

New Benzofuran Oligomers from the Roots of Eupatorium heterophyllum Collected in China


Abstract

1. Introduction
2. Results and Discussion
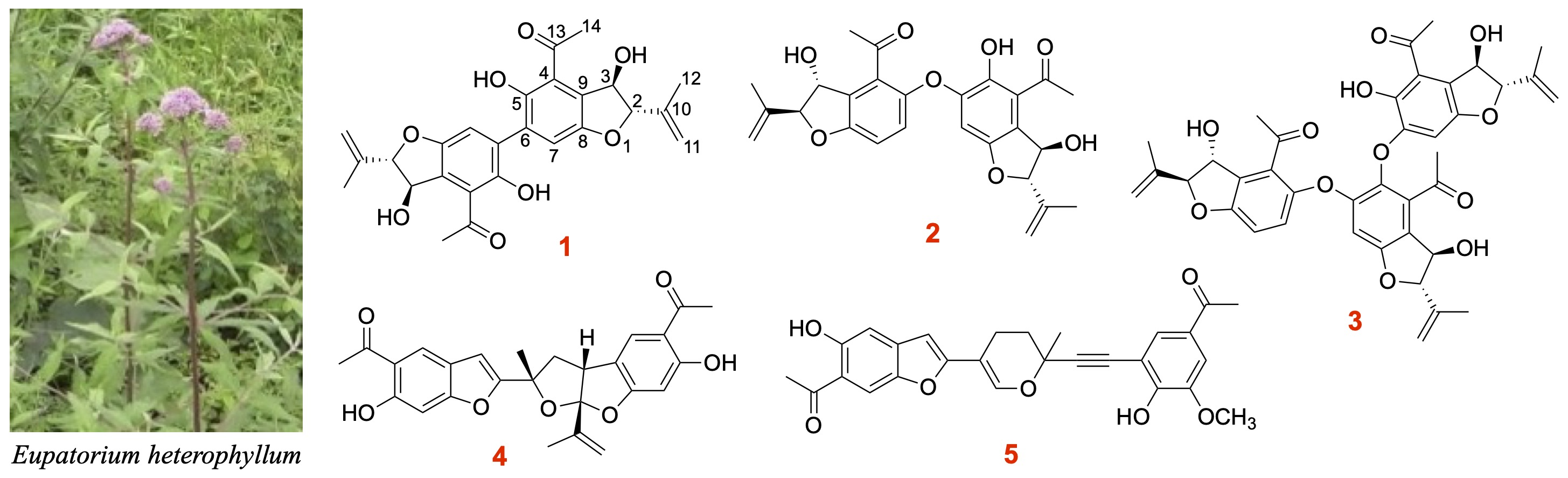
2.1. Samples
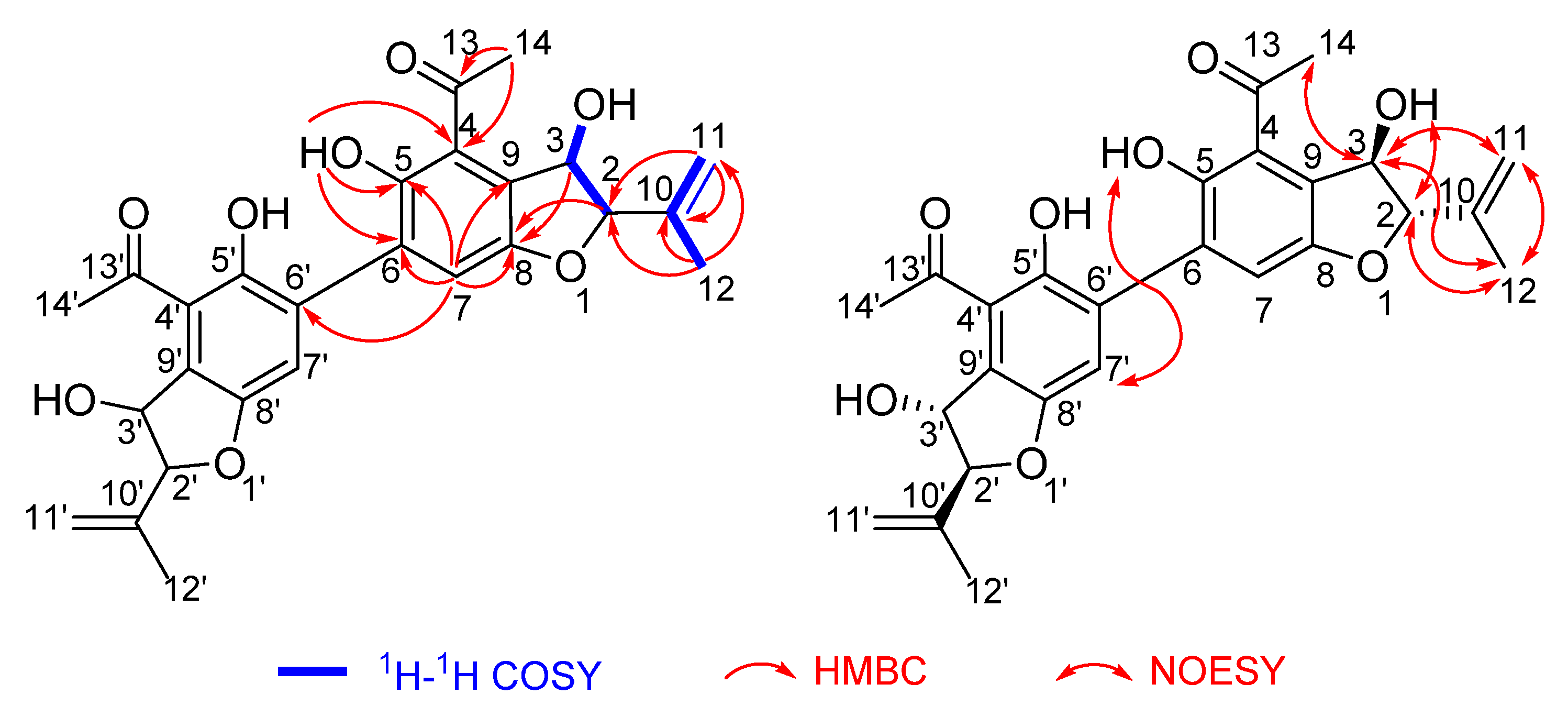
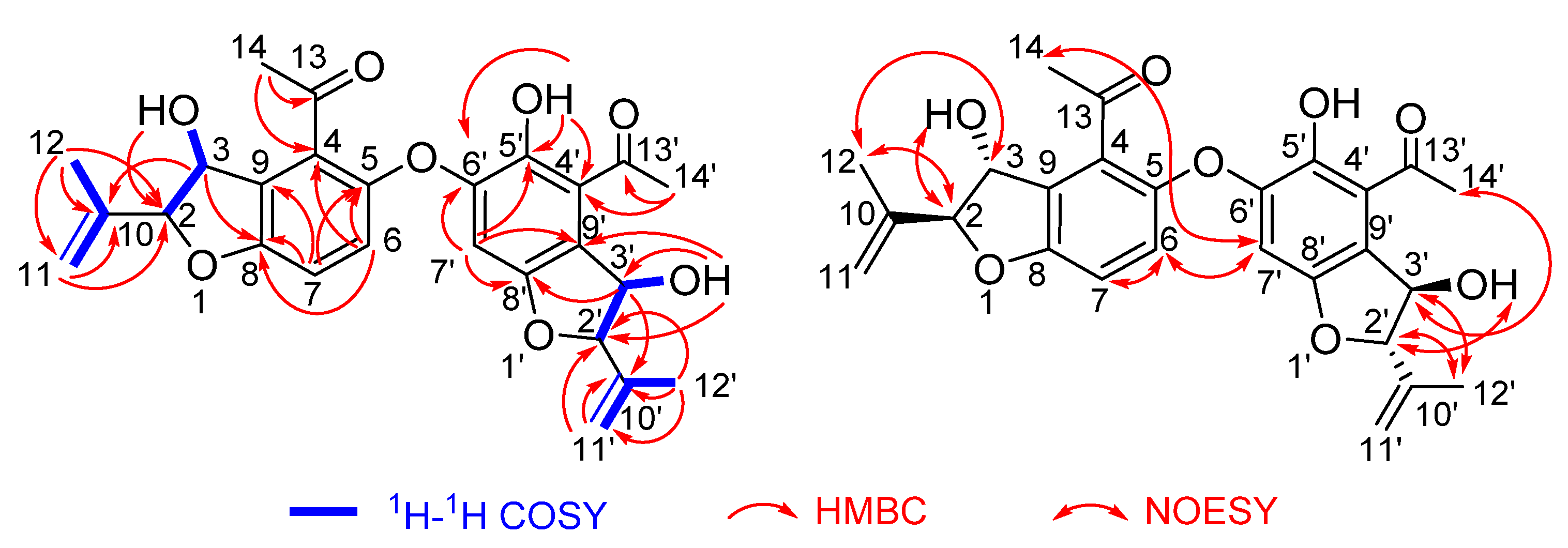
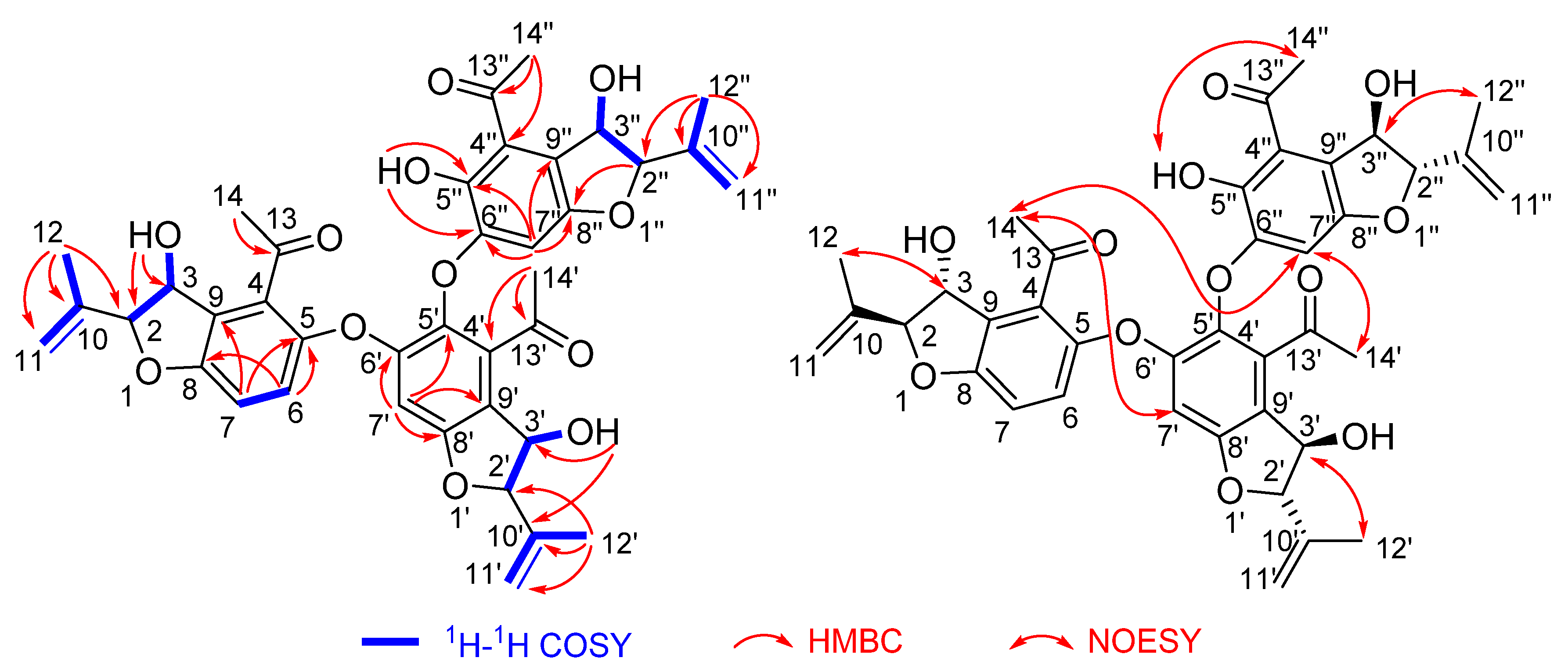
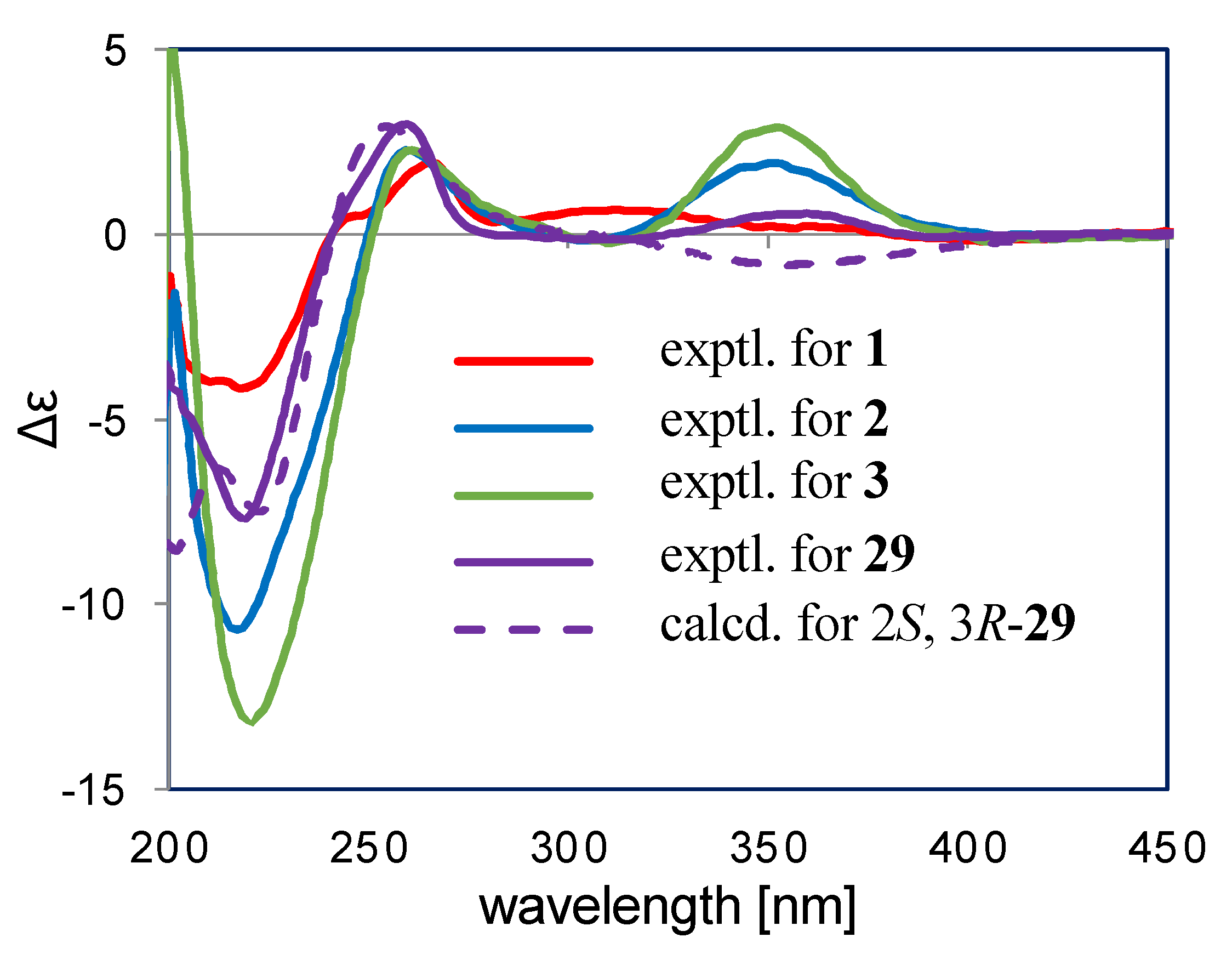
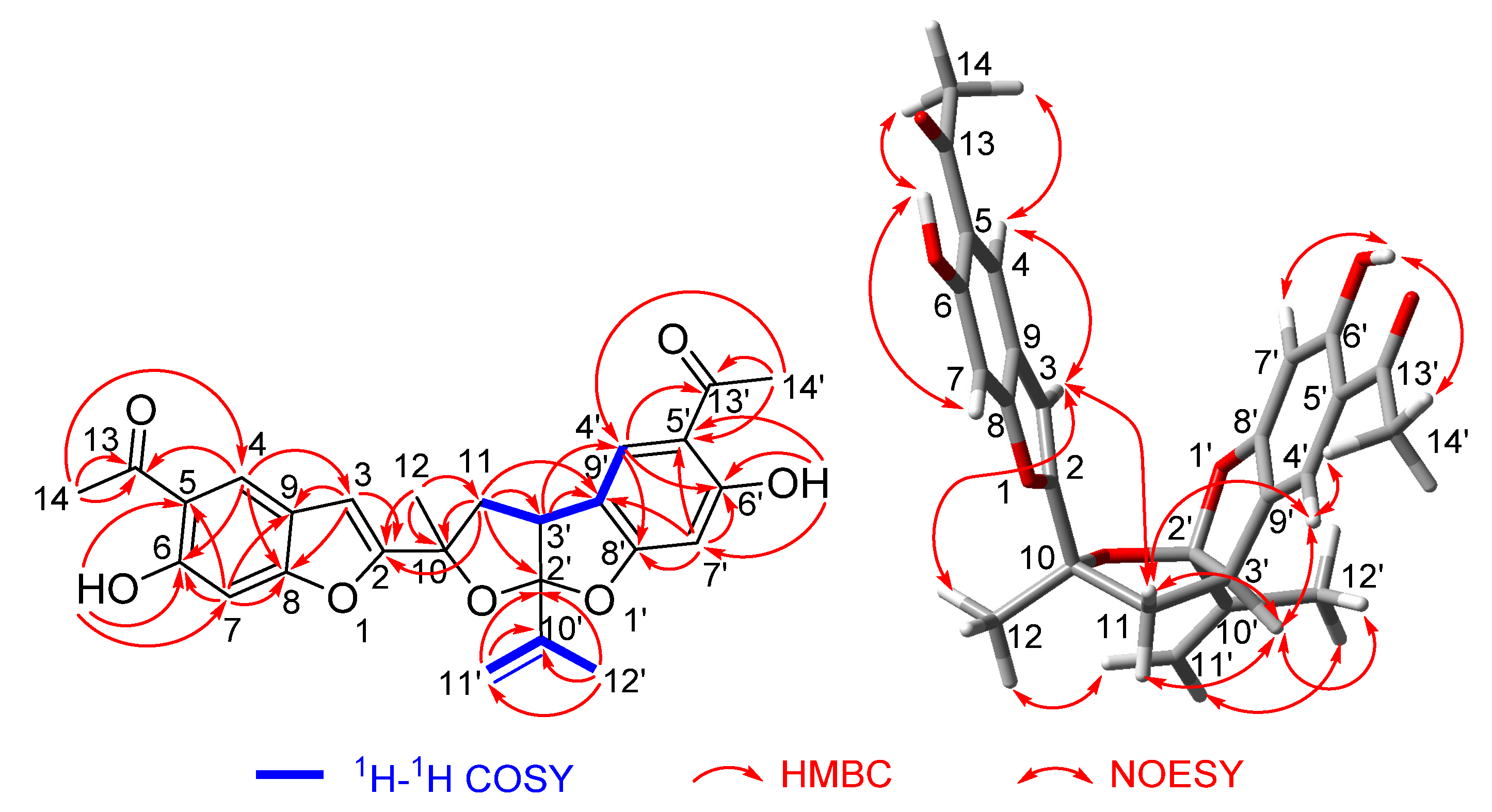
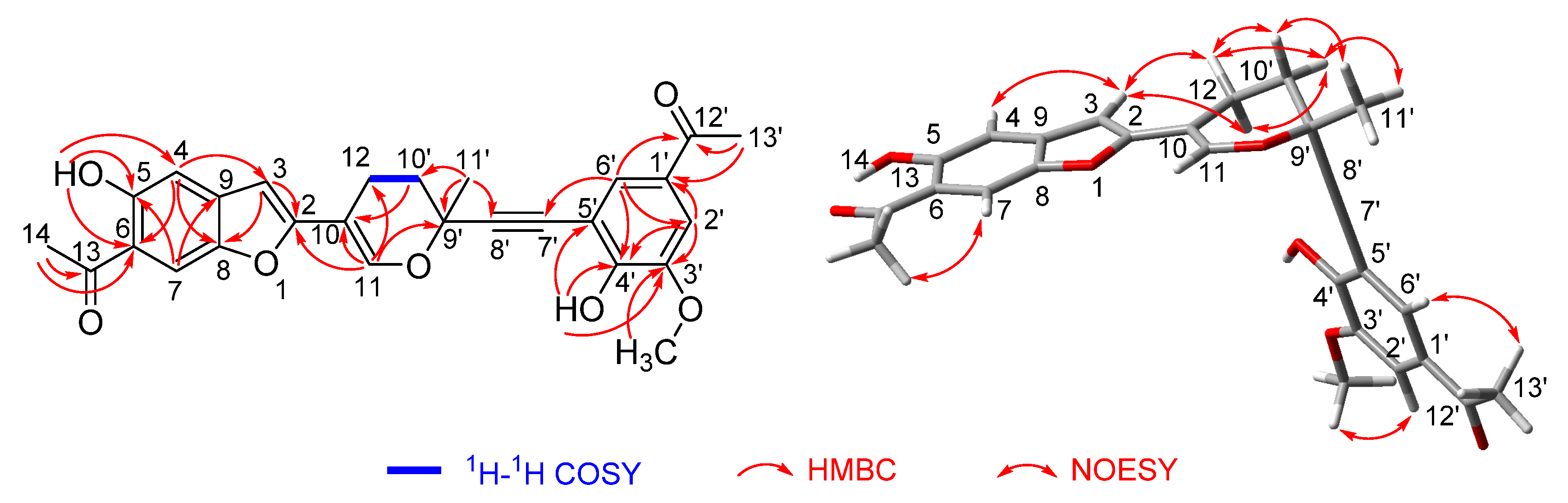
2.2. Structure Elucidation
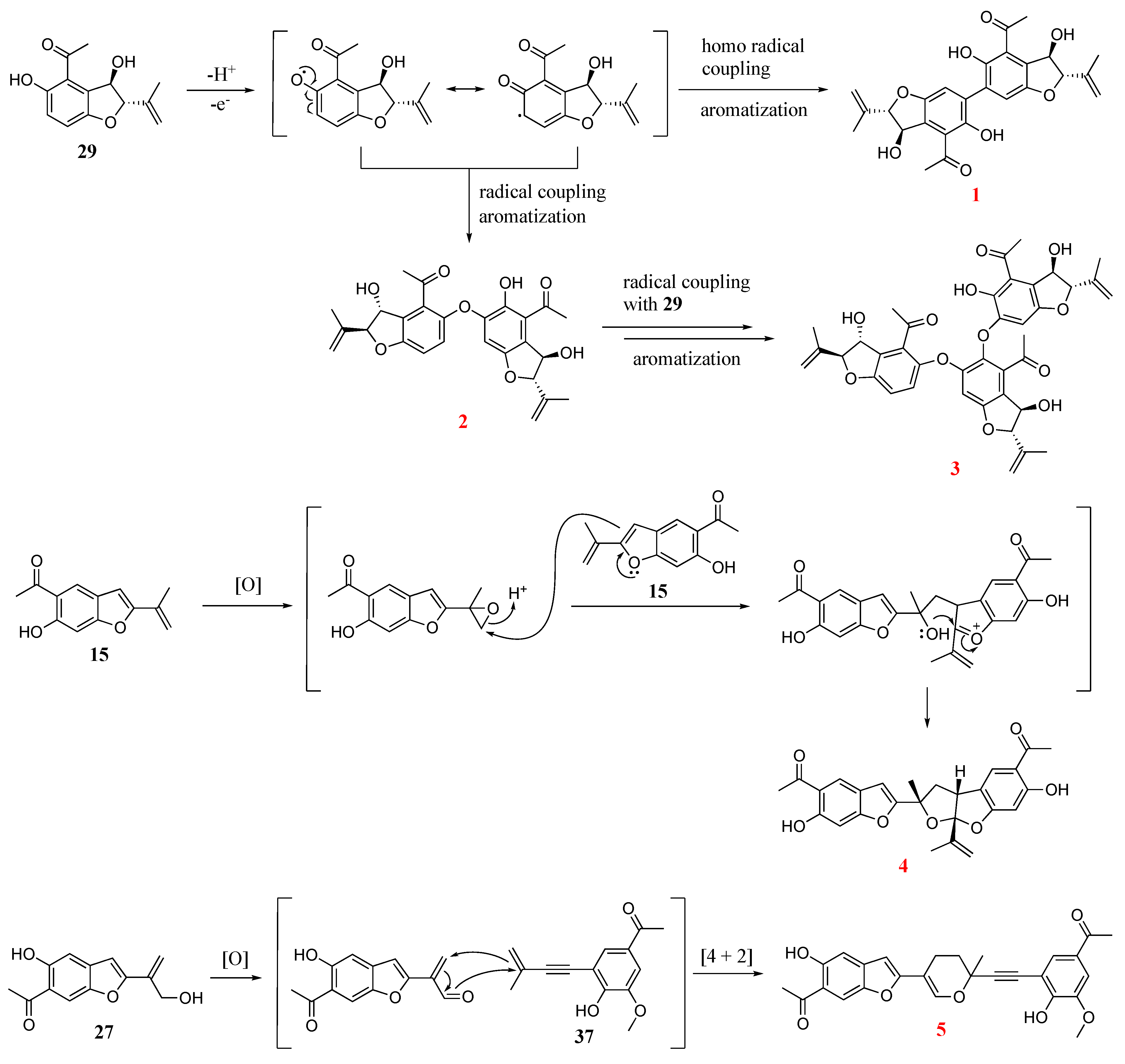
2.3. Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Materials
3.3. Extraction and Isolation
3.4. Chiral HPLC Analyses of Compounds 4 and 5
3.5. Calculation of ECD Spectra
3.6. Compound Data
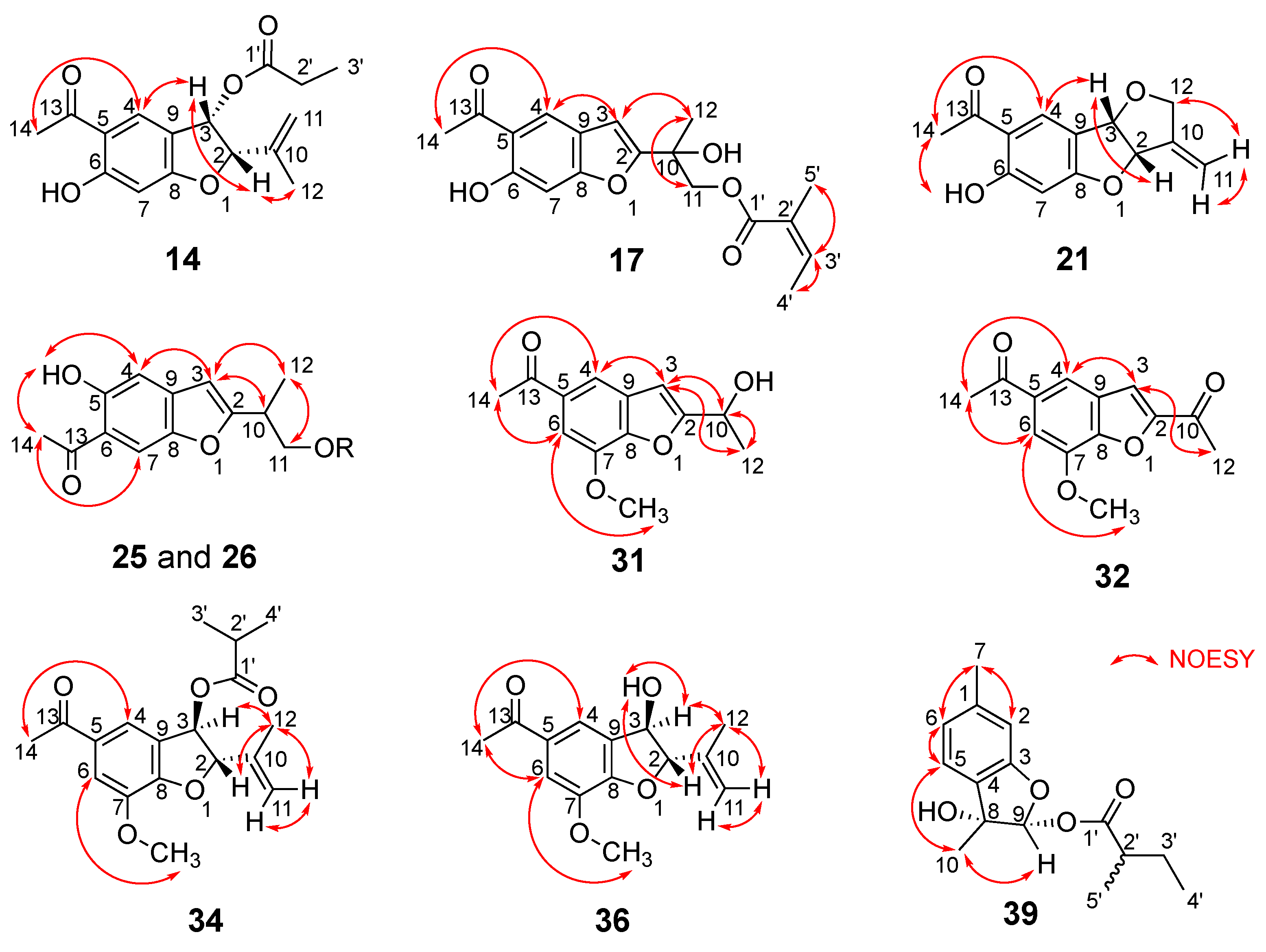
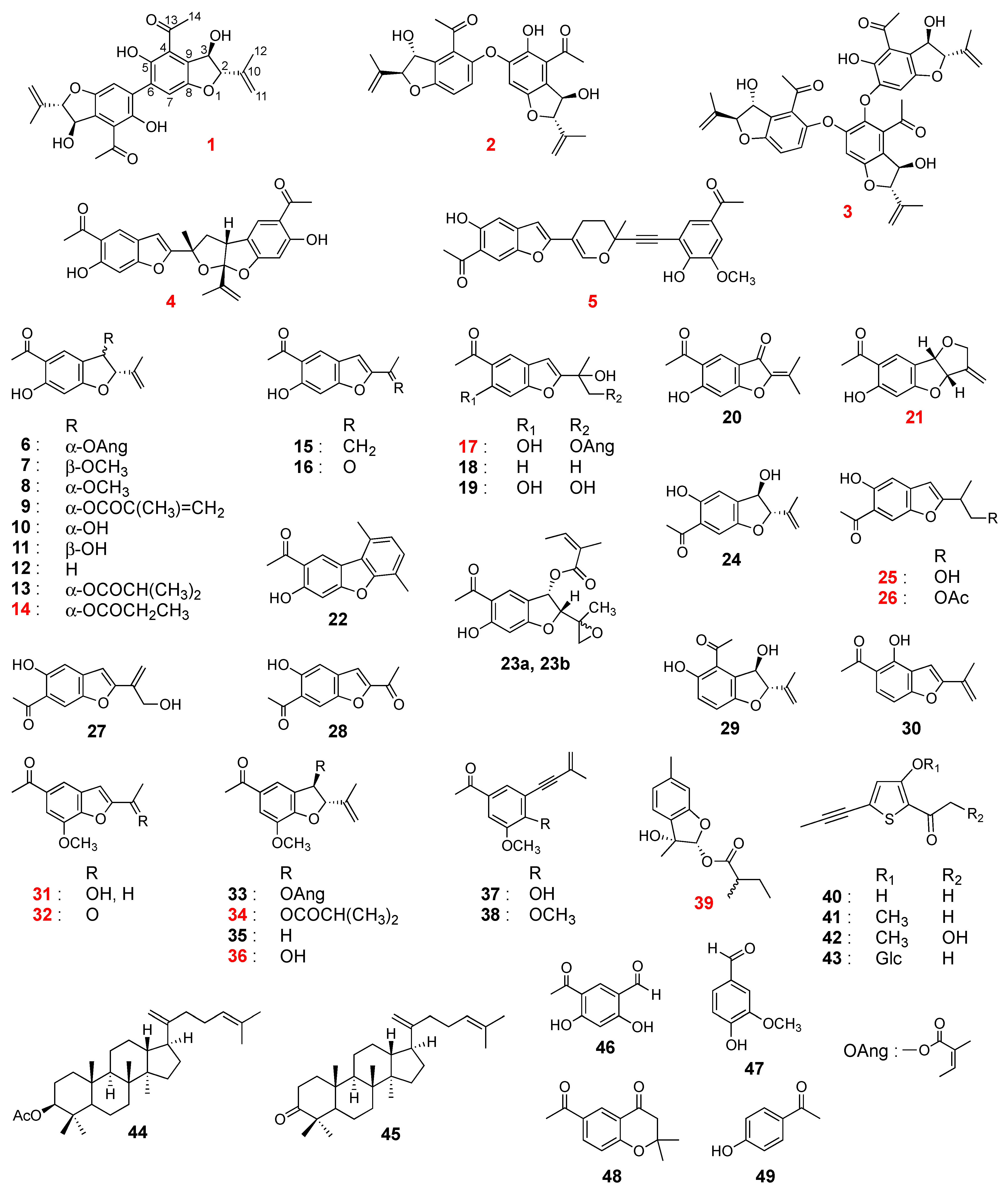
- Compound 1
- Compound 2
- Compound 3
- Compound 4
- Compound 5
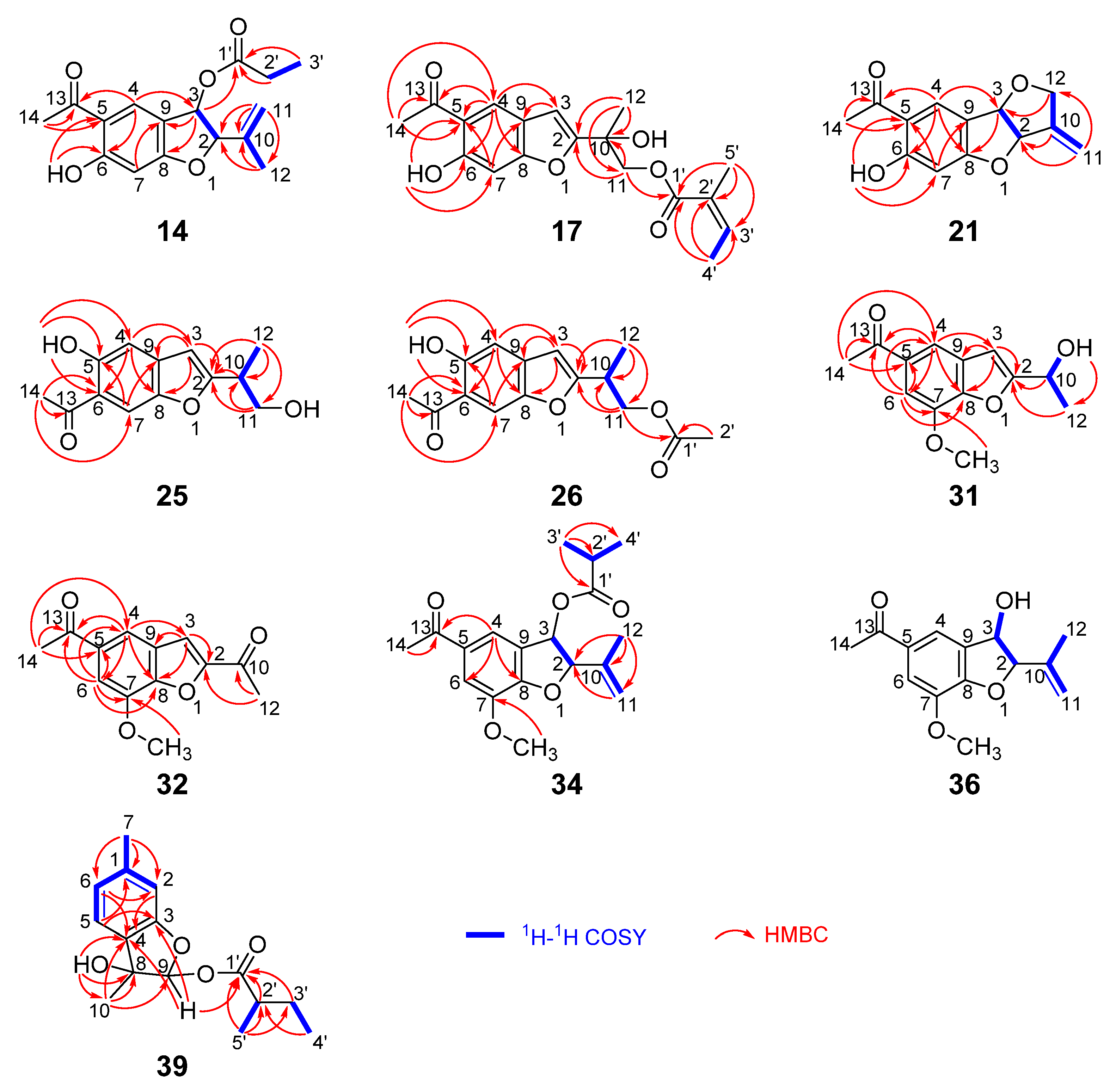
- Compound 14
- Compound 17
- Compound 21
- Compound 25
- Compound 26
- Compound 31
- Compound 32
- Compound 34
- Compound 36
- Compound 39
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kuroda, C.; Hanai, R.; Nagano, H.; Tori, M.; Gong, X. Diversity of Furanoeremophilanes in Major Ligularia Species in the Hengduan Mountains. Nat. Prod. Commun. 2012, 7, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, C.; Hanai, R.; Tori, M.; Okamoto, Y.; Saito, Y.; Nagano, H.; Ohsaki, A.; Hirota, H.; Kawahara, T.; Gong, X. Diversity in Furanoeremophilane Composition Produced by Ligularia Species (Asteraceae) in the Hengduan Mountains Area of China. J. Synth. Org. Chem. Jpn. 2014, 72, 717–725. [Google Scholar] [CrossRef]
- Tori, M.; Saito, Y.; Gong, X.; Kuroda, C. Chemical Studies of Cremanthodium (Asteraceae) Species; Sesquiterpenoids and Related Compounds. Nat. Prod. Commun. 2019, 14, 1934578X19878594. [Google Scholar] [CrossRef]
- Saito, Y.; Iwamoto, Y.; Okamoto, Y.; Gong, X.; Kuroda, C.; Tori, M. Eight New Alkyne and Alkene Derivatives from Four Saussurea Species Collected in China. Nat. Prod. Commun. 2013, 8, 631–634. [Google Scholar] [CrossRef]
- Saito, Y.; Iwamoto, Y.; Okamoto, Y.; Gong, X.; Kuroda, C.; Tori, M. Four New Guaianolides and Acetylenic Alcohol from Saussurea katochaete Collected in China. Nat. Prod. Commun. 2012, 7, 447–450. [Google Scholar] [CrossRef]
- Saito, Y.; Takiguchi, K.; Gong, X.; Kuroda, C.; Tori, M. Thiophene, Furans, and Related Aromatic Compounds from Eupatorium heterophyllum. Nat. Prod. Commun. 2011, 6, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Mukai, T.; Iwamoto, Y.; Baba, M.; Takiguchi, K.; Okamoto, Y.; Gong, X.; Kawahara, T.; Kuroda, C.; Tori, M. Germacranolides and Their Diversity of Eupatorium heterophyllum Collected in P. R. China. Chem. Pharm. Bull. 2014, 62, 1092–1099. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, Y.; Saito, Y.; Gong, X.; Matsuo, Y.; Tanaka, T. Dihydrobenzofurans and Propynylthiophenes From the Roots of Eupatorium heterophyllum. Nat. Prod. Commun. 2022, 17, 1934578X211072331. [Google Scholar] [CrossRef]
