. Oncol. 2013, 42, 1061–1069. [Google Scholar] [CrossRef][Green Version]
- Chowdhury, P.; Nagesh, P.; Khan, S.; Hafeez, B.B.; Chauhan, S.C.; Jaggi, M.; Yallapu, M.M. Development of polyvinylpyrrolidone/paclitaxel self-assemblies for breast cancer. Acta Pharm. Sin. B 2018, 8, 602–614. [Google Scholar] [CrossRef]
- Horwitz, S.B. Taxol (paclitaxel): Mechanisms of action. Ann. Oncol. 1994, 5 (Suppl. 6), S3–S6. [Google Scholar]
- Wang, T.H.; Wang, H.S.; Soong, Y.K. Paclitaxel-induced cell death: Where the cell cycle and apoptosis come together. Cancer-Am. Cancer Soc. 2000, 88, 2619–2628. [Google Scholar] [CrossRef]
- Liu, Q.; Hirohashi, Y.; Du, X.; Greene, M.I.; Wang, Q. Nek2 targets the mitotic checkpoint proteins mad2 and cdc20: A mechanism for aneuploidy in cancer. Exp. Mol. Pathol. 2010, 88, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Cui, Z.; Zhu, X.; Gao, Q.; Xiong, B. Supplementation of nicotinamide mononucleotide improves the quality of postovulatory aged porcine oocytes. J. Mol. Cell Biol. 2022, 14, mjac025. [Google Scholar] [CrossRef] [PubMed]
- Sivakumaran, N.; Samarakoon, S.R.; Adhikari, A.; Ediriweera, M.K.; Tennekoon, K.H.; Malavige, N.; Thabrew, I.; Shrestha, R. Cytotoxic and apoptotic effects of govaniadine isolated from corydalis govaniana wall. Roots on human breast cancer (mcf-7) cells. Biomed Res. Int. 2018, 2018, 3171348. [Google Scholar] [CrossRef] [PubMed]
- Pagliara, P.; Carla, E.C.; Caforio, S.; Chionna, A.; Massa, S.; Abbro, L.; Dini, L. Kupffer cells promote lead nitrate-induced hepatocyte apoptosis via oxidative stress. Comp. Hepatol. 2003, 2, 8. [Google Scholar] [CrossRef][Green Version]
- Pan, G.; Mao, A.; Liu, J.; Lu, J.; Ding, J.; Liu, W. Circular rna hsa_circ_0061825 (circ-tff1) contributes to breast cancer progression through targeting mir-326/tff1 signalling. Cell Prolif. 2020, 53, e12720. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, S.Z.; Zhang, X.; Jia, H.R.; Zhu, Y.X.; Zhang, X.; Gao, G.; Jiang, Y.W.; Li, C.; Chen, X.; et al. In situ generation of micrometer-sized tumor cell-derived vesicles as autologous cancer vaccines for boosting systemic immune responses. Nat. Commun. 2022, 13, 6534. [Google Scholar] [CrossRef]
- Yerushalmi, R.; Woods, R.; Ravdin, P.M.; Hayes, M.M.; Gelmon, K.A. Ki67 in breast cancer: Prognostic and predictive potential. Lancet Oncol. 2010, 11, 174–183. [Google Scholar] [CrossRef]
- Venkatasubbu, G.D.; Ramasamy, S.; Gaddam, P.R.; Kumar, J. Acute and subchronic toxicity analysis of surface modified paclitaxel attached hydroxyapatite and titanium dioxide nanoparticles. Int. J. Nanomed. 2015, 10 (Suppl. 1), 137–148. [Google Scholar] [CrossRef]
- Mathew, A.E.; Mejillano, M.R.; Nath, J.P.; Himes, R.H.; Stella, V.J. Synthesis and evaluation of some water-soluble prodrugs and derivatives of taxol with antitumor activity. J. Med. Chem. 1992, 35, 145–151. [Google Scholar] [CrossRef]
- Liu, C.; Strobl, J.S.; Bane, S.; Schilling, J.K.; Mccracken, M.; Chatterjee, S.K.; Rahim-Bata, R.; Kingston, D.G. Design, synthesis, and bioactivities of steroid-linked taxol analogues as potential targeted drugs for prostate and breast cancer. J. Nat. Prod. 2004, 67, 152–159. [Google Scholar] [CrossRef]
- Ali, S.M.; Hoemann, M.Z.; Aube, J.; Mitscher, L.A.; Georg, G.I.; Mccall, R.; Jayasinghe, L.R. Novel cytotoxic 3′-(tert-butyl) 3′-dephenyl analogs of paclitaxel and docetaxel. J. Med. Chem. 1995, 38, 3821–3828. [Google Scholar] [CrossRef]
