3.1. Chemistry
All the reactions were monitored by thin-layer chromatography (TLC) on Merck 60 F254 (0.25 mm) plates, which were visualized by UV inspection (254 nm) and/or by spraying KMnO
4 (0.5 g in 100 mL 0.1 N NaOH). Na
2SO
4 was used as drying agent for the organic phases. Flash chromatography (FC) purifications were performed using silica gel Merck with 60 mesh particles. Unless otherwise specified, all reagents were used as received without further purification. Dichloromethane was dried over P
2O
5 and freshly distilled under nitrogen prior to use. DMF was stored over 3Å molecular sieves. Anhydrous THF was freshly distilled under nitrogen from Na/benzophenone ketyl.
1H and
13C-NMR spectra were registered on JEOL ECZR600 spectrometer, at 600 and 151 MHz. Coupling constants (J) are given in Hertz (Hz) and chemical shifts (δ) are given in ppm, calibrated to solvent signal as internal standard. The following abbreviations are used to describe multiplicities: s = singlet, d = doublet, t = triplet, q = quadruplet, m = multiplet and br = broad signal. The following abbreviation is used to identify the exact proton: ArH = aromatic proton. ESI-mass spectra were recorded on a Waters Micromass Quattro Micro equipped with an ESI source. Semi-preparative HPLC purifications were carried out on a Varian Pro-Star 210 chromatograph equipped with a variable wavelength detector (Prostar 325). The chromatography was performed using a 5 μM particle size Hibar LiChrosper C18 endcapped prepacked column (250 × 25 mm), with a flow rate of 20 mL/min; UV detection was performed at 226 and 240 nm. Samples of the mixture were dissolved (ca 0.5%) in mobile phase. The purity of target compounds was checked by RP-HPLC. Analyses were performed on an HP1100 chromatograph system (Agilent Technologies, Palo Alto, CA, USA) equipped with a quaternary pump (G1311A), a membrane degasser (G1379A) and a diode-array detector (DAD) (G1315B) integrated in the HP1100 system. Data analyses were processed by HP ChemStation system (Agilent Technologies). The analytical column was a LiChrospher
® 100 C18-e (250 × 4.6 mm, 5 µm) (Merck KGaA, 64271 Darmstadt, Germany) eluted with acetonitrile/0.1% TFA in a ratio depending on the characteristics of the compound. All compounds were dissolved in the mobile phase at a concentration of about 0.1 mg/mL and injected through a 20 μL loop. HPLC retention times (t
R) were obtained at flow rates of 1.0 mL min
−1, and the column effluent was monitored at 226, 254 and 300 nm, referenced against 800 nm. The purity of the compounds was evaluated as a percentage ratio between the areas of the main peak and of possible impurities at the three wavelengths, and also using DAD purity analysis of the chromatographic peak. The purity of all the target compounds was found to be ≥95% (
Table S4 and
Figure S7). Compound P5B was a kind gift of Prof. Frank Sicheri (Center for Molecular, Cell and Systems Biology, Lunenfeld-Tanenbaum Research Institute, Sinai Health System, Toronto, Ontario, Canada).
Encorafenib (1; 0.350 g; 6.49 mmol) was dissolved in ethanol (10 mL), and KOH 6M aqueous solution (10 mL) was added. The mixture was stirred at 80 °C for 7 h. The reaction mixture was neutralized with HCl 6M to pH 7 and the organic solvent evaporated under reduced pressure. The obtained precipitate was filtered, and the filtrate was extracted with THF (3 × 15 mL). The organic phases and the collected precipitate were combined and purified by flash chromatography using DCM/MeOH 9/1+ 0.1% NH3 as the eluent. Compound 16 was obtained in 85% yield as a white solid. 1H NMR (CD3OD) δ: 8.35 (s, 1H), 8.14 (d, J = 5.3 Hz, 1H), 7.51 (dd, J = 6.5, 2.5 Hz, 1H), 7.17 (s, 1H), 6.74 (s, 1H), 4.62–4.58 (m, 1H), 3.32 (s, 1H), 3.28 (dd, J = 3.1, 1.5 Hz, 1H), 2.96 (s, 3H), 1.55 (d, J = 6.7 Hz, 6H), 1.15 (s, 3H). 13C NMR (CD3OD) δ: 162.27 (s), 160.51 (s), 157.64 (s), 151.79 (d, J = 245.9 Hz), 143.27 (s), 129.61 (s), 128.57 (d, J = 2.7 Hz), 124.44 (d, J = 16.1 Hz), 123.63 (d, J = 12.6 Hz), 119.58 (s), 107.41 (s), 54.68 (s), 44.57 (s), 39.27 (s), 21.70 (s), 15.34 (s). MS (ESI−): m/z 480/482 [M-H]−; MS (ESI+): m/z 482/484 [MH+].
