
Atoms-In-Molecules’ Faces of Chemical Hardness by Conceptual Density Functional Theory



{kind=link}
Abstract
1. Introduction
2. Chemical Hardness-Softness Driving Chemical Bonding
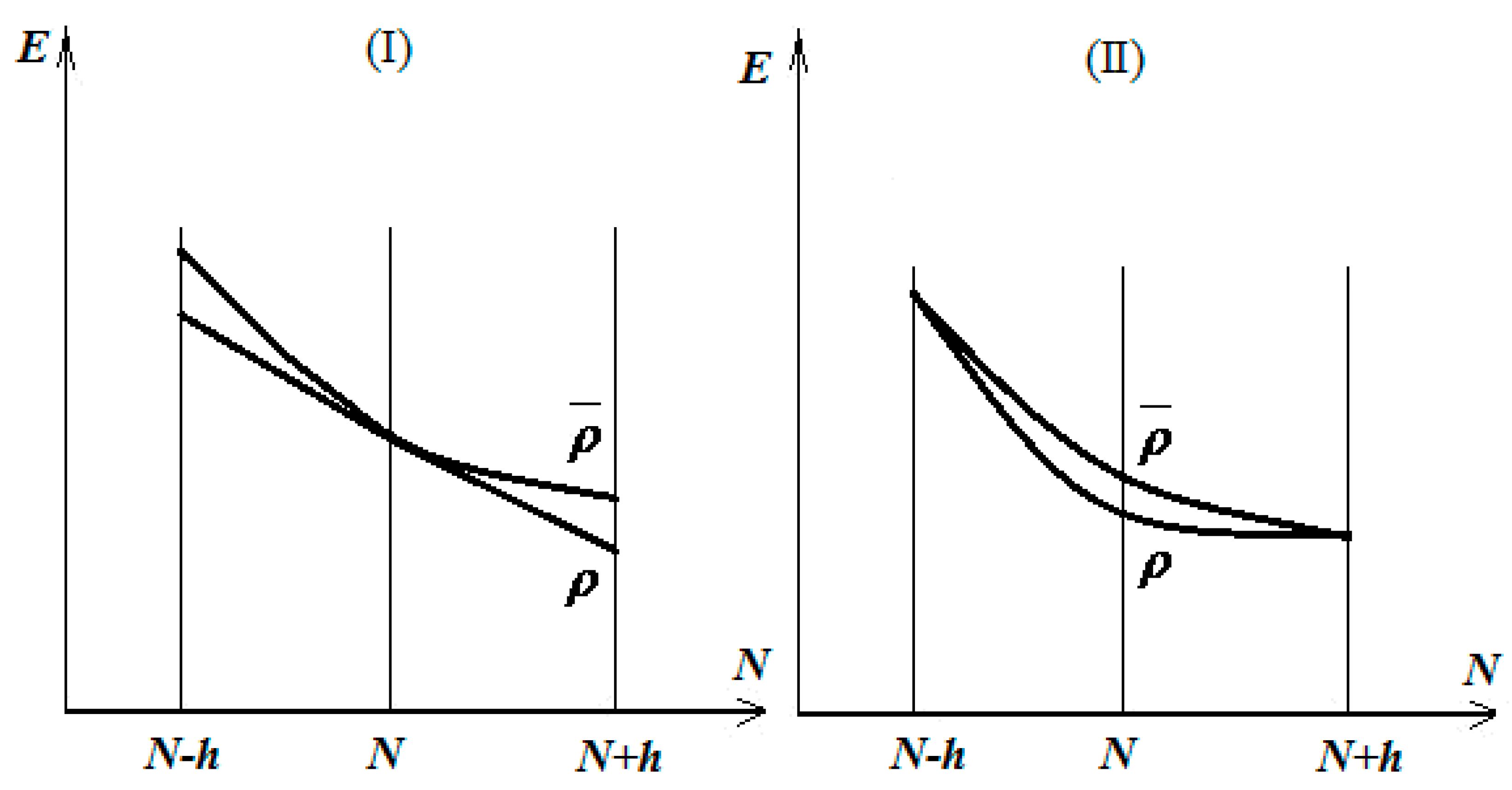
2.1. The Electonic Sharing Ansatz
2.2. Additive Equalization of Atoms in Molecules
2.3. Geometrical Equalization of Atoms in Molecules
3. Applications of Chemical Hardness in Solid-State Chemistry
4. Bond Force Constant and Chemical Hardness
5. Conclusions
- The chemical information contained within the basic density functional of the total number of electrons in terms of simple electronic density may also be regarded at the level of the exchange–correlation density, enriching the inter-electronic effects.