- Hu, Y.; Saito, Y.; Matsuo, Y.; Gong, X.; Tanaka, T. Two new dimeric benzofuran diastereomers from the roots of Eupatorium heterophyllum. Tetrahedron Lett. 2022, 102, 153924. [Google Scholar] [CrossRef]
- Chen, Y.; Kawahara, T.; Hind, D.J.N. Tribe Eupatorieae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2011; Volume 20–21, pp. 879–891. [Google Scholar]
- Yu, X.; Zhang, Q.; Tian, L.; Guo, Z.; Liu, C.; Chen, J.; Ebrahim, W.; Liu, Z.; Proksch, P.; Zou, K. Germacrane-Type Sesquiterpenoids with Antiproliferative Activities from Eupatorium chinense. J. Nat. Prod. 2018, 81, 85–91. [Google Scholar] [CrossRef]
- Yang, S.-P.; Huo, J.; Wang, Y.; Lou, L.-G.; Yue, J.-M. Cytotoxic Sesquiterpenoids from Eupatorium chinense. J. Nat. Prod. 2004, 67, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.-Y.; Zhang, Y.-J.; Wang, Y.-F.; Wang, D.; Yang, C.-R. Chemical Constituents of Guangdong Tu-Niu-Xi (Eupatorium chinense, Compositae). Acta Bot. Yunnan 2010, 32, 183–188. [Google Scholar] [CrossRef]
- Dwarakanath, D.; Gaonkar, S.L. Advances in Synthetic Strategies and Medicinal Importance of Benzofurans: A Review. Asian J. Org. Chem. 2022, 11, e202200282. [Google Scholar] [CrossRef]
- Liu, M.-Y.; Yu, L.-J.; Li, Y.-C.; Tian, K.; Li, L.-J.; Wu, Z.-Z. Chemical constituents from Eupatorium chinense L. root and their in vitro antibacterial activity. Nat. Prod. Res. Dev. 2015, 27, 1905–1909. [Google Scholar]
- Zheng, G.; Luo, S.; Li, S.; Hua, J.; Li, W.; Li, S. Specialized metabolites from Ageratina Adenophora and their inhibitory activities against pathogenic fungi. Phytochemistry 2018, 148, 57–62. [Google Scholar] [CrossRef]
- Ruiz-Vásquez, L.; Ruiz-Mesia, L.; Reina-Artiles, M.; López-Rodríguez, M.; González-Platas, J.; Giménez, C.; Cabrera, R.; González-Coloma, A. Benzofurans, benzoic acid derivatives, diterpenes and pyrrolizidine alkaloids from Peruvian Senecio. Phytochem. Lett. 2018, 28, 47–54. [Google Scholar] [CrossRef]
- Faini, F.; Labbe, C.; Salgado, I.; Coll, J. Chemistry, Toxicity and Antifeedant Activity of the Resin of Flourensia thurifera. Biochem. Syst. Ecol. 1997, 25, 189–193. [Google Scholar] [CrossRef]
- Morimoto, M.; Urakawa, M.; Fujitaka, T.; Komai, K. Structure-activity Relationship for the Insect Antifeedant Activity of Benzofuran Derivatives. Biosci. Biotechnol. Biochem. 1999, 63, 840–846. [Google Scholar] [CrossRef]
- Zalkow, L.H.; Keinan, E.; Steindel, S.; Kalyanaraman, A.R.; Bertrand, J.A. On the absolute configuration of toxol at C-3. Vicinal H-H coupling constants in 2-alkyl-3-hydroxydihydrobenzofurans. Tetrahedron Lett. 1972, 13, 2873–2876. [Google Scholar] [CrossRef]