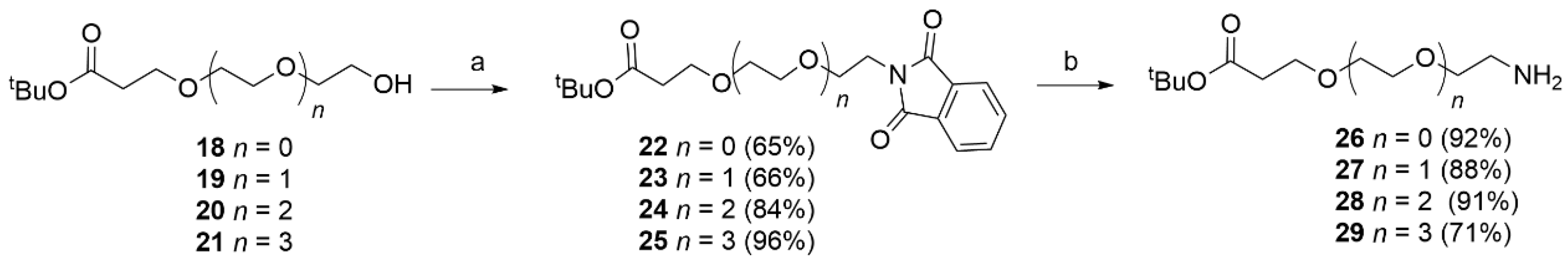
Phthalimide (1.4 eq), the appropriate alcohol (18–21) (1 eq) and triphenylphosphine (1.4 eq) were dissolved in dry THF (20 mL). After 15 min, DIAD (1.4 eq) was added dropwise at rt. After 12 h under stirring, EtOH (1 mL) was added to quench the reaction. The solvent was removed and the obtained residue purified by flash chromatography (PE/EtOAc 7.5/2.5 to 1/1) to afford the desired product. The obtained compounds were characterized by MS and used directly in the next step.
Alcohol 18 (1.39 g, 7.30 mmol) was reacted with phthalimide (1.5 g, 0.01 mmol), PPh3 (2.68 g, 0.01 mol) and DIAD (2.06 g, 0.01 mol, 2 mL) in dry THF (25 mL) using the general procedure described above to obtain 22 (1.53 g, 4.79 mmol, 65%) as a white solid. MS (ESI+): m/z 320 [MH+].
Alcohol 19 (1.5 g, 6.40 mmol) was reacted with phthalimide (1.14 g, 7.80 mmol), PPh3 (2.04 g, 7.8 mmol) and DIAD (1.5 g, 7.8 mmol, 1.46 mL) in dry THF (25 mL) using the general procedure described above to obtain 23 (1.98 g, 5.46 mmol, 70%) as a white solid. MS (ESI+): m/z 364 [MH+].
Alcohol 20 (0.694 g, 2.49 mmol) was reacted with phthalimide (0.513 g, 3.49 mmol) PPh3 (0.915 g, 3.49 mmol) and DIAD (0.706 g, 3.49 mmol, 0.687 mL) in dry THF (10 mL) using the general procedure reported above to obtained 24 (0.095 g, 9.54 mmol, 94%) as a white solid. MS (ESI+): m/z 408 [MH+].
Tert-butyl 1-(1,3-dioxoisoindolin-2-yl)-3,6,9,12-tetraoxapentadecan-15-oate (25).
Alcohol 21 (1.43 g, 4.44 mmol) was reacted with phthalimide (0.914 g, 6.21 mmol) PPh3 (2.25 g, 6.21 mmol) and DIAD (1.25 g, 6.21 mmol, 1.25 mL) in dry THF (20 mL) using the general procedure reported above to give 25 (1.92 g, 4.25 mmol, 96%) as a white solid. MS (ESI+): m/z 452 [MH+].
