
Investigation of the Effect of Substituents on Electronic and Charge Transport Properties of Benzothiazole Derivatives


Abstract
1. Introduction
2. Methods
2.1. Computational Details
2.2. Experimental Details
2.2.1. Synthesis of Comp1
2.2.2. Synthesis of Comp2
2.2.3. Crystal Structure of Comp2
2.2.4. Synthesis of Comp3
2.2.5. Synthesis of Comp4
3. Results and Discussion
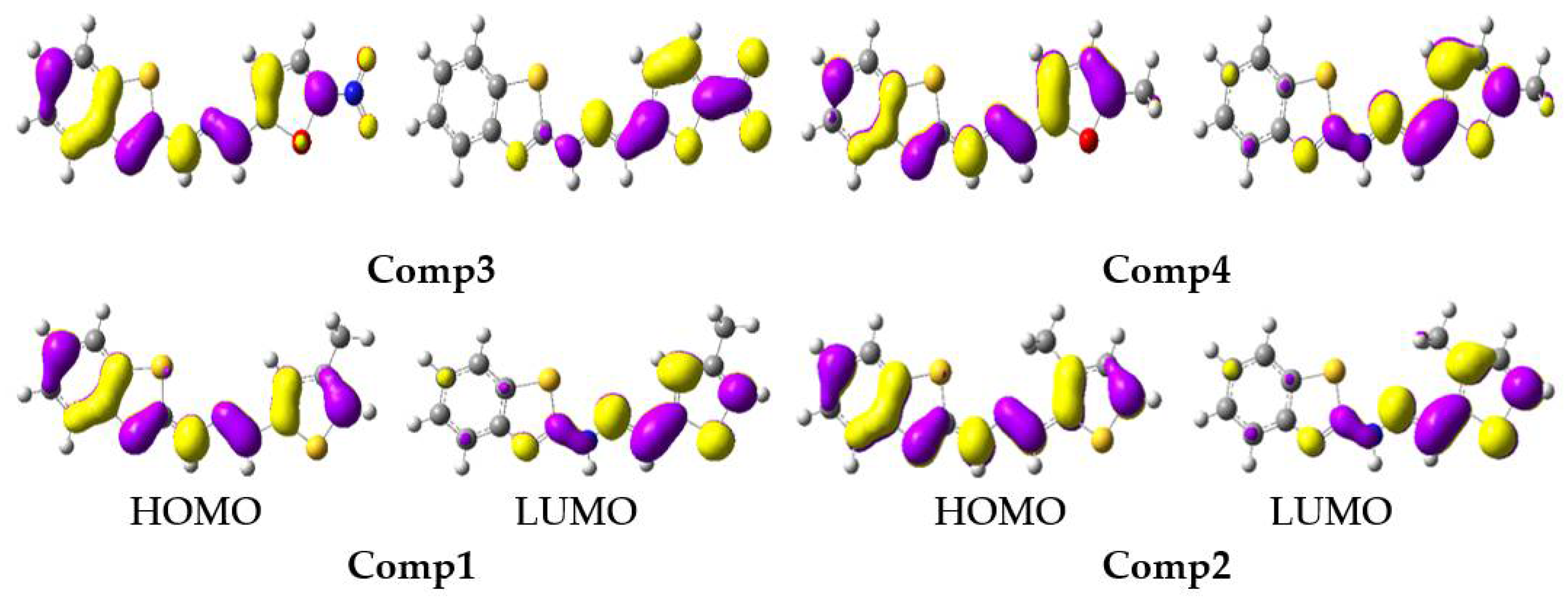
3.1. Electronic Properties
3.2. Absorption Spectra
3.3. Charge Transport Properties
3.4. Molecular Electrostatic Potential
3.5. UV–vis Experiment, Thin Film Preparation and Characterization
3.5.1. UV–vis Experiment
3.5.2. Thin Film Preparation
3.5.3. Thin Film Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reimers, J.R.; Picconnatto, C.A.; Ellenbogen, J.C.; Shashidhar, R. (Eds.) Molecular Electronics iii; New York Academy of Sciences: New York, NY, USA, 2003. [Google Scholar]
- Tour, J. Molecular Electronics; World Scientific: Singapore, 2003. [Google Scholar]
- Cunimberti, G.; Fagas, G.; Richter, K. (Eds.) Introducing Molecular Electronics. In Lecture Notes in Physics; Springer: Berlin, Germany, 2005. [Google Scholar]
- Habib, M.; Ghosh, N.N.; Sarkar, R.; Pramanik, A.; Sarkar, P.; Pal, S. Controlling the charge transfer and recombination dynamics in hollow zno qd based dye sensitized solar cell: An insight from ab initio simulation. Chem. Phys. Lett. 2018, 709, 21–25. [Google Scholar] [CrossRef]
- Pramanik, A.; Sarkar, S.; Pal, S.; Sarkar, P. Pentacene–fullerene bulk-heterojunction solar cell: A computational study. Phys. Lett. A 2015, 379, 1036–1042. [Google Scholar] [CrossRef]
- Bouit, P.-A.; Infantes, L.; Calbo, J.; Viruela, R.; Ortí, E.; Delgado, J.L.; Martín, N. Efficient light harvesters based on the 10-(1,3-dithiol-2-ylidene)anthracene core. Org. Lett. 2013, 15, 4166–4169. [Google Scholar] [CrossRef] [PubMed]
- Habib; Saha, S.; Sarkar, R.; Pramanik, A.; Sarkar, P.; Pal, S. Computational design of some TTF-substituted acene-based dyes for solar cell application using hollow ZnO quantum dot as acceptor. Comput. Theor. Chem. 2018, 1136–1137, 10–17. [Google Scholar] [CrossRef]
- Roy, P.; Biswas, S.; Pramanik, A.; Sarkar, P. Substitution induced carrier switching in s,n-heteroacene molecular junctions: A first principle analysis. Chem. Phys. Lett. 2018, 708, 87–93. [Google Scholar] [CrossRef]
- Irfan, A.; Muhammad, S.; Al-Sehemi, A.G.; Al-Assiri, M.S.; Kalam, A.; Chaudhry, A.R. The effect of anchoring groups on the electro-optical and charge injection in triphenylamine derivatives@ti6o12. J. Theor. Comput. Chem. 2015, 14, 1550027. [Google Scholar] [CrossRef]
- Irfan, A.; Al-Sehemi, A.G.; Al-Assiri, M.S. Push–pull effect on the electronic, optical and charge transport properties of the benzo [2,3-b]thiophene derivatives as efficient multifunctional materials. Comput. Theor. Chem. 2014, 1031, 76–82. [Google Scholar] [CrossRef]