- Mertes, M.P.; Powers, L.J.; Shefter, E. Isolation and identification of the cis-trans stereoisomers of substituted 3-hydroxy-(or acetoxy) 2-methyl-2,3-dihydrobenzofurans. Dihydrobenzofurans which obey the Karplus equation. J. Org. Chem. 1971, 36, 1805–1807. [Google Scholar] [CrossRef]
- Kamthory, B.; Robertson, A. Furano-compounds. Part V. The synthesis of tetrahydroeuparin and the structure of euparin. J. Chem. Soc. 1939, 933–936. [Google Scholar] [CrossRef]
- Bohlmann, F.; Zdero, C. Polyacetylenverbindungen, 193 Notiz über die Inhaltsstoffe von Carelia cistifolia Less. Chem. Ber. 1971, 104, 964–966. [Google Scholar] [CrossRef]
- Breuer, M.; Budzikiewicz, H.; Siebertz, R.; Proksch, P. Benzofuran derivatives from Ageratum houstonianum. Phytochemistry 1987, 26, 3055–3057. [Google Scholar] [CrossRef]
- Zdero, C.; Bohlmann, F. Eupatoriopicrin 19-O-Linolenoate and Other Constituents from Eupatorium cannabium. Planta Med. 1987, 53, 169–172. [Google Scholar] [CrossRef]
- Hussein, N.S. Benzofurans from Senecio desfontainei. Pharmazie 1992, 47, 468–469. [Google Scholar]
- Paredes, L.; Jakupovic, J.; Bohlmann, F.; King, R.M.; Robinsona, H. p-Hydroxyacetophenone derivatives of the monotypic genus Platypodanthera. Phytochemistry 1988, 27, 3329–3330. [Google Scholar] [CrossRef]
- Bohlmann, F.; Ahmed, M.; Robinson, H.; King, R.M. A kolavane derivative from Liatris scariosa. Phytochemistry 1981, 20, 1439–1440. [Google Scholar] [CrossRef]
- Bohlmann, F.; Jakupovic, J.; Lonitz, M. Natürlich vorkommende Terpen-Derivate, 76. Über Inhaltsstoffe der Eupatorium-Gruppe. Chem. Ber. 1977, 110, 301–314. [Google Scholar] [CrossRef]
- Zhou, Z.-Y.; Liu, W.-X.; Pei, G.; Ren, H.; Wang, J.; Xu, Q.-L.; Xie, H.-H.; Wan, F.-H.; Tan, J.-W. Phenolics from Ageratina adenophora Roots and Their Phytotoxic Effects on Arabidopsis thaliana Seed Germination and Seedling Growth. J. Agric. Food Chem. 2013, 61, 11792–11799. [Google Scholar] [CrossRef]
- Zdero, C.; Bohlmann, F.; King, R.M. Diterpenes and norditerpenes from the Aristeguetia group. Phytochemistry 1991, 30, 2991–3000. [Google Scholar] [CrossRef]
- Bohlmann, F.; Zdero, C.; King, R.M.; Robinson, H. New sesquiterpene lactones and other constituents from Fitchia speciosa. Phytochemistry 1980, 19, 1141–1143. [Google Scholar] [CrossRef]
- Zalkow, L.H.; Gelbaum, L.; Ghosal, M.; Fleischmann, T.J. The co-occurrence of desmethylencecalin and hydroxytremetone in Eupatorium rugosum. Phytochemistry 1977, 16, 1313. [Google Scholar] [CrossRef]
- Mahmoud, A.E.; Norman, J.D.; Maynard, W.Q.; Joseph, E.K.; David, J.S.; Paul, L.S. Euparone, a New Benzofuran from Ruscus Aculeatus L. J. Pharm. Sci. 1974, 63, 1623–1624. [Google Scholar]