Phthalimide derivatives 22–25 and 40 were dissolved in EtOH and NH2NH2·2H2O was added at rt. The reaction mixture was stirred overnight. The solvent was removed and chloroform was added to the residue and stirred for 15 min. The formed precipitate was filtered off. The filtrate was concentrated under reduced pressure and the crude product was purified using flash chromatography (DCM/MeOH 95/5 to 90/10 + 0.1% NH3).
Compound
22 (0.50 g, 1.55 mmol) was treated with NH
2NH
2·2H
2O (0.39 g, 7.82 mmol) in EtOH (15 mL) using the general procedure reported above to obtain
26 (0.27 g, 1.44 mmol, 92%) as a colorless oil. MS (ESI
+):
m/
z 452 [MH
+].
1H NMR (600 MHz, CDCl
3)
δ 3.69 (t,
J = 6.5 Hz, 2H), 3.44 (t,
J = 5.2 Hz, 2H), 2.83 (t,
J = 5.2 Hz, 2H), 2.48 (t,
J = 6.5 Hz, 2H), 1.44 (s, 9H), in agreement with that reported in the literature [
32].
Compound
23 (0.70 g, 1.92 mmol) was treated with NH
2NH
2·2H
2O (0.48 g, 9.63 mmol) in EtOH (15 mL) using the general procedure described above to obtain
27 (0.41 g, 1.78 mmol, 93%) as a colorless oil. MS (ESI
+):
m/
z [MH
+] 234.
1H NMR (CDCl
3) δ: 3.71–3.66 (m, 2H), 3.64–3.57 (m, 4H), 3.55–3.49 (m, 2H), 2.87 (t,
J = 4.9 Hz, 2H), 2.47 (dd,
J = 12.2, 5.9 Hz, 2H), 2.37 (s, 2H), 1.41 (s, 9H).
13C NMR (151 MHz, CDCl
3) δ 170.9, 80.7, 73.6, 70.5, 70.3, 66.9, 41.6, 36.3, 28.2; in agreement with the reported data [
32].
Compound 24 (0.93 g, 2.28 mmol) was treated with NH2NH2·2H2O (1.14 g, 0.022 mol) in EtOH (15 mL) using the general procedure described above to obtain 28 (0.578 g, 2.07 mmol, 91%) as a colorless oil. 1H NMR (CDCl3) δ: 3.71–3.66 (m, 2H), 3.64–3.57 (m, 8H), 3.55–3.49 (m, 2H), 2.87 (t, J = 4.9 Hz, 2H), 2.47 (dd, J = 12.2, 5.9 Hz, 2H), 2.37 (s, 2H), 1.41 (d, J = 4.8 Hz, 9H). 13C NMR (151 MHz, CDCl3) δ 170.8, 80.7, 72.8, 70.5 (multiple overlapping peaks), 70.3, 67.0, 41.6, 36.3, 28.2. MS (ESI+): m/z 279 [MH+].
Compound
25 (0.959 g, 2.12 mmol) was treated with NH
2NH
2·2H
2O (0.53 g, 0.01 mol) in EtOH (15 mL) using the general procedure described above to obtain
29 (0.49 g, 1.52 mmol, 71%) as a colorless oil. MS (ESI
+):
m/
z 322 [MH
+].
1H NMR (400 MHz, CDCl3)
δ 3.70 (t,
J = 6.5 Hz, 2H), 3.66−3.57 (m, 8H), 3.50 (t,
J = 5.2 Hz, 2H), 2.86 (t,
J = 5.2 Hz, 2H), 2.49 (t,
J = 6.5 Hz, 2H), 1.43 (s, 9H); in agreement with that reported in the literature [
32].
Compound 40 (0.235 g, 0.528 mmol) was treated with NH2NH2·2H2O (0.264 g, 5.28 mol) in EtOH (4 mL) using the general procedure described above to obtain 41 (0.125 g, 0.398 mmol, 75%) as a colorless oil. MS (ESI+): m/z 315 [MH+]. 1H NMR (400 MHz, CDCl3) δ 7.66 (s, 1H), 4.44 (m, 2H), 3.96 (s, 2H), 3.78 (m, 2H), 3.62 (m, 2H), 3.52 (m, 4H), 3.35 (m, 2H), 2.42 (t, J = 6.4 Hz, 2H), 1.37 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 170.9, 148.0, 122.5, 80.7, 70.5, 70.3, 69.5, 66.9, 50.2, 37.2, 36.3, 28.1.