- As a consequence, the related electronic covariance in the bond may eventually be equated with the charge transfer in bonding in what is considered the first-order level of the structure–reactivity density functional connection.
- Assuming the previous structure–reactivity connection, the variation in the electronic covariance respecting the equalized electronegativity of atoms in molecules behaves like the softness of the partners in bonding and the entire bonded system for additive and product (geometrical mean) models of atoms in molecules, respectively; it may thus be viewed as the second-order level of the structure–reactivity connection.
- The maximum hardness and minimum polarizability principles have important applications in solid-state chemistry. These applications can be accepted as strong linkages between solid-state chemistry and the conceptual density functional theory.
- The chemical hardness and Fukui potential provide important hints about the stabilities of inorganic ionic systems and these descriptors can be easily used in the calculation of the lattice energies of inorganic crystals.
- The chemical hardness and molar volume of inorganic ionic systems can be used in the calculation of their bond force constants.
- The previously questionable observable characteristics of chemical hardness based on the second quantification framework [92] are partly determined here as being positive for the limiting cases of unitary electronic density on the frontier states, i.e., for ionization energy/HOMO and affinity energy/LUMO, or on the valence and conduction levels in the solid-state bands of chemical solids, respectively.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3807. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electronic gas. Phys. Rev. 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Yang, W.; Parr, R.G.; Pucci, R. Electron density, Kohn-Sham frontier orbitals, and Fukui functions. J. Chem. Phys. 1984, 81, 2862–2863. [Google Scholar] [CrossRef]
- Gázquez, J.L.; Vela, A.; Galvan, M. Fukui function, electronegativity and hardness in the Kohn-Sham Theory. Struct. Bond. 1987, 66, 79–98. [Google Scholar]
- Putz, M.V. Electronegativity and chemical hardness: Different patterns in quantum chemistry. Curr. Phys. Chem. 2011, 1, 111–139. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Cedillo, A.; Parr, R.G. Variational method for determining the Fukui function and chemical hardness of an electronic system. J. Chem Pys. 1995, 103, 7645–7646. [Google Scholar] [CrossRef]
- Matito, E.; Putz, M.V. New link between conceptual density functional theory and electron delocalization. J. Phys. Chem. A 2011, 115, 12459–12462. [Google Scholar] [CrossRef]
- Torrent-Sucarrat, M.; Salvador, P.; Solà, M.; Geerlings, P. The hardness kernel as the basis for global and local reactivity indices. J. Comput. Chem. 2007, 29, 1064–1074. [Google Scholar] [CrossRef]
- Torrent-Sucarrat, M.; De Proft, F.; Geerlings, P.; Ayers, P.W. Do the local softness and hardness indicate the softest and hardest regions of a molecule? Chem. Eur. J. 2008, 14, 8652–8660. [Google Scholar] [CrossRef]
- Fuentealba, P. A local model for the hardness kernel and related reactivity parameters in density functional theory. J. Chem. Phys. 1995, 103, 6571–6575. [Google Scholar] [CrossRef]