- Takasugi, M.; Masuda, T. Three 4′-hydroxyacetophenone-related phytoalexins from Polymnia sonchifolia. Phytochemistry 1996, 43, 1019–1021. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, Z.; Yang, L. Sinapyl alcohol derivatives and other constituents from Ligularia nelumbifolia. Phytochemistry 1994, 37, 1149–1152. [Google Scholar]
- Elsohly, M.A.; Slatkin, D.J.; Knapp, J.E.; Doorenbos, N.J.; Quimby, M.W.; Schiff, P.L.; Gopalakrishna, E.M.; Watson, W.H. Ruscodibenzofuran, a new dibenzofuran from Ruscus aculeatus l. (liliaceae). Tetrahedron 1977, 33, 1711–1715. [Google Scholar] [CrossRef]
- Clavel, J.-M.; Guillaumel, J.; Demerseman, P.; Royer, R. Recherches sur le benzofuranne. LVI. Synthèses dans la série de l′Euparone par acétylation de benzofurannes acétylés et méthoxylés sur l′homocycle. J. Heterocycl. Chem. 1977, 14, 219–224. [Google Scholar] [CrossRef]
- Sütfeld, R.; Balza, F.; Neil Towers, G.H. A benzofuran from Tagetes patula seedlings. Phytochemistry 1985, 24, 876–877. [Google Scholar] [CrossRef]
- Bohlmann, F.; Grenz, M. Neue Isopentenyl-acetophenon-Derivate aus Helianthella uniflora. Chem. Ber. 1970, 103, 90–96. [Google Scholar] [CrossRef]
- Bohlmann, F.; Kleine, K.-M.; Bornowski, H. Polyacetylenverbindungen, XLI. Über zwei Thiophenketone aus Artemisia arborescens L. Chem. Ber. 1962, 95, 2934–2938. [Google Scholar] [CrossRef]
- Liu, R.; Wang, X.-B.; Kong, L.-Y. Dammaradienyl acetate. Acta Cryst. 2006, E62, o3544–o3546. [Google Scholar] [CrossRef]
- Phongmaykin, J.; Kumamoto, T.; Ishikawa, T.; Suttisri, R.; Saifah, E. A new sesquiterpene and other terpenoid constituents of Chisocheton penduliflorus. Arch. Pharm. Res. 2008, 31, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.R.; Seshadri, T.R.; Sood, G.R. The structure and synthesis of neobavachalcone, a new component of Psoralea corylifolia. Phytochemistry 1977, 16, 1995–1997. [Google Scholar] [CrossRef]
- Ren, Y.-L.; Tang, Q.-R.; Zhang, Z.; Chen, L.; He, H.-P.; Hao, X.-J. Chemical constituents from Tinospora sinensis. Nat. Prod. Res. Dev. 2008, 2, 278–279. [Google Scholar]
- Rocha, D.D.; Dantas, I.N.F.; Albuquerque, M.R.J.R.; Montenegro, R.C.; Pessoa, C.; de Moraes, M.O.; Pessoa, O.D.L.; Silveira, E.R.; Costa-Lotufo, L.V. Studies on the cytotoxicity of miscellaneous compounds from Eupatorium betonicaeforme (D.C.) Baker (Asteraceae). Chem. Biodivers. 2007, 4, 2835–2844. [Google Scholar] [CrossRef]
- Dhanuskodi, S.; Manikadan, S. EPR investigations on γ-irradiated 4-hydroxyacetophenone single crystals: An NLO material. Radiat. Eff. Defects Solids 2005, 160, 197–205. [Google Scholar] [CrossRef]
- Kawahara, T.; Yahara, T.; Watanabe, K. Distribution of Sexual and Agamospermous Populations of Eupatorium (Compositae) in Asia. Plant Spec. Biol. 1989, 4, 37–46. [Google Scholar] [CrossRef]