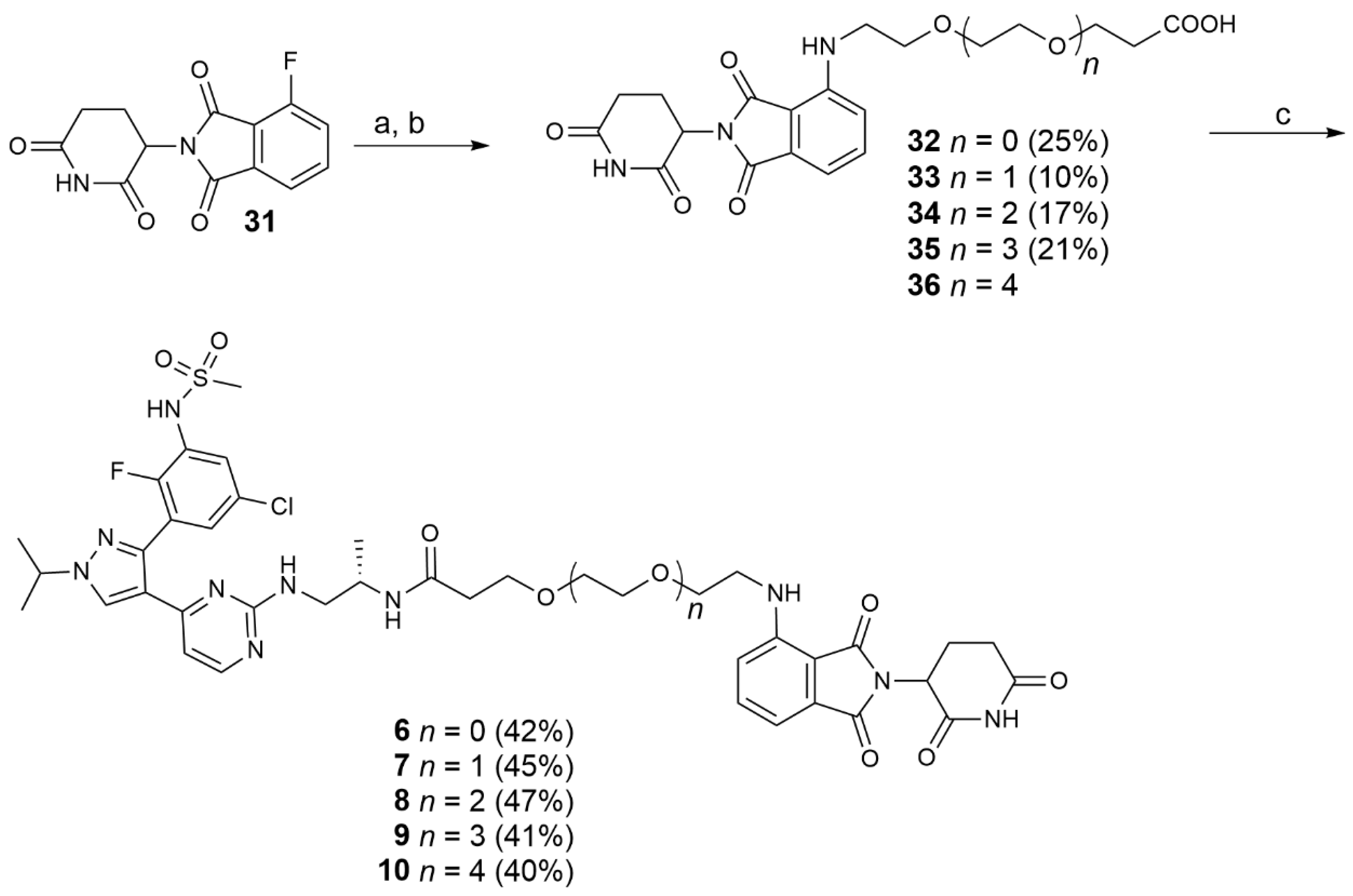
Amines 26–29 and 41(1 eq) were dissolved in DMF, DIPEA (2 eq) and 31 (1 eq) were added and the reaction mixture was stirred at 90 °C for 16 h. When the reaction was complete (TLC), the solvent was removed and the obtained residue treated with water (5 mL), extracted with DCM (3 × 20 mL) and the combined organic phases washed with brine (20 mL). After evaporation of the solvent, the protected intermediate was purified by flash chromatography (PE/EtOAc 1/1) to afford the tert-butyl protected acids. The purified protected intermediates were dissolved in a solution of TFA 10% in DCM and stirred overnight at rt. The solvent was evaporated and the precipitate washed several times with DCM. The product was purified by flash chromatography (DCM/MeOH 98/2 to 95/5) to afford the desired acids 32–35.