- Torrent-Sucarrat, M.; De Proft, F.; Ayers, P.W.; Geerlings, P. On the applicability of local softness and hardness. Phys. Chem. Chem. Phys. 2010, 12, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Ayers, P.W.; Parr, R.G. Variational principles for describing chemical reactions: The Fukui function and chemical hardness revisited. J. Am. Chem. Soc. 2000, 122, 2010–2018. [Google Scholar] [CrossRef]
- Putz, M.V. Contributions within Density Functional Theory with Applications to Chemical Reactivity Theory and Electronegativity; Dissertation.com: Parkland, FL, USA, 2003. [Google Scholar]
- Pearson, R.G. Chemical Hardness; Wiley-VCH: Weinheim, Germany, 1997. [Google Scholar]
- Mortier, W.J.; Ghosh, S.K.; Shankar, S. Electronegativity equalization method for the calculation of atomic charge in molecules. J. Am. Chem. Soc. 1986, 108, 4315–4320. [Google Scholar] [CrossRef]
- Mortier, W.J.; van Genechten, K.; Gasteiger, J. Electronegativity equalization: Application and parameterization. J. Am. Chem. Soc. 1985, 107, 829–835. [Google Scholar] [CrossRef]
- Mortier, W.J. Electronegativity equalization and its applications. Struct. Bond. 1987, 66, 125–143. [Google Scholar]
- Ray, K.; Samuels, L.; Parr, R.G. Studies of electronegativity equalization. J. Chem. Phys. 1979, 70, 3680–3684. [Google Scholar]
- Sanderson, R.T. Principles of electronegativity Part I. General nature. J. Chem. Edu. 1988, 65, 112–119. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Berkowitz, M. Density functional approach to frontier controlled reactions. J. Am. Chem. Soc. 1987, 109, 4823–4825. [Google Scholar] [CrossRef]
- Lewis, G.N. The atom and the molecule. J. Am. Chem. Soc. 1916, 38, 762–785. [Google Scholar] [CrossRef]
- Langmuir, I. The arrangement of electrons in atoms and molecules. J. Am. Chem. Soc. 1919, 41, 868–934. [Google Scholar] [CrossRef]
- Thomson, J.J. On the structure of the molecule and chemical combination. Philos. Mag. 1921, 41, 510–538. [Google Scholar] [CrossRef]
- Heitler, W.; London, F. Wechselwirkung neutraler Atome und homöopolare Bindung nach der Quantenmechanik. Z. Phys. 1927, 44, 455–472. [Google Scholar] [CrossRef]
- Hückel, E. Quantentheoretische beiträge zum benzolproblem. Z. Phys. 1931, 70, 204–286. [Google Scholar] [CrossRef]
- Pauling, L. Quantum mechanics and the chemical bond. Phys. Rev. 1931, 37, 1185–1186. [Google Scholar] [CrossRef]
- Roothaan, C.C.J. New developments in molecular orbital theory. Rev. Mod. Phys. 1951, 23, 69–89. [Google Scholar] [CrossRef]
- Pariser, R.; Parr, R. A semi-empirical theory of the electronic spectra and electronic structure of complex unsaturated molecules. I. J. Chem. Phys. 1953, 21, 466–471. [Google Scholar] [CrossRef]
- Pariser, R.; Parr, R. A semi-empirical theory of the electronic spectra and electronic structure of complex unsaturated molecules. II. J. Chem. Phys. 1953, 21, 767–776. [Google Scholar] [CrossRef]
- Pople, J.A. Electron interaction in unsaturated hydrocarbons. Trans. Faraday Soc. 1953, 49, 1375–1385. [Google Scholar] [CrossRef]
- Löwdin, P.O. Quantum theory of many-particle systems. I. Physical interpretations by means of density matrices, natural spin-orbitals, and convergence problems in the method of configurational interaction. Phys. Rev. 1955, 97, 1474–1489. [Google Scholar] [CrossRef]
- Löwdin, P.O. Quantum theory of many-particle systems. II. Study of the ordinary Hartree-Fock approximation. Phys. Rev. 1955, 97, 1490–1508. [Google Scholar] [CrossRef]