- Şengez, B.; Doğruyol, Z.; San, S.E.; Kösemen, A.; Yılmaz, F.; Okutan, M.; Yerli, Y.; Demir, A.; Başaran, E. Use of side chain thiophene containing copolymer as a non-ionic gel-dielectric material for sandwich ofet assembly. Microelectron. Eng. 2013, 103, 111–117. [Google Scholar] [CrossRef]
- Szlachcic, P.; Danel, K.S.; Gryl, M.; Stadnicka, K.; Usatenko, Z.; Nosidlak, N.; Lewińska, G.; Sanetra, J.; Kuźnik, W. Organic light emitting diodes (oled) based on helical structures containing 7-membered fused rings. Dyes Pigm. 2015, 114, 184–195. [Google Scholar] [CrossRef]
- Jungsuttiwong, S.; Tarsang, R.; Surakhot, Y.; Khunchalee, J.; Sudyoadsuk, T.; Promarak, V.; Namuangruk, S. Theoretical study of α-fluorenyl oligothiophenes as color tunable emissive materials for highly efficient electroluminescent device. Org. Electron. 2012, 13, 1836–1843. [Google Scholar] [CrossRef]
- Marks, T.J.; Hersam, M.C. Materials science: Semiconductors grown large and thin. Nature 2015, 520, 631–632. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, A.; Prakasam, M.; Anbarasan, P.M. Influence of donor substitution at d-π-aarchitecture in efficient sensitizers for dye-sensitized solar cells: First-principle study. Bull. Mater. Sci. 2017, 40, 1389–1396. [Google Scholar] [CrossRef]
- Ghosh, N.N.; Habib, M.; Pramanik, A.; Sarkar, P.; Pal, S. Tuning the bodipy core for its potential use in dssc: A quantum chemical approach. Bull. Mater. Sci. 2018, 41, 56. [Google Scholar] [CrossRef]
- Kumar, M.S.; Charanadhar, N.; Srikanth, V.V.; Rao, K.B.S.; Raj, B. Materials in harnessing solar power. Bull. Mater. Sci. 2018, 41, 62. [Google Scholar] [CrossRef]
- Wang, D.; Huang, S.; Wang, C.; Yue, Y.; Zhang, Q. Computational prediction for oxidation and reduction potentials of organic molecules used in organic light-emitting diodes. Org. Electron. 2019, 64, 216–222. [Google Scholar] [CrossRef]
- Li, M.; Kou, L.; Diao, L.; Zhang, Q.; Li, Z.; Wu, Q.; Lu, W.; Pan, D. Theoretical Study of Acene-Bridged Dyes for Dye-Sensitized Solar Cells. J. Phys. Chem. A 2015, 119, 3299–3309. [Google Scholar] [CrossRef]
- Biswas, S.; Pramanik, A.; Pal, S.; Sarkar, P. A theoretical perspective on the photovoltaic performance of s,n-heteroacenes: An even–odd effect on the charge separation dynamics. J. Phys. Chem. C 2017, 121, 2574–2587. [Google Scholar] [CrossRef]
- Tarsang, R.; Promarak, V.; Sudyoadsuk, T.; Namuangruk, S.; Kungwan, N.; Khongpracha, P.; Jungsuttiwong, S. Triple bond-modified anthracene sensitizers for dye-sensitized solar cells: A computational study. RSC Adv. 2015, 5, 38130–38140. [Google Scholar] [CrossRef]
- Al-Horaibi, S.A.; Alrabie, A.A.; Alghamdi, M.T.; Al-Ostoot, F.H.; Garoon, E.M.; Rajbhoj, A.S. Novel hemicyanine sensitizers based on benzothiazole-indole for dye-sensitized solar cells: Synthesis, optoelectrical characterization and efficiency of solar cell. J. Mol. Struct. 2020, 1224, 128836. [Google Scholar] [CrossRef]