- Greenwald, R.B.; Pendri, A.; Bolikal, D. Highly water soluble taxol derivatives: 7-polyethylene glycol carbamates and carbonates. J. Org. Chem. 1995, 60, 331–336. [Google Scholar] [CrossRef]
- Robledo-Cadena, D.X.; Gallardo-Perez, J.C.; Davila-Borja, V.; Pacheco-Velazquez, S.C.; Belmont-Diaz, J.A.; Ralph, S.J.; Blanco-Carpintero, B.A.; Moreno-Sanchez, R.; Rodriguez-Enriquez, S. Non-steroidal anti-inflammatory drugs increase cisplatin, paclitaxel, and doxorubicin efficacy against human cervix cancer cells. Pharmaceuticals 2020, 13, 463. [Google Scholar] [CrossRef]
- Wei, W. Experimental Methodology of Pharmacology (Version 4); People’s Medical Publishing House(PMPH): Beijing, China, 2010. [Google Scholar]
- Li, J.; Zeng, L.; Wang, Z.; Chen, H.; Fang, S.; Wang, J.; Cai, C.Y.; Xing, E.; Liao, X.; Li, Z.W.; et al. Cycloruthenated self-assembly with metabolic inhibition to efficiently overcome multidrug resistance in cancers. Adv. Mater. 2022, 34, e2100245. [Google Scholar] [CrossRef]
- Inoue, T.; Li, C.; Yang, D.J.; Higuchi, T.; Oriuchi, N.; Yu, D.; Milas, L.; Hunter, N.; Wallace, S.; Kim, E.E.; et al. Evaluation of in-111 dtpa-paclitaxel scintigraphy to predict response on murine tumors to paclitaxel. Ann. Nucl. Med. 1999, 13, 169–174. [Google Scholar] [CrossRef]
- Li, C.; Yu, D.; Inoue, T.; Yang, D.J.; Tansey, W.; Liu, C.; Milas, L.; Hunter, N.R.; Kim, E.E.; Wallace, S. Synthesis, biodistribution and imaging properties of indium-111-dtpa-paclitaxel in mice bearing mammary tumors. J. Nucl. Med. 1997, 38, 1042–1047. [Google Scholar]
- Green, M.C.; Buzdar, A.U.; Smith, T.; Ibrahim, N.K.; Valero, V.; Rosales, M.F.; Cristofanilli, M.; Booser, D.J.; Pusztai, L.; Rivera, E.; et al. Weekly paclitaxel improves pathologic complete remission in operable breast cancer when compared with paclitaxel once every 3 weeks. J. Clin. Oncol. 2005, 23, 5983–5992. [Google Scholar] [CrossRef]
- Shetti, D.; Zhang, B.; Fan, C.; Mo, C.; Lee, B.H.; Wei, K. Low dose of paclitaxel combined with xav939 attenuates metastasis, angiogenesis and growth in breast cancer by suppressing wnt signaling. Cells 2019, 8, 892. [Google Scholar] [CrossRef]
- Liang, L.; Lin, S.W.; Dai, W.; Lu, J.K.; Yang, T.Y.; Xiang, Y.; Zhang, Y.; Li, R.T.; Zhang, Q. Novel cathepsin b-sensitive paclitaxel conjugate: Higher water solubility, better efficacy and lower toxicity. J. Control. Release 2012, 160, 618–629. [Google Scholar] [CrossRef]
- Huang, S.; Wang, D.; Zhang, S.; Huang, X.; Wang, D.; Ijaz, M.; Shi, Y. Tunicamycin potentiates paclitaxel-induced apoptosis through inhibition of pi3k/akt and mapk pathways in breast cancer. Cancer Chemother Pharmacol 2017, 80, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, B.; Zhang, X.; Ye, L. The oncoprotein hbxip promotes migration of breast cancer cells via gcn5-mediated microtubule acetylation. Biochem. Biophys. Res. Commun. 2015, 458, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Torres, K.; Horwitz, S.B. Mechanisms of taxol-induced cell death are concentration dependent. Cancer Res. 1998, 58, 3620–3626. [Google Scholar] [PubMed]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef]
- Bai, Z.; Ding, N.; Ge, J.; Wang, Y.; Wang, L.; Wu, N.; Wei, Q.; Xu, S.; Liu, X.; Zhou, G. Esomeprazole overcomes paclitaxel-resistance and enhances anticancer effects of paclitaxel by inducing autophagy in a549/taxol cells. Cell Biol. Int. 2021, 45, 177–187. [Google Scholar] [CrossRef]