- Xu, F.; Zhang, L.; Zhou, C.; Mo, J.; Shen, S.; Zhang, T.; Li, J.; Lin, L.; Wu, R.; Gan, L. Alkyl-benzofuran dimers from Eupatorium chinense with insulin-sensitizing and anti-inflammatory activities. Bioorg. Chem. 2021, 113, 105030. [Google Scholar] [CrossRef]
- Zhang, Q.-Q.; Zhou, J.-H.; Chen, Y.; Zhang, Z.-M.; Liu, Z.-X.; Guo, Z.-Y.; Liu, C.-X.; Zou, K. Seven new chemical constituents from the underground parts of Eupatorium chinense. Fitoterapia 2020, 146, 104674. [Google Scholar] [CrossRef]
- Ke, J.-H.; Zhang, L.-S.; Chen, S.-X.; Shen, S.-N.; Zhang, T.; Zhou, C.-X.; Mo, J.-X.; Lin, L.-G.; Gan, L.-S. Benzofurans from Eupatorium chinense enhance insulin-stimulated glucose uptake in C2C12 myotubes and suppress inflammatory response in RAW264.7 macrophages. Fitoterapia 2019, 134, 346–354. [Google Scholar] [CrossRef]
- Wang, W.-J.; Wang, L.; Liu, Z.; Jiang, R.-W.; Liu, Z.-W.; Li, M.-M.; Zhang, Q.-W.; Dai, Y.; Li, Y.-L.; Zhang, X.-Q.; et al. Antiviral benzofurans from Eupatorium chinense. Phytochemistry 2016, 122, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-J.; Wang, L.; Huang, X.-J.; Jiang, R.-W.; Yang, X.-L.; Zhang, D.-M.; Chen, W.-M.; Tang, B.-Q.; Wang, Y.; Zhang, X.-Q.; et al. Two pairs of new benzofuran enantiomers with unusual skeletons from Eupatorium chinense. Tetrahedron Lett. 2013, 54, 3321–3324. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6.1.1; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | δH | (mult., J in Hz) | δC | Position | δH | (mult., J in Hz) | δC | Position | δH | (mult., J in Hz) | δC |
| 2, 2′ | 5.00 | (1H, br s) | 92.9 | 2 | 5.04 | (1H, br s) | 92.2 | 2 | 4.97 | (1H, br s) | 91.86 |
| 3, 3′ | 5.34 | (1H, br s) | 77.7 | 3 | 5.21 | (1H, br s) | 76.3 | 3 | 5.06 | (1H, br s) | 76.22 |
| 4, 4′ | 119.2 | 4 | 128.1 | 4 | 126.31 | ||||||
| 5, 5′ | 153.4 | 5 | 148.8 | 5 | 149.28 | ||||||
| 6, 6′ | 130.3 | 6 | 6.91 | (1H, d, 8.8) | 122.1 | 6 | 6.76 | (1H, d, 8.8) | 118.89 | ||
| 7, 7′ | 7.14 | (1H, s) | 119.8 | 7 | 7.03 | (1H, d, 8.8) | 115.5 | 7 | 6.88 | (1H, d, 8.8) | 114.94 |
| 8, 8′ | 153.4 | 8 | 157.2 | 8 | 156.35 | ||||||
| 9, 9′ | 126.6 | 9 | 130.3 | 9 | 129.56 | ||||||
| 10, 10′ | 141.1 | 10 | 141.4 | 10 | 141.24 (a) | ||||||
| 11, 11′ | 5.10 | (1H, s) | 112.6 | 11 | 5.10 | (1H, s) | 112.0 | 11 | 5.04 | (1H, s) | 112.14 |
| 4.94 | (1H, s) | 4.92 | (1H, s) | 4.88 | (1H, s) | ||||||
| 12, 12′ | 1.79 | (3H, s) | 17.8 | 12 | 1.78 | (3H, s) | 17.6 | 12 | 1.72 | (3H, s) | 17.51 |
| 13, 13′ | 204.6 | 13 | 202.0 | 13 | 202.48 | ||||||
| 14, 14′ | 2.88 | (3H, s) | 31.1 | 14 | 2.69 | (3H, s) | 32.1 | 14 | 2.49 | (3H, s) | 32.23 |
| 3-OH, 3′-OH | 2.84 | (1H, br s) | 3-OH | 4.37 | (1H, br s) | 3-OH | 4.46 | (1H, d, 1.2) | |||
| 5-OH, 5′-OH | 11.72 | (1H, s) | 2′ | 4.95 | (1H, br s) | 93.0 | 2′ | 5.06 | (1H, br s) | 92.72 | |