- Löwdin, P.O. Quantum theory of many-particle systems. III. Extension of the Hartree-Fock scheme to include degenerate systems and correlation effects. Phys. Rev. 1955, 97, 1509–1520. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Pople, J.A.; Binkley, J.S.; Seeger, R. Theoretical models incorporating electron correlation. Int. J. Quantum Chem. 1976, 10, 1–19. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. Quadratically convergent simultaneous optimization of wavefunction and geometry. Int. J. Quantum Chem. 1989, 36, 291–303. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F. Conceptual and computational DFT in the study of aromaticity. Chem. Rev. 2001, 101, 1451–1464. [Google Scholar]
- Pyykkö, P.; Zhao, L.-B. Search for effective local model potentials for simulation of QED effects in relativistic calculations. J. Phys. B 2003, 36, 1469–1478. [Google Scholar] [CrossRef]
- Szekeres, Z.; Exner, T.; Mezey, P.G. Fuzzy fragment selection strategies, basis set dependence and HF–DFT comparisons in the applications of the ADMA method of macromolecular quantum chemistry. Int. J. Quantum Chem. 2005, 104, 847–860. [Google Scholar] [CrossRef]
- Richard, M.M. Electronic Structure: Basic Theory and Practical Methods; Cambridge University Press: New York, NY, USA, 2004. [Google Scholar]
- Kohanoff, J. Electronic Structure Calculations for Solids and Molecules: Theory and Computational Methods; Cambridge University Press: New York, NY, USA, 2006. [Google Scholar]
- Sholl, D.; Steckel, J.A. Density Functional Theory: A Practical Introduction; Wiley-Interscience: Hoboken, NJ, USA, 2009. [Google Scholar]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Putz, M.V. Density functionals of chemical bonding. Int. J. Mol. Sci. 2008, 9, 1050–1095. [Google Scholar] [CrossRef]
- Capelle, K. A bird’s-eye view of density-functional theory. Braz. J. Phys. 2006, 36, 1318–1343. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. Principle of stationary action and the definition of a proper open system. Phys. Rev. B 1994, 49, 13348–13356. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A bond path: A universal indicator of bonded interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Fradera, X.; Solà, M. Electron localization and delocalization in open-shell molecules. J. Comput. Chem. 2002, 23, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Matito, E.; Solà, M.; Salvador, P.; Duran, M. Electron sharing indexes at the correlated level. Application to aromaticity calculations. Faraday Discuss 2007, 135, 325–345. [Google Scholar] [CrossRef]
- Putz, M.V. On relationship between electronic sharing in bonding and electronegativity equalization of atoms in molecules. Int. J. Chem. Model. 2011, 3, 371–384. [Google Scholar]
- Fuentealba, P. Reactivity indices and response functions in density functional theory. J. Mol. Struct. (Q.) 1998, 433, 113–118. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Kohn-Sham description of equilibria and charge transfer in reactive systems. Int. J. Quantum Chem. 1998, 69, 591–605. [Google Scholar] [CrossRef]
- Parr, R.G.; Bartolotti, L.J. On the geometric mean principle of electronegativity equalization. J. Am. Chem. Soc. 1982, 104, 3801–3803. [Google Scholar] [CrossRef]
- Kaya, S.; Kaya, C. A new method for calculation of molecular hardness: A theoretical study. Comput. Theor. Chem. 2015, 1060, 66–70. [Google Scholar] [CrossRef]