- Paczkowski, I.M.; Coelho, F.L.; Campo, L.F. 2,1,3-Benzothiadiazole dyes conjugated with benzothiazole and benzoxazole: Synthesis, solvatochromism and solid-state properties. J. Mol. Liq. 2020, 319, 114277. [Google Scholar] [CrossRef]
- Dutta, G.K.; Guha, S.; Patil, S. Synthesis of liquid crystalline benzothiazole based derivatives: A study of their optical and electrical properties. Org. Electron. 2010, 11, 1–9. [Google Scholar] [CrossRef]
- Al-Horaibi, S.A.; Asiri, A.M.; El-Shishtawy, R.M.; Gaikwad, S.T.; Rajbhoj, A.S. Indoline and benzothiazole-based squaraine dye-sensitized solar cells containing bis-pendent sulfonate groups: Synthesis, characterization and solar cell performance. J. Mol. Struct. 2019, 1195, 591–597. [Google Scholar] [CrossRef]
- Shehzad, R.A.; Iqbal, J.; Khan, M.U.; Hussain, R.; Javed, H.M.A.; Rehman, A.U.; Alvi, M.U.; Khalid, M. Designing of benzothiazole based non-fullerene acceptor (NFA) molecules for highly efficient organic solar cells. Comput. Theor. Chem. 2020, 1181, 112833. [Google Scholar] [CrossRef]
- Aziz, H.; Popovic, Z.D.; Hu, N.-X.; Hor, A.-M.; Xu, G. Degradation Mechanism of Small Molecule-Based Organic Light-Emitting Devices. Science 1999, 283, 1900–1902. [Google Scholar] [CrossRef]
- Grimsdale, A.C.; Chan, K.L.; Martin, R.E.; Jokisz, P.G.; Holmes, A.B. Synthesis of Light-Emitting Conjugated Polymers for Applications in Electroluminescent Devices. Chem. Rev. 2009, 109, 897–1091. [Google Scholar]
- Hughes, G.; Bryce, M.R. Electron-transporting materials for organic electroluminescent and electrophosphorescent devices. J. Mater. Chem. 2004, 15, 94–107. [Google Scholar] [CrossRef]
- Yoon, K.R.; Ko, S.-O.; Lee, S.M.; Lee, H. Synthesis and characterization of carbazole derived nonlinear optical dyes. Dye. Pigment. 2007, 75, 567–573. [Google Scholar] [CrossRef]
- Garnier, F. Organic-based electronics à la carte. Acc. Chem. Res. 1999, 32, 209–215. [Google Scholar] [CrossRef]
- Katz, H.E.; Bao, Z.; Gilat, S.L. Synthetic Chemistry for Ultrapure, Processable, and High-Mobility Organic Transistor Semiconductors. Acc. Chem. Res. 2001, 34, 359–369. [Google Scholar] [CrossRef]
- Shirota, Y.; Kageyama, H. Charge Carrier Transporting Molecular Materials and Their Applications in Devices. Chem. Rev. 2007, 107, 953–1010. [Google Scholar] [CrossRef]
- Izumi, T.; Kobashi, S.; Takimiya, K.; Aso, Y.; Otsubo, T. Synthesis and spectroscopic properties of a series of β-blocked long oligothiophenes up to the 96-mer: Revaluation of effective conjugation length. J. Am. Chem. Soc. 2003, 125, 5286–5287. [Google Scholar] [CrossRef] [PubMed]