| 3′ | 5.29 | (1H, dd, 7.1, 2.6) | 77.5 | 3′ | 5.24 | (1H, dd, 3.4, 1.7) | 75.98 | ||||
| 4′ | 119.2 | 4′ | 130.41 | ||||||||
| 5′ | 148.0 | 5′ | 147.93 | ||||||||
| 6′ | 147.2 | 6′ | 139.88 | ||||||||
| 7′ | 6.66 | (1H, s) | 106.4 | 7′ | 6.81 | (1H, s) | 107.59 | ||||
| 8′ | 153.0 | 8′ | 157.70 | ||||||||
| 9′ | 120.6 | 9′ | 126.03 | ||||||||
| 10′ | 141.0 | 10′ | 141.09 | ||||||||
| 11′ | 5.04 | (1H, s) | 112.5 | 11′ | 5.13 | (1H, s) | 112.23 | ||||
| 4.92 | (1H, s) | 4.96 | (1H, s) | ||||||||
| 12′ | 1.75 | (3H, s) | 17.7 | 12′ | 1.82 | (3H, s) | 17.74 | ||||
| 13′ | 203.6 | 13′ | 201.64 | ||||||||
| 14′ | 2.86 | (3H, s) | 31.3 | 14′ | 2.67 | (3H, s) | 30.98 | ||||
| 3′-OH | 2.93 | (1H, d, 7.1) | 3′-OH | 4.32 | (1H, d, 1.7) | ||||||
| 5′-OH | 10.49 | (1H, s) | 2″ | 4.91 | (1H, br s) | 93.22 | |||||
| 3″ | 5.14 | (1H, dd, 9.3, 2.4) | 77.28 | ||||||||
| 4″ | 118.39 | ||||||||||
| 5″ | 146.83 | ||||||||||
| 6″ | 146.83 | ||||||||||
| 7″ | 6.58 | (1H, s) | 104.86 | ||||||||
| 8″ | 152.60 | ||||||||||
| 9″ | 120.32 | ||||||||||
| 10″ | 141.09 (a) | ||||||||||
| 11″ | 4.98 | (1H, s) | 112.06 | ||||||||
| 4.87 | (1H, s) | ||||||||||
| 12″ | 1.71 | (3H, s) | 17.67 | ||||||||
| 13″ | 204.18 | ||||||||||
| 14″ | 2.78 | (3H, s) | 32.22 | ||||||||
| 3″-OH | 4.04 | (1H, d, 9.3) | |||||||||
| 5″-OH | 11.25 | (1H, s) | |||||||||
| 4 | 5 | ||||||
|---|---|---|---|---|---|---|---|
| Position | δH | (mult., J in Hz) | δC | Position | δH | (mult., J in Hz) | δC |
| 2 | 160.2 | 2 | 161.7 | ||||
| 3 | 6.31 | (1H, d, 0.8) | 102.3 | 3 | 6.35 | (1H, s) | 99.0 |
| 4 | 7.70 | (1H, s) | 123.5 | 4 | 6.94 | (1H, s) | 106.5 |
| 5 | 116.8 | 5 | 158.7 | ||||
| 6 | 160.8 | 6 | 115.4 | ||||
| 7 | 6.79 | (1H, br s) | 99.1 | 7 | 7.70 | (1H, s) | 111.0 |
| 8 | 159.2 | 8 | 147.3 | ||||
| 9 | 120.8 | 9 | 137.5 | ||||
| 10 | 84.6 | 10 | 104.7 | ||||
| 11 | 2.84 | (1H, dd, 13.2, 1.2) | 43.1 | 11 | 7.37 | (1H, s) | 144.4 |
| 2.45 | (1H, dd, 13.2, 8.8) | 12 | 2.82 | (1H, dddd, 16.2, 11.2, 6.0, 1.8) | 18.9 | ||
| 12 | 1.74 | (3H, s) | 27.9 | 2.44 | (1H, dddd, 16.2, 5.4, 2.6, 1.0) | ||
| 13 | 204.0 | 13 | 203.4 | ||||
| 14 | 2.64 | (3H, s) | 26.9 | 14 | 2.66 | (3H, s) | 26.7 |
| 6-OH | 12.42 | (1H, s) | 5-OH | 12.26 | (1H, s) | ||
| 2′ | 122.8 | 1′ | 129.6 | ||||
| 3′ | 3.87 | (1H, d, 8.8) | 49.8 | 2′ | 7.47 | (1H, d, 1.9) | 110.0 |
| 4′ | 7.30 | (1H, d, 1.0) | 126.0 | 3′ | 146.7 | ||
| 5′ | 113.9 | 4′ | 151.6 | ||||
| 6′ | 165.9 | 5′ | 108.1 | ||||
| 7′ | 6.17 | (1H, s) | 97.9 | 6′ | 7.58 | (1H, d, 1.9) | 127.1 |
| 8′ | 165.5 | 7′ | 78.4 | ||||
| 9′ | 120.9 | 8′ | 94.8 | ||||
| 10′ | 141.7 | 9′ | 71.9 | ||||
| 11′ | 5.46 | (1H, s) | 113.8 | 10′ | 2.29 | (1H, ddd, 13.5, 6.0, 2.6) | 33.2 |
| 5.10 | (1H, t, 1.5) | 2.02 | (1H, ddd, 13.5, 11.2, 5.4) | ||||
| 12′ | 1.86 | (3H, s) | 18.2 | 11′ | 1.79 | (3H, s) | 28.1 |