- Sampath, D.; Greenberger, L.M.; Beyer, C.; Hari, M.; Liu, H.; Baxter, M.; Yang, S.; Rios, C.; Discafani, C. Preclinical pharmacologic evaluation of mst-997, an orally active taxane with superior in vitro and in vivo efficacy in paclitaxel- and docetaxel-resistant tumor models. Clin. Cancer Res. 2006, 12, 3459–3469. [Google Scholar] [CrossRef]
- Gallego-Jara, J.; Lozano-Terol, G.; Sola-Martinez, R.A.; Canovas-Diaz, M.; de Diego, P.T. A compressive review about taxol((r)): History and future challenges. Molecules 2020, 25, 5986. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Targeting apoptosis pathways in cancer therapy. Curr. Cancer Drug Targets 2004, 4, 569–576. [Google Scholar] [CrossRef]
- Su, J.; Wang, J.; Luo, J.; Li, H. Ultrasound-mediated destruction of vascular endothelial growth factor (vegf) targeted and paclitaxel loaded microbubbles for inhibition of human breast cancer cell mcf-7 proliferation. Mol. Cell. Probes 2019, 46, 101415. [Google Scholar] [CrossRef]
- Liu, T.J. Preparation Method and Application of a Water-Soluble Paclitaxel Compound. CN102702140A, 3 October 2012. [Google Scholar]
- Xu, Y.; Chen, W.; Liang, J.; Zeng, X.; Ji, K.; Zhou, J.; Liao, S.; Wu, J.; Xing, K.; He, Z.; et al. The mir-1185-2-3p-golph3l pathway promotes glucose metabolism in breast cancer by stabilizing p53-induced serpine1. J. Exp. Clin. Cancer Res. 2021, 40, 47. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, Y.; Zeng, H.; Wang, C.; Xu, C.; Wang, Q.; Wang, H.; Li, S.; Chen, J.; Xiao, C.; et al. Mechano-boosting nanomedicine antitumour efficacy by blocking the reticuloendothelial system with stiff nanogels. Nat. Commun. 2023, 14, 1437. [Google Scholar] [CrossRef]
- Wu, W.; Yang, J.; Feng, X.; Wang, H.; Ye, S.; Yang, P.; Tan, W.; Wei, G.; Zhou, Y. Microrna-32 (mir-32) regulates phosphatase and tensin homologue (pten) expression and promotes growth, migration, and invasion in colorectal carcinoma cells. Mol. Cancer 2013, 12, 30. [Google Scholar] [CrossRef]
- Gao, W.; Ji, L.; Li, L.; Cui, G.; Xu, K.; Li, P.; Tang, B. Bifunctional combined au-fe(2)o(3) nanoparticles for induction of cancer cell-specific apoptosis and real-time imaging. Biomaterials 2012, 33, 3710–3718. [Google Scholar] [CrossRef]
- Huang, S.; Tang, Y.; Liu, T.; Zhang, N.; Yang, X.; Yang, D.; Hong, G. A novel antioxidant protects against contrast medium-induced acute kidney injury in rats. Front. Pharmacol. 2020, 11, 599577. [Google Scholar] [CrossRef]
- Luo, W.; Xu, Y.; Liu, R.; Liao, Y.; Wang, S.; Zhang, H.; Li, X.; Wang, H. Retinoic acid and rargamma maintain satellite cell quiescence through regulation of translation initiation. Cell Death Dis. 2022, 13, 838. [Google Scholar] [CrossRef]
- Wang, Z.; Ho, P.C. A nanocapsular combinatorial sequential drug delivery system for antiangiogenesis and anticancer activities. Biomaterials 2010, 31, 7115–7123. [Google Scholar] [CrossRef]
- Zhou, S.; Huang, Y.; Chen, Y.; Liu, Y.; Xie, L.; You, Y.; Tong, S.; Xu, J.; Jiang, G.; Song, Q.; et al. Reprogramming systemic and local immune function to empower immunotherapy against glioblastoma. Nat. Commun. 2023, 14, 435. [Google Scholar] [CrossRef]
- Zhou, B.; Ge, Y.; Shao, Q.; Yang, L.; Chen, X.; Jiang, G. Long noncoding rna linc00284 facilitates cell proliferation in papillary thyroid cancer via impairing mir-3127-5p targeted e2f7 suppression. Cell Death Discov. 2021, 7, 156. [Google Scholar] [CrossRef]