Compound 31 (0.357 g, 1.44 mmol) was reacted with 26 (0.273 g, 1.44 mmol) and DIPEA (0.558 g; 4.32 mmol) in DMF (3 mL) using the general procedure reported above to give, after purification by flash chromatography (PE/EtOAc 1/1), the desired protected acid as a yellow oil. This intermediate was hydrolyzed with a 10% TFA in DCM solution (10 mL). The crude product was purified by flash chromatography (DCM/MeOH 98/2) to give 32 (0.141 g, 0.363 mmol, 25% yield) as a yellow solid. 1H NMR (CDCl3) δ: 9.03 (s, 1H), 7.44 (dd, J = 8.5, 7.1 Hz, 1H), 7.05 (d, J = 7.1 Hz, 1H), 6.87 (d, J = 8.6 Hz, 1H), 6.44 (s, 1H), 5.00–4.85 (m, 1H), 3.73 (t, J = 6.0 Hz, 2H), 3.67 (t, J = 5.3 Hz, 2H), 3.42 (s, 2H), 2.88–2.66 (m, 3H), 2.58 (t, J = 6.0 Hz, 2H), 2.12–2.01 (m, 1H). 13C NMR (CDCl3) δ: 175.8, 172.2, 169.4, 167.79 (s), 146.92 (s), 136.09 (s), 132.51 (s), 117.05 (s), 111.77 (s), 110.35 (s), 69.30 (s), 66.31 (s), 48.90 (s), 42.26 (s), 34.76 (s), 31.40 (s), 22.78 (s). MS (ESI−): m/z 388 [M-H].
Compound 31 (0.389 g, 1.41 mmol) was reacted with 27 (0.329 g, 1.41 mmol) and DIPEA (0.364 g, 2,82 mmol) in DMF (3 mL) using the general procedure reported above to give, after purification by flash chromatography (PE/EtOAc 1/1), the desired protected acid as a yellow oil. This intermediate was hydrolyzed with 10% TFA in DCM solution (10 mL). The crude product was purified by flash chromatography (DCM/MeOH 98/2) to give 33 (0.06 g, 0.138 mmol, 10% yield) as a yellow solid. 1H NMR (CD3CN) δ: 9.06 (s, 1H), 7.56–7.44 (m, 1H), 7.05–6.93 (m, 2H), 6.45 (s, 1H), 4.98–4.84 (m, 1H), 3.65–3.59 (m, 5H), 3.54 (td, J = 5.6, 2.8 Hz, 5H), 3.42 (t, J = 4.5 Hz, 3H), 2.80–2.57 (m, 4H), 2.51–2.41 (m, 2H), 2.06 (dd, J = 8.6, 3.7 Hz, 1H), 1.91 (dd, J = 4.8, 2.4 Hz, 1H). 13C NMR (CD3CN) δ: 172.6, 172.3, 169.8, 169.5, 167.7, 146.9, 136.2, 132.6, 117.3, 110.9, 109.9, 70.1, 70.0, 69.1, 66.4, 48.9, 42.0, 34.4, 31.1, 22.4. MS (ESI−): m/z 432 [M-H]−.
Compound 31 (0.337 g, 1.22 mmol) was reacted with 28 (0.339 g, 1.22 mmol) and DIPEA (0.315 g, 2.44 mmol) in DMF (3 mL) using the general procedure reported above to give, after purification by flash chromatography (PE/EtOAc 1/1), the desired protected acid as a yellow oil. This intermediate was hydrolyzed with 10% TFA in DCM solution (10 mL). The crude product was purified by flash chromatography (DCM/MeOH 98/2) to give 34 (0.101 g, 0.211 mmol, 17% yield) as a yellow solid. 1H NMR (CDCl3) δ: 8.94 (s, 1H), 7.46 (dd, J = 8.5, 7.1 Hz, 1H), 7.07 (d, J = 7.1 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 4.92 (dd, J = 12.3, 5.4 Hz, 1H), 3.78–3.58 (m, 13H), 3.49–3.40 (m, 2H), 2.76–2.60 (m, 3H), 2.59 (t, J = 6.2 Hz, 2H), 2.10 (m, 1H). 13C NMR (CDCl3) δ: 175.5, 172, 169.4, 169.1, 167.7, 146.9, 136.2, 132.6, 116.9, 111.7, 110.3, 70.8, 70.7, 70.5, 69.5, 66.4, 48.9, 42.4, 34.8, 31.4, 22.9. MS (ESI−): m/z 476 [M−H]−.