- Meier, H.; Gerold, J.; Kolshorn, H.; Mühling, B. Extension of Conjugation Leading to Bathochromic or Hypsochromic Effects in OPV Series. Chem.-A Eur. J. 2004, 10, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.; Li, Z.H.; Tao, A.Y.; D’Iorio, M. Synthesis and Functional Properties of Donor−Acceptor π-Conjugated Oligomers. Chem. Mater. 2003, 15, 1198–1203. [Google Scholar] [CrossRef]
- Schweikart, K.-H.; Hanack, M.; Lüer, L.; Oelkrug, D. Synthesis, absorption and luminescence of a new series of soluble distyrylbenzenes featuring cyano substituents at the peripheral rings. Eur. J. Org. Chem. 2001, 2001, 293–302. [Google Scholar] [CrossRef]
- Lee, T.H.; Tong, K.L.; So, S.K.; Leung, L.M. Synthesis and electroluminescence of thiophene-based bipolar small molecules with different arylamine moieties. Synth. Met. 2005, 155, 116–124. [Google Scholar] [CrossRef]
- Cao, D.-X.; Fang, Q.; Wang, D.; Liu, Z.-Q.; Xue, G.; Xu, G.-B.; Yu, W.-T. Synthesis and two-photon-excited fluorescence of benzothiazole-based compounds with various π-electron donors. Eur. J. Org. Chem. 2003, 2003, 3628–3636. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Q.; Ren, H.; Yan, H.; Zhang, J.; Zhang, H.; Gu, J. Calculation of band gap in long alkyl-substituted heterocyclic-thiophene-conjugated polymers with electron donor–acceptor fragment. Sol. Energy Mater. Sol. Cells 2008, 92, 581–587. [Google Scholar] [CrossRef]
- Hutchison, G.R.; Ratner, M.A.; Marks, T.J. Accurate Prediction of Band Gaps in Neutral Heterocyclic Conjugated Polymers. J. Phys. Chem. A 2002, 106, 10596–10605. [Google Scholar] [CrossRef]
- Kwon, O.; McKee, M.L. Theoretical Calculations of Band Gaps in the Aromatic Structures of Polythieno[3,4-b]benzene and Polythieno[3,4-b]pyrazine. J. Phys. Chem. A 2000, 104, 7106–7112. [Google Scholar] [CrossRef]
- Hassanali, A.A.; Li, T.; Zhong, D.; Singer, S.J. A Molecular Dynamics Study of Lys-Trp-Lys: Structure and Dynamics in Solution Following Photoexcitation. J. Phys. Chem. B 2006, 110, 10497–10508. [Google Scholar] [CrossRef]
- Irfan, A.; Imran, M.; Thomas, R.; Basra, M.A.R.; Ullah, S.; Al-Sehemi, A.G.; Assiri, M.A. Exploring the effect of oligothiophene and acene cores on the optoelectronic properties and enhancing p- and n-type ability of semiconductor materials. J. Sulfur Chem. 2020, 42, 180–192. [Google Scholar] [CrossRef]
- Irfan, A. Push-pull effect on the charge transport characteristics in V-shaped organic semiconductor materials. Bull. Mater. Sci. 2021, 44, 43. [Google Scholar] [CrossRef]
- Wazzan, N.; Irfan, A. Promising architectures modifying the d-π-a architecture of 2,3-dipentyldithieno[3,2-f:2′,3′-h]quinoxaline-based dye as efficient sensitizers in dye-sensitized solar cells: A dft study. Mater. Sci. Semicond. Process. 2020, 120, 105260. [Google Scholar] [CrossRef]