- Gomez, B.; Fuentealba, P.; Contreras, R. The maximum hardness and minimum polarizability principles as the basis for the study of reaction profiles. Theor. Chem. Acc. 2003, 110, 421–427. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Sengupta, S. Popular electronic structure principles in a dynamical context. J. Phys. Chem. 1996, 100, 16126–16130. [Google Scholar] [CrossRef]
- Glasser, L. Lattice energies of crystals with multiple ions: A generalized Kapustinskii equation. Inorg. Chem. 1995, 34, 4935–4936. [Google Scholar] [CrossRef]
- Kaya, S.; Kaya, C. A new equation for calculation of chemical hardness of groups and molecules. Mol. Phys. 2015, 113, 1311–1319. [Google Scholar] [CrossRef]
- von Szentpály, L. Hardness maximization or equalization? New insights and quantitative relations between hardness increase and bond dissociation energy. J. Mol. Model. 2017, 23, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ghanty, T.K.; Ghosh, S.K. Correlation between hardness, polarizability, and size of atoms, molecules, and clusters. J. Phys. Chem. 1993, 97, 4951–4953. [Google Scholar] [CrossRef]
- Glasser, L.; Jenkins, H.D.B. Predictive thermodynamics for condensed phases. Chem. Soc. Rev. 2005, 34, 866–874. [Google Scholar] [CrossRef]
- Jenkins, H.D.B.; Tudela, D.; Glasser, L. Lattice potential energy estimation for complex ionic salts from density measurements. Inorg. Chem. 2002, 41, 2364–2367. [Google Scholar] [CrossRef]
- Mallouk, T.E.; Rosenthal, G.L.; Mueller, G.; Brusasco, R.; Bartlett, N. Fluoride ion affinities of germanium tetrafluoride and boron trifluoride from thermodynamic and structural data for (SF3) 2GeF6, ClO2GeF5, and ClO2BF4. Inorg. Chem. 1984, 23, 3167–3173. [Google Scholar] [CrossRef]
- Jenkins, H.D.B.; Roobottom, H.K.; Passmore, J.; Glasser, L. Relationships among ionic lattice energies, molecular (formula unit) volumes, and thermochemical radii. Inorg. Chem. 1999, 38, 3609–3620. [Google Scholar] [CrossRef] [PubMed]
- Kaya, S.; Kaya, C. A simple method for the calculation of lattice energies of inorganic ionic crystals based on the chemical hardness. Inorg. Chem. 2015, 54, 8207–8213. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, E.; Chattaraj, P.K.; Fuentealba, P. Variation of the electrophilicity index along the reaction path. J. Phys. Chem. A 2003, 107, 7068–7072. [Google Scholar] [CrossRef] [PubMed]
- von Szentpály, L.; Kaya, S.; Karakuş, N. Why and when is electrophilicity minimized? New theorems and guiding rules. J. Phys. Chem. A 2020, 124, 10897–10908. [Google Scholar] [CrossRef]
- Cárdenas, C. The Fukui potential is a measure of the chemical hardness. Chem. Phys. Lett. 2011, 513, 127–129. [Google Scholar] [CrossRef]
- Cárdenas, C.; Tiznado, W.; Ayers, P.W.; Fuentealba, P. The Fukui potential and the capacity of charge and the global hardness of atoms. J. Phys. Chem. A 2011, 115, 2325–2331. [Google Scholar] [CrossRef]
- Kaya, S.; Robles-Navarro, A.; Mejía, E.; Gómez, T.; Cardenas, C. On the Prediction of Lattice Energy with the Fukui Potential: Some Supports on Hardness Maximization in Inorganic Solids. J. Phys. Chem. A 2022, 126, 4507–4516. [Google Scholar] [CrossRef]
- Badger, R.M. A relation between internuclear distances and bond force constants. J. Chem. Phys. 1934, 2, 128–131. [Google Scholar] [CrossRef]
- Pearson, R.G. A simple model for vibrational force constants. J. Am. Chem. Soc. 1977, 99, 4869–4875. [Google Scholar] [CrossRef]