1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12-tetraoxapentadecan-15-oic acid (35).
Compound 31 (0.429 g, 1.55 mmol) was reacted with 29 (0.500 g, 1.55 mmol) and DIPEA (0.400 g, 3.1 mmol) in DMF (3 mL) using the general procedure reported above to afford, after purification by flash chromatography (PE/EtOAc 1/1), the desired protected acid as a yellow oil. This intermediate was hydrolyzed with 10% TFA in DCM solution (10 mL). The crude product was purified by flash chromatography (DCM/MeOH 98/2) to give 35 (0.178 g, 0.341 mmol, 21% yield) as a yellow oil. 1H NMR (DMSO-D6) δ: 11.07 (s, 1H), 7.54 (dd, J = 8.6, 7.1 Hz, 1H), 7.11 (d, J = 8.6 Hz, 1H), 7.00 (d, J = 7.1 Hz, 1H), 6.56 (m, 1H), 5.02 (dd, J = 12.9, 5.4 Hz, 1H), 3.77–3.17 (m, 18H), 2.84 (m, 1H), 2.60–2.44 (m, 2H), 2.37 (m, 2H), 2.03–1.93 (m, 1H). 13C NMR (DMSO-D6) δ: 173.3, 170.9, 170.6, 169.4, 167.8, 146.9, 136.8, 132.6, 118, 111.2, 109.7, 72.9, 70.3, 69.4, 66.7, 60.7, 49.1, 42.2, 36.3, 31.5, 22.7. MS (ESI−): m/z 520 [M−H]−.
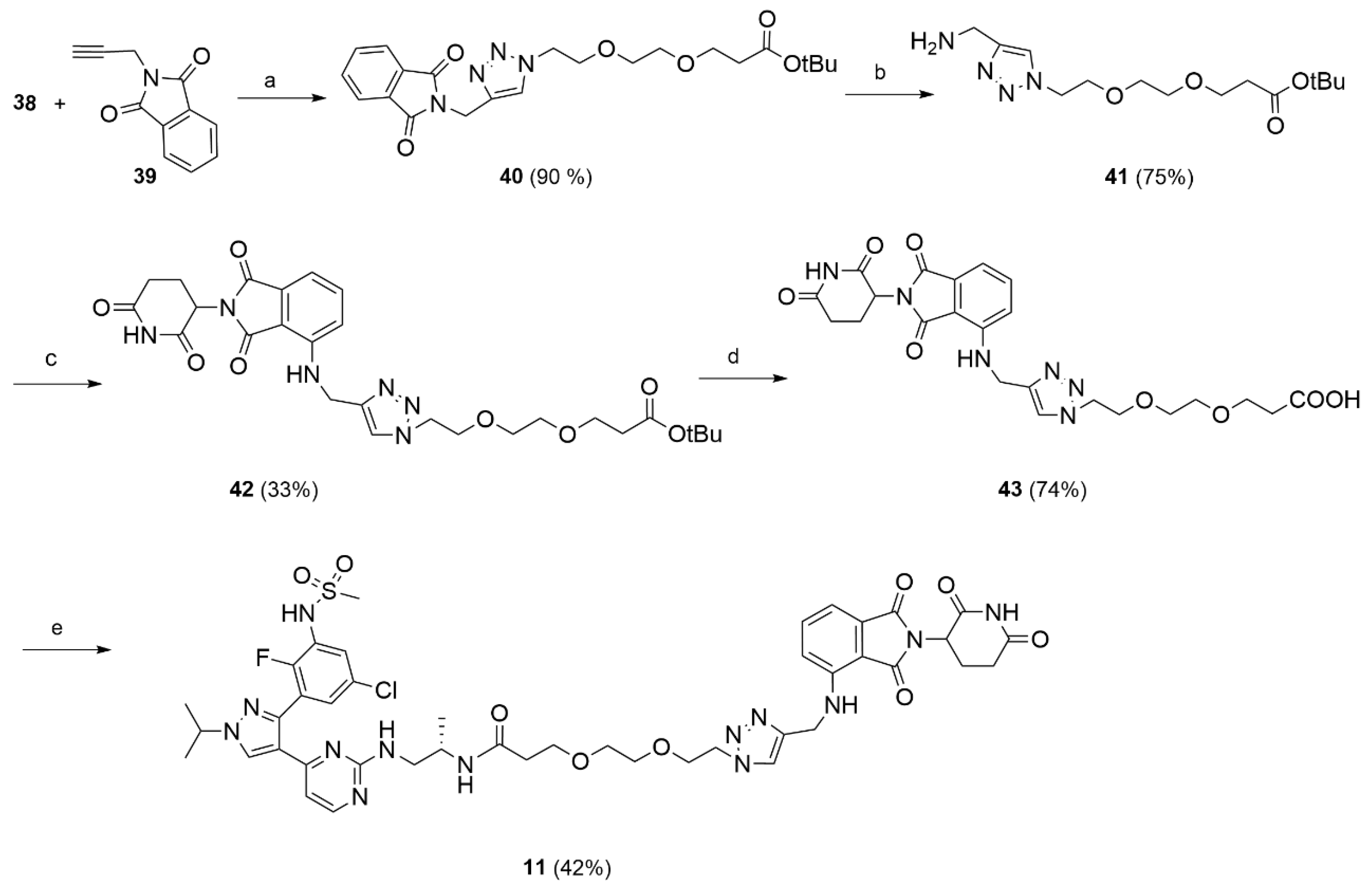
Compound 19 (1.10 g, 4.69 mmol, 1 eq) was dissolved in THF under nitrogen atmosphere. CBr4 (3.08 g, 9.39 mmol, 2 eq) and PPh3 (2.46 g, 9.39 mmol, 2 eq) were successively added and the reaction mixture was stirred at rt for 4 h. The formed white precipitate was filtered off, the solvent removed under reduced pressure and the residue purified by flash chromatography (PE/EtoAc 8/2) to afford 37 (1.38 g, 4.64 mmol, 100%) as a white oil. The compound was characterized by MS and used in the next step without further characterization. MS (ESI+): m/z 297/299 [MH+].