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R.; El-Kerdawy, M.M.; Ni, N.; Shaaban, M.I. Synthesis, antimicrobial, antiquorum-sensing and cytotoxic activities of new series of benzothiazole derivatives. Chin. Chem. Lett. 2015, 26, 1522–1528. [Google Scholar] [CrossRef]
- Soni, B.; Ranawat, M.S.; Sharma, R.; Bhandari, A.; Sharma, S. Synthesis and evaluation of some new benzothiazole derivatives as potential antimicrobial agents. Eur. J. Med. Chem. 2010, 45, 2938–2942. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. Iii. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Tang, S.-S.; Liu, J.-B.; Chen, G.; Jin, R.-F. Theoretical study on electronic and charge transfer properties of oligo[8]thiophene and its circular, hooped, and helical derivatives. Chin. J. Struct. Chem. 2014, 33, 104–114. [Google Scholar]
- Li, P.; Bu, Y.; Ai, H. Theoretical Determinations of Ionization Potential and Electron Affinity of Glycinamide Using Density Functional Theory. J. Phys. Chem. A 2004, 108, 1200–1207. [Google Scholar] [CrossRef]
- Furche, F.; Ahlrichs, R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 2002, 117, 7433–7447. [Google Scholar] [CrossRef]
- Frisch, G.W.T.M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A. 01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Takano, Y.; Houk, K.N. Benchmarking the Conductor-like Polarizable Continuum Model (CPCM) for Aqueous Solvation Free Energies of Neutral and Ionic Organic Molecules. J. Chem. Theory Comput. 2004, 1, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www2.chemistry.msu.edu/faculty/harrison/cem483/work_functions.pdf (accessed on 25 April 2021).
- Sonar, P.; Singh, S.P.; Leclère, P.; Surin, M.; Lazzaroni, R.; Lin, T.T.; Dodabalapur, A.; Sellinger, A. Synthesis, characterization and comparative study of thiophene–benzothiadiazole based donor–acceptor–donor (d–a–d) materials. J. Mater. Chem. 2009, 19, 3228–3237. [Google Scholar] [CrossRef]
- Nikolka, M.; Nasrallah, I.; Rose, B.; Ravva, M.K.; Broch, K.; Sadhanala, A.; Harkin, D.; Charmet, J.; Hurhangee, M.; Brown, A.; et al. High operational and environmental stability of high-mobility conjugated polymer field-effect transistors through the use of molecular additives. Nat. Mater. 2017, 16, 356–362. [Google Scholar] [CrossRef]
- De Leeuw, D.; Simenon, M.; Brown, A.; Einerhand, R. Stability of n-type doped conducting polymers and consequences for polymeric microelectronic devices. Synth. Met. 1997, 87, 53–59. [Google Scholar] [CrossRef]
- Abbaszadeh, D.; Kunz, A.; Kotadiya, N.B.; Mondal, A.; Andrienko, D.; Michels, J.J.; Wetzelaer, G.-J.A.H.; Blom, P.W.M. Electron trapping in conjugated polymers. Chem. Mater. 2019, 31, 6380–6386. [Google Scholar] [CrossRef]
- Usta, H.; Risko, C.; Wang, Z.; Huang, H.; Deliomeroglu, M.K.; Zhukhovitskiy, A.; Facchetti, A.; Marks, T.J. Design, synthesis, and characterization of ladder-type molecules and polymers. Air-stable, solution-processable n-channel and ambipolar semiconductors for thin-film transistors via experiment and theory. J. Am. Chem. Soc. 2009, 131, 5586–5608. [Google Scholar] [CrossRef]