- Nalewajski, R.F. A simple relation between the internuclear distances and force constants of diatomic molecules. J. Phys. Chem. 1979, 83, 2677–2682. [Google Scholar] [CrossRef]
- Kaya, S.; Kaya, C.; Obot, I.B.; Islam, N. A novel method for the calculation of bond stretching force constants of diatomic molecules. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 154, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Kaya, S.; Chamorro, E.; Petrov, D.; Kaya, C. New insights from the relation between lattice energy and bond stretching force constant in simple ionic compounds. Polyhedron 2017, 123, 411–418. [Google Scholar] [CrossRef]
- Kovács, A.; Esterhuysen, C.; Frenking, G. The Nature of the Chemical Bond Revisited: An Energy-Partitioning Analysis of Nonpolar Bonds. Chem. Eur. J. 2005, 11, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Comment on the Comparative Use of the Electron Density and Its Laplacian. Chem. Eur. J. 2006, 12, 7769–7772. [Google Scholar] [CrossRef] [PubMed]
- Frenking, G.; Esterhuysen, C.; Kovács, A. Reply to “Comment on the Comparative Use of the Electron Density and Its Laplacian”. Chem. Eur. J. 2006, 12, 7773–7774. [Google Scholar] [CrossRef]
- Poater, J.; Sola, M.; Bickelhaupt, F.M. Hydrogen–Hydrogen Bonding in Planar Biphenyl, Predicted by Atoms-In-Molecules Theory, Does Not Exist. Chem. Eur. J. 2006, 12, 2889–2895. [Google Scholar] [CrossRef]
- Bader, R.F.W. Pauli Repulsions Exist Only in the Eye of the Beholder. Chem. Eur. J. 2006, 12, 2896–2901. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Bickelhaupt, F.M. A Model of the Chemical Bond Must Be Rooted in Quantum Mechanics, Provide Insight, and Possess Predictive Power. Chem. Eur. J. 2006, 12, 2902–2905. [Google Scholar] [CrossRef]
- Putz, M.V. Graphenic Nanospace: Bondonic Entanglement Perspectives. Fuller. Nanotub. Carbon Nanostruct. 2022, 30. [Google Scholar] [CrossRef]
- Frenking, G.; Shaik, S. (Eds.) The Chemical Bond. Fundamental Aspects of Chemical Bonding; Wiley-VCH Verlag GmbH & Co.KGaA: Weinheim, Germany, 2014. [Google Scholar]
- Putz, M.V. Quantum Nanochemistry. A Fully Integrated Approach: Vol III. Quantum Molecules and Reactivity; Apple Academic Press & CRC Press: Toronto, ON, Canada; Waretown, NJ, USA, 2016; p. 579+index. ISBN 978-1-771881-35-7. [Google Scholar]
- Putz, M.V. Density Functional Theory of Bose-Einstein Condensation: Road to Chemical Bonding Quantum Condensate. Struct. Bond. 2012, 149, 1–50. [Google Scholar]
- Putz, M.V. Electronegativity: Quantum Observable. Int. J. Quantum Chem. 2009, 109, 733–738. [Google Scholar] [CrossRef]
- Putz, M.V. Chemical Hardness: Quantum Observable? Stud. Univ. Babeş-Bolyai-Ser. Chem. 2010, 55, 47–50. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaya, S.; Putz, M.V. Atoms-In-Molecules’ Faces of Chemical Hardness by Conceptual Density Functional Theory. Molecules 2022, 27, 8825. https://doi.org/10.3390/molecules27248825
Kaya S, Putz MV. Atoms-In-Molecules’ Faces of Chemical Hardness by Conceptual Density Functional Theory. Molecules. 2022; 27(24):8825. https://doi.org/10.3390/molecules27248825
Chicago/Turabian StyleKaya, Savas, and Mihai V. Putz. 2022. "Atoms-In-Molecules’ Faces of Chemical Hardness by Conceptual Density Functional Theory" Molecules 27, no. 24: 8825. https://doi.org/10.3390/molecules27248825
APA StyleKaya, S., & Putz, M. V. (2022). Atoms-In-Molecules’ Faces of Chemical Hardness by Conceptual Density Functional Theory. Molecules, 27(24), 8825. https://doi.org/10.3390/molecules27248825