Compound 37 (0.42 g, 1.41 mmol, 1 eq) was dissolved in DMF (2 mL). NaN3 (0.46 g, 7.06 mmol, 5 eq) was added, followed by few drops of water in order to allow complete dissolution of the reagents. The reaction mixture was stirred at 70 °C for 16 h. When the reaction was complete (TLC), the mixture was saturated with NaCl and extracted with DCM (3 × 20 mL). The solvent was removed under reduced pressure to give 38 (0.38 g, 1.46 mmol, 85% yield) as a white oil. The obtained product was used in the following steps without further purification. 1H NMR (CDCl3) δ: 3.70 (t, J = 6.5 Hz, 2H), 3.67–3.58 (m, 5H), 3.41–3.34 (m, 2H), 2.49 (t, J = 6.5 Hz, 2H), 1.43 (s, 9H). MS (ESI): m/z 282 [MNa+].
Tert-butyl 3-(2-(2-(4-((1,3-dioxoisoindolin-2-yl)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)propanoate (40).
N-propargyl phthalimide (39, 0.109 g, 0.0593 mmol) and the previously synthesized azide 38 (0.154 g, 0.0593 mmol) were dissolved in t-BuOH/H2O 1/1 (10 mL) at rt. To the obtained mixture, CuSO4 (0.009 g, 0.0059 mmol) and sodium ascorbate (0.023 g; 0.0118 mmol) were added, and the reaction was stirred at 90 °C for 3 h. After cooling to rt, the mixture was treated with an aqueous solution of EDTA (5 mL). The aqueous phase was extracted with DCM (3 × 20 mL), the organic phase was washed with brine (5 mL), dried, and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (DCM/MeOH 98/2) to afford 40 (0.261 g; 0. 587 mmol; 90% yield) as a white solid. 1H NMR (CDCl3) δ: 7.82 (dd, J = 5.4, 3.0 Hz, 2H), 7.74 (s, 1H), 7.68 (dd, J = 5.4, 3.0 Hz, 2H), 4.96 (s, 2H), 4.45 (t, J = 5.1 Hz, 2H), 3.83–3.74 (m, 2H), 3.65 (t, J = 6.4 Hz, 2H), 3.53 (s, 4H), 2.45 (d, J = 6.4 Hz, 2H), 1.40 (s, 9H). 13C NMR (CDCl3) δ: 170.9, 167.7, 142.7, 134.1, 132.2, 124.0, 123.5, 80.6, 70.6, 70.3, 69.5, 67.0, 50.3, 36.3, 33.2, 28.2. MS (ESI+): m/z 244 [MH+].