- Hing, Z.A.; Walker, J.S.; Whipp, E.C.; Brinton, L.; Cannon, M.; Zhang, P.; Sher, S.; Cempre, C.B.; Brown, F.; Smith, P.L.; et al. Dysregulation of prmt5 in chronic lymphocytic leukemia promotes progression with high risk of richter’s transformation. Nat. Commun. 2023, 14, 97. [Google Scholar] [CrossRef]
- Lin, Y.; Lin, F.; Zhang, Z.; Peng, L.; Yang, W.; Yang, M.; Luo, B.; Wu, T.; Li, D.; Li, X.; et al. The fgfr1 signaling pathway upregulates the oncogenic transcription factor foxq1 to promote breast cancer cell growth. Int. J. Biol. Sci. 2023, 19, 744–759. [Google Scholar] [CrossRef]
- Gajjala, P.R.; Kasam, R.K.; Soundararajan, D.; Sinner, D.; Huang, S.K.; Jegga, A.G.; Madala, S.K. Dysregulated overexpression of sox9 induces fibroblast activation in pulmonary fibrosis. JCI Insight 2021, 6, e152503. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, G.; Kang, J.; Niu, M.; Wang, Z.; Wang, H.; Ma, J.; Wang, X. Paclitaxel nanosuspensions coated with p-gp inhibitory surfactants: I. Acute toxicity and pharmacokinetics studies. Colloids Surf. B Biointerfaces 2013, 111, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Oda, C.; Silva, J.O.; Fernandes, R.S.; Braga, A.V.; Machado, R.R.; Coelho, M.M.; Cassali, G.D.; Reis, D.C.; de Barros, A.; Leite, E.A. Encapsulating paclitaxel in polymeric nanomicelles increases antitumor activity and prevents peripheral neuropathy. Biomed. Pharmacother. 2020, 132, 110864. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Qu, Y.; Huang, Y.; Zhang, L.; Chen, X.; Long, C.; He, Y.; Ou, C.; Qian, Z. Peg-derivatized octacosanol as micellar carrier for paclitaxel delivery. Int. J. Pharm. 2016, 500, 345–359. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MDA-MB-231 | MCF-7 | 4T1 | MCF 10A | |
|---|---|---|---|---|
| IC50 PTX (nM) | 8.68 ± 0.75 | 9.99 ± 0.94 | 9.45 ± 1.34 | 69.40 ± 2.48 |
| SIPTX | 7.99 | 6.94 | 7.34 | |
| IC50 PTX-TTHA(nM) | 12.67 ± 1.34 | 15.83 ± 1.04 | 17.19 ± 1.96 | 367.10 ± 8.20 |
| SIPTX-TTHA | 28.97 | 23.19 | 21.36 |
| Heart (mg/g) | Liver (mg/g) | Spleen (mg/g) | Lung (mg/g) | Kidney (mg/g) | |
|---|---|---|---|---|---|
| Ctrl (n = 6) | 5.64 ± 0.53 | 63.66 ± 2.80 | 6.26 ± 0.58 | 7.22 ± 0.50 | 16.10 ± 0.19 |
| PTX (n = 6) | 5.60 ± 0.61 | 59.89 ± 1.67 * | 5.78 ± 0.42 | 7.53 ± 0.29 | 15.89 ± 0.21 |
| LPTX-TTHA (n = 6) | 5.56 ± 0.63 | 63.63 ± 4.54 | 6.35 ± 0.51 | 7.29 ± 0.79 | 16.03 ± 0.49 |
| MPTX-TTHA (n = 6) | 5.49 ± 0.15 | 63.20 ± 5.17 | 6.16 ± 1.05 | 7.16 ± 0.61 | 16.05 ± 0.47 |
| HPTX-TTHA (n = 6) | 5.56 ± 0.49 | 63.73 ± 5.63 | 6.20 ± 0.65 | 7.28 ± 0.73 | 16.09 ± 0.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Hong, G.; Mao, L.; Su, Z.; Liu, T.; Liu, H. A Novel Paclitaxel Derivative for Triple-Negative Breast Cancer Chemotherapy. Molecules 2023, 28, 3662. https://doi.org/10.3390/molecules28093662
Liu Y, Hong G, Mao L, Su Z, Liu T, Liu H. A Novel Paclitaxel Derivative for Triple-Negative Breast Cancer Chemotherapy. Molecules. 2023; 28(9):3662. https://doi.org/10.3390/molecules28093662
Chicago/Turabian StyleLiu, Yuetong, Ge Hong, Lina Mao, Zhe Su, Tianjun Liu, and Hong Liu. 2023. "A Novel Paclitaxel Derivative for Triple-Negative Breast Cancer Chemotherapy" Molecules 28, no. 9: 3662. https://doi.org/10.3390/molecules28093662
APA StyleLiu, Y., Hong, G., Mao, L., Su, Z., Liu, T., & Liu, H. (2023). A Novel Paclitaxel Derivative for Triple-Negative Breast Cancer Chemotherapy. Molecules, 28(9), 3662. https://doi.org/10.3390/molecules28093662