| 13′ | 201.8 | 12′ | 196.1 | ||||
| 14′ | 2.44 | (3H, s) | 26.1 | 13′ | 2.53 | (3H, s) | 26.2 |
| 5′-OH | 12.76 | (1H, s) | 3′-OCH3 | 3.92 | (3H, s) | 56.3 | |
| 4′-OH | 6.28 | (1H, s) | |||||
| 14 | 17 | 21 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Position | δH | (mult., J in Hz) | δC | δH | (mult., J in Hz) | δC | δH | (mult., J in Hz) | δC |
| 2 | 5.14 | (1H, d, 6.4) | 89.2 | 160.6 | 5.47 | (1H, d, 6.1) | 88.4 | ||
| 3 | 6.26 | (1H, d, 6.4) | 72.2 | 6.64 | (1H, d, 1.0) | 102.5 | 5.67 | (1H, d, 6.1) | 81.9 |
| 4 | 7.81 | (1H, s) | 130.0 | 7.93 | (1H, s) | 123.7 | 7.81 | (1H, s) | 129.2 |
| 5 | 114.7 | 117.0 | 114.8 | ||||||
| 6 | 166.7 | 161.1 | 167.0 | ||||||
| 7 | 6.46 | (1H, s) | 98.9 | 6.99 | (1H, br s) | 99.8 | 6.38 | (1H, s) | 98.5 |
| 8 | 166.7 | 159.5 | 166.8 | ||||||
| 9 | 117.8 | 120.8 | 118.3 | ||||||
| 10 | 138.0 | 71.3 | 145.3 | ||||||
| 11 | 5.20 | (1H, d, 1.0) | 114.7 | 4.53 | (1H, d, 11.5) | 69.7 | 4.36 | (1H, d, 12.5) | 69.2 |
| 5.12 | (1H, d, 1.0) | 4.38 | (1H, d, 11.5) | 4.22 | (1H, d, 12.5) | ||||
| 12 | 1.81 | (3H, s) | 19.2 | 1.67 | (3H, s) | 23.9 | 5.56 | (1H, s) | 112.9 |
| 5.38 | (1H, s) | ||||||||
| 13 | 202.7 | 204.0 | 202.5 | ||||||
| 14 | 2.57 | (3H, s) | 26.4 | 2.69 | (3H, s) | 26.9 | 2.58 | (3H, s) | 26.3 |
| 1′ | 174.1 | 167.9 | |||||||
| 2′ | 2.31 | (2H, q, 7.6) | 27.7 | 127.1 | |||||
| 3′ | 1.12 | (3H, t, 7.6) | 9.0 | 6.09 | (1H, qq, 7.3, 1.4) | 139.5 | |||
| 4′ | 1.89 | (3H, dq, 7.3, 1.4) | 15.8 | ||||||
| 5′ | 1.83 | (3H, quint, 1.4) | 20.5 | ||||||
| 6-OH | 13.04 | (1H, s) | 12.46 | (1H, s) | 12.99 | (1H, s) | |||
| 25 | 26 | 31 | 32 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | δH | (mult., J in Hz) | δC | δH | (mult., J in Hz) | δC | δH | (mult., J in Hz) | δC | δH | (mult., J in Hz) | δC |
| 2 | 166.8 | 165.7 | 162.0 | 154.2 | ||||||||
| 3 | 6.46 | (1H, s) | 102.8 | 6.42 | (1H, s) | 102.6 | 6.70 | (1H, s) | 102.7 | 7.56 | (1H, s) | 112.9 |
| 4 | 7.01 | (1H, s) | 107.4 | 7.01 | (1H, s) | 107.5 | 7.80 | (1H, d, 1.5) | 115.9 | 7.93 | (1H, d, 1.5) | 117.4 |
| 5 | 158.4 | 158.4 | 133.8 | 134.7 | ||||||||
| 6 | 115.8 | 115.9 | 7.48 | (1H, d, 1.5) | 105.5 | 7.62 | (1H, d, 1.5) | 108.0 | ||||
| 7 | 7.78 | (1H, s) | 111.8 | 7.78 | (1H, s) | 111.8 | 145.3 | 146.2 | ||||
| 8 | 147.8 | 147.7 | 146.8 | 147.8 | ||||||||
| 9 | 136.7 | 136.7 | 129.4 | 128.3 | ||||||||
| 10 | 3.18 | (1H, sext, 6.5) | 36.9 | 3.32 | (1H, sext, 6.8) | 33.7 | 5.06 | (1H, m) | 64.0 | 188.6 | ||
| 11 | 3.88 | (2H, m) | 66.0 | 4.36 | (1H, dd, 11.0, 6.8) | 66.3 | ||||||
| 4.27 | (1H, dd, 11.0, 6.3) | |||||||||||
| 12 | 1.39 | (3H, d, 7.1) | 14.9 | 1.39 | (3H, d, 7.1) | 15.4 | 1.66 | (3H, d, 6.6) | 21.4 | 2.66 | (3H, s) | 26.7 |
| 13 | 203.7 | 203.8 | 197.6 | 197.1 | ||||||||