- Felscia, U.R.; Rajkumar, B.J.M.; Mary, M.B. Charge transport properties of pyrene and its derivatives: Optoelectronic and nonlinear optical applications. J. Mater. Sci. 2018, 53, 15213–15225. [Google Scholar] [CrossRef]
- Yang, G.; Su, Z.; Qin, C. Theoretical study on the second-order nonlinear optical properties of asymmetric spirosilabifluorene derivatives. J. Phys. Chem. A 2006, 110, 4817–4821. [Google Scholar] [CrossRef]
- Wazzan, N.; El-Shishtawy, R.M.; Irfan, A. DFT and TD–DFT calculations of the electronic structures and photophysical properties of newly designed pyrene-core arylamine derivatives as hole-transporting materials for perovskite solar cells. Theor. Chim. Acta 2017, 137, 9. [Google Scholar] [CrossRef]
- Wazzan, N.; Irfan, A. Theoretical study of triphenylamine-based organic dyes with mono-, di-, and tri-anchoring groups for dye-sensitized solar cells. Org. Electron. 2018, 63, 328–342. [Google Scholar] [CrossRef]
- Brédas, J.-L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Charge-Transfer and Energy-Transfer Processes in π-Conjugated Oligomers and Polymers: A Molecular Picture. Chem. Rev. 2004, 104, 4971–5004. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, W.; Ma, Y. Molecular design toward good hole transport materials based on anthra[2,3-c]thiophene: A theoretical investigation. Comput. Theor. Chem. 2013, 1010, 25–31. [Google Scholar] [CrossRef]
- Chai, W.; Jin, R. Theoretical investigations into optical and charge transfer properties of donor-acceptor 1,8-naphthalimide derivatives as possible organic light-emitting materials. J. Mol. Struct. 2016, 1103, 177–182. [Google Scholar] [CrossRef]
- Gruhn, N.E.; Filho, D.A.D.S.; Bill, T.G.; Malagoli, M.; Coropceanu, V.; Kahn, A.; Brédas, J.-L. The Vibrational Reorganization Energy in Pentacene: Molecular Influences on Charge Transport. J. Am. Chem. Soc. 2002, 124, 7918–7919. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | EHOMO | ELUMO | Egap | EHOMO | ELUMO | Egap |
|---|---|---|---|---|---|---|
| Ground State | Excited State | |||||
| Comp1 | −5.59 | −1.95 | 3.64 | −4.77 | −2.61 | 2.16 |
| Comp2 | −5.58 | −1.88 | 3.70 | −4.76 | −2.55 | 2.21 |
| Comp3 | −6.18 | −3.35 | 2.83 | −5.13 | −4.01 | 1.12 |
| Comp4 | −5.52 | −1.92 | 3.60 | −4.73 | −2.56 | 2.17 |
| Compounds | HIE (Ag) | EIE (Ag) | HIE (Al) | EIE (Al) |
|---|---|---|---|---|
| Comp1 | 0.85 | 2.79 | 1.51 | 2.13 |
| Comp2 | 0.84 | 2.86 | 1.50 | 2.20 |
| Comp3 | 1.44 | 1.39 | 2.10 | 0.73 |
| Comp4 | 0.78 | 1.82 | 1.44 | 2.16 |
| Comp | λmax | f | Tran | %Con | λmax | f | Tran | %Con |
|---|---|---|---|---|---|---|---|---|
| Gas phase | In Ethanol | |||||||
| Comp1 | 358 | 0.8839 | H→L | 70% | 363 | 1.1034 | H→L | 70% |