| 14 | 2.68 | (3H, s) | 26.7 | 2.68 | (3H, s) | 26.7 | 2.66 | (3H, s) | 26.6 | 2.68 | (3H, s) | 26.6 |
| 1′ | 170.9 | |||||||||||
| 2′ | 2.05 | (3H, s) | 20.9 | |||||||||
| 5-OH | 12.16 | (1H, s) | 12.16 | (1H, s) | ||||||||
| 7-OCH3 | 4.06 | (3H, s) | 56.1 | 4.08 | (3H, s) | 56.2 | ||||||
| 10-OH | 2.18 | (1H, d, 4.9) | ||||||||||
| 34 | 36 (a) | 39 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Position | δH | (mult., J in Hz) | δC | δH | (mult., J in Hz) | δH (Major/Minor) | (mult., J in Hz) | δC (Major/Minor) | ||
| 1 | 141.60 | |||||||||
| 2 | 5.13 | (1H, d, 2.8) | 92.1 | 5.04 | (1H, d, 4.0) | 6.761 | (1H, d, 0.5) | 111.67 | ||
| 3 | 6.17 | (1H, d, 2.8) | 77.3 | 5.22 | (1H, dd, 7.8, 4.0) | 158.47 | ||||
| 4 | 7.62 | (1H, d, 1.5) | 120.7 | 7.65 | (1H, d, 1.5) | 127.58 | ||||
| 5 | N.D. (b) | 7.222 | (1H, d, 7.6) | 122.68 | ||||||
| 6 | 7.59 | (1H, d, 1.5) | 113.0 | 7.57 | (1H, d, 1.5) | 6.848 | (1H, dd, 7.6, 0.5) | 123.07 | ||
| 7 | 144.8 | 2.353 | (3H, s) | 21.72 | ||||||
| 8 | 154.0 | 79.73 | ||||||||
| 9 | N.D. (b) | 6.506 | (1H, s) | 104.85/104.80 | ||||||
| 10 | 140.5 | 1.626 | (3H, s) | 20.12 | ||||||
| 11 | 5.10 | (1H, s) | 114.1 | 5.13 | (1H, d, 0.8) | |||||
| 4.98 | (1H, s) | 4.97 | (1H, d, 0.8) | |||||||
| 12 | 1.76 | (3H, s) | 17.5 | 1.76 | (3H, s) | |||||
| 13 | 196.4 | |||||||||
| 14 | 2.56 | (3H, s) | 26.4 | 2.58 | (3H, s) | |||||
| 1′ | 176.8 | 175.28 | ||||||||
| 2′ | 2.57 | (1H, sept, 7.1) | 33.9 | 2.407/2.398 | (1H, sext, 7.0) | 41.02/40.97 | ||||
| 3′ | 1.20 | (3H, d, 7.1) | 18.9 | 1.688 | (1H, m) | 26.49/26.35 | ||||
| 1.498 | (1H, m) | |||||||||
| 4′ | 1.17 | (3H, d, 7.1) | 18.8 | 0.913/0.907 | (3H, t, 7.0) | 11.42 | ||||
| 5′ | 1.157/1.164 | (3H, d, 7.0) | 16.07/16.38 | |||||||
| 3-OH | 2.16 | (1H, d, 7.8) | ||||||||
| 7-OCH3 | 3.96 | (3H, s) | 56.2 | 3.96 | (3H, s) | |||||
| 8-OH | 2.033/2.036 | (1H, s) | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Saito, Y.; Matsuo, Y.; Gong, X.; Tanaka, T. New Benzofuran Oligomers from the Roots of Eupatorium heterophyllum Collected in China. Molecules 2022, 27, 8856. https://doi.org/10.3390/molecules27248856
Hu Y, Saito Y, Matsuo Y, Gong X, Tanaka T. New Benzofuran Oligomers from the Roots of Eupatorium heterophyllum Collected in China. Molecules. 2022; 27(24):8856. https://doi.org/10.3390/molecules27248856
Chicago/Turabian StyleHu, Yiming, Yoshinori Saito, Yosuke Matsuo, Xun Gong, and Takashi Tanaka. 2022. "New Benzofuran Oligomers from the Roots of Eupatorium heterophyllum Collected in China" Molecules 27, no. 24: 8856. https://doi.org/10.3390/molecules27248856
APA StyleHu, Y., Saito, Y., Matsuo, Y., Gong, X., & Tanaka, T. (2022). New Benzofuran Oligomers from the Roots of Eupatorium heterophyllum Collected in China. Molecules, 27(24), 8856. https://doi.org/10.3390/molecules27248856