| 281 | 0.2282 | H→L + 1 | 30% | 281 | 0.1313 | H→L + 1 | 29% | |
| Comp2 | 357 | 0.8301 | H→L | 70% | 359 | 1.0638 | H→L | 70% |
| 278 | 0.2280 | H→L + 1 | 35% | 277 | 0.1452 | H→L + 1 | 39% | |
| Comp3 | 451 | 0.6867 | H→L | 70% | 481 | 0.8210 | H→L | 70% |
| 323 | 0.4853 | H→L + 1 | 27% | 335 | 0.4279 | H→L + 1 | 23% | |
| Comp4 | 362 | 0.9571 | H→L | 70% | 367 | 1.1864 | H→L | 70% |
| 284 | 0.2341 | H→L + 1 | 29% | 283 | 0.1312 | H→L + 1 | 31% | |
| Comp | λmax | f | Tran | %Con | λmax | f | Tran | %Con |
| In acetone | In DMF | |||||||
| Comp1 | 363 | 1.1018 | H→L | 70% | 364 | 1.1213 | H→L | 70% |
| 281 | 0.1323 | H→L + 1 | 29% | 281 | 0.1280 | H→L + 1 | 29% | |
| Comp2 | 359 | 1.0620 | H→L | 70% | 360 | 1.0840 | H→L | 70% |
| 277 | 0.1461 | H→L + 1 | 39% | 277 | 0.1429 | H→L + 1 | 39% | |
| Comp3 | 481 | 0.8202 | H→L | 70% | 484 | 0.8388 | H→L | 70% |
| 335 | 0.4287 | H→L + 1 | 23% | 336 | 0.4224 | H→L + 1 | 23% | |
| Comp4 | 367 | 1.1847 | H→L | 70% | 368 | 1.2041 | H→L | 70% |
| 283 | 0.1322 | H→L + 1 | 31% | 283 | 0.1280 | H→L + 1 | 31% | |
| Comp | λmax | f | Tran | %Con | ||||
| In DMSO | ||||||||
| Comp1 | 364 | 1.1189 | H→L | 70% | ||||
| 281 | 0.1278 | H→L + 1 | 29% | |||||
| Comp2 | 360 | 1.0813 | H→L | 70% | ||||
| 277 | 0.1425 | H→L + 1 | 39% | |||||
| Comp3 | 484 | 0.8357 | H→L | 70% | ||||
| 336 | 0.4228 | H→L + 1 | 23% | |||||
| Comp4 | 368 | 1.2018 | H→L | 70% | ||||
| 283 | 0.1278 | H→L + 1 | 31% | |||||
| Compounds | IPa | EAa | IPv | EAv | |||
|---|---|---|---|---|---|---|---|
| Comp1 | 7.07 | 0.41 | 6.96 | 0.52 | 0.235 | 0.327 | 0.092 |
| Comp2 | 7.05 | 0.36 | 6.95 | 0.46 | 0.233 | 0.340 | 0.107 |
| Comp3 | 7.64 | 2.09 | 7.52 | 1.85 | 0.216 | 0.487 | 0.271 |
| Comp4 | 6.98 | 0.39 | 6.88 | 0.51 | 0.249 | 0.320 | 0.071 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irfan, A.; Kalam, A.; Al-Sehemi, A.G.; Dubey, M. Investigation of the Effect of Substituents on Electronic and Charge Transport Properties of Benzothiazole Derivatives. Molecules 2022, 27, 8672. https://doi.org/10.3390/molecules27248672
Irfan A, Kalam A, Al-Sehemi AG, Dubey M. Investigation of the Effect of Substituents on Electronic and Charge Transport Properties of Benzothiazole Derivatives. Molecules. 2022; 27(24):8672. https://doi.org/10.3390/molecules27248672
Chicago/Turabian StyleIrfan, Ahmad, Abul Kalam, Abdullah G. Al-Sehemi, and Mrigendra Dubey. 2022. "Investigation of the Effect of Substituents on Electronic and Charge Transport Properties of Benzothiazole Derivatives" Molecules 27, no. 24: 8672. https://doi.org/10.3390/molecules27248672
APA StyleIrfan, A., Kalam, A., Al-Sehemi, A. G., & Dubey, M. (2022). Investigation of the Effect of Substituents on Electronic and Charge Transport Properties of Benzothiazole Derivatives. Molecules, 27(24), 8672. https://doi.org/10.3390/molecules27248672