The appropriate amine 41 or 44 (1 eq) was dissolved in DMF (3 mL) then DIPEA (2 eq) and 31 (1 eq) were added. The reaction mixture was stirred for 16 h at 90 °C. When the reaction was complete (TLC), the solvent was removed and the residue dissolved in water and extracted with DCM (3 × 15 mL). The organic phase was washed with brine, dried and evaporated under reduced pressure. The product was isolated by flash chromatography (PE/EtOac 1/1).
Tert-butyl 3-(2-(2-(4-(((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)propanoate (42).
Compound 31 (0.109 g, 0.397 mmol) was reacted with amine 41 (0.125 g, 0.397 mmol) and DIPEA (0,102 g, 0.795 mmol) using the general procedure described above to give 42 (0.075 g, 0.131 mmol, 33% yield) as a yellow oil. 1H NMR (CDCl3) δ: 8.91 (s, 1H), 8.45 (s, 1H), 7.69 (s, 1H), 7.48–7.44 (m, 1H), 7.12–7.07 (m, 1H), 7.02 (d, J = 8.5 Hz, 1H), 6.70 (t, J = 5.9 Hz, 1H), 4.95–4.83 (m, 1H), 4.61 (d, J = 5.9 Hz, 2H), 4.51–4.46 (m, 2H), 3.83–3.79 (m, 2H), 3.64 (m, 2H), 3.58–3.49 (m, 4H), 2.87–2.67 (m, 3H), 2.50–2.42 (m, 2H), 2.15–2.06 (m, 1H), 1.41 (s, 9H). 13C NMR (CDCl3) δ: 171.2, 171.0, 169.4, 168.5, 167.6, 146.3, 144.9, 136.2, 132.5, 123.1, 117.3, 112.2, 110.8, 80.7, 70.7, 70.5, 70.4, 70.3, 69.4, 67.0, 50.4, 49.0, 38.7, 36.3, 31.5, 28.2, 22.8, 21.0. MS (ESI−): m/z 569 [M−H]−.
Tert-butyl 3-(2-(2-(4-(((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)methyl)-1H-1,2,3-triazol-1 yl)ethoxy)ethoxy)propanoate (46).
Compound 31 (0.136 g, 0.492 mmol) was reacted with amine 44 (0.07 g, 0.492 mmol) and DIPEA (0.187 g, 1.44 mmol) using the general procedure described above to give 46 (0.04 g, 0.100 mmol, 20% yield) as a yellow oil. 1H NMR (CDCl3) δ 8.59 (s, 1H), 7.45 (m, 1H), 7.06 (d, J = 6.9 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.45 (m, 1H), 4.94–4.85 (m, 1H), 4.17 (s, 2H), 3.73–3.62 (m, 6H), 3.45 (m, 2H), 2.85–2.73 (m, 3H), 2.42 (s, 2H), 2.11–2.03 (m, 1H). 13C NMR (CDCl3) δ 171.6, 169.4, 168.7, 167.7, 146.9, 136.1, 132.6, 116.9, 111.7, 110.3, 79.7, 74.7, 70.5, 69.7, 69.2, 58.5, 48.9, 42.4, 31.5, 22.8. MS (ESI−): m/z 398 [M−H]−.
3-(2-(2-(4-(((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)propanoic acid (43).
Compound 42 (0.075 g, 0.122 mmol) was treated with a solution of TFA 10% in DCM (10 mL) and the reaction mixture was stirred at rt for 16 h. The solvent was evaporated and the obtained residue purified by flash chromatography (DCM/MeOH 95/5) to give 43 (0.05 g; 0.097 mmol; 74%) as a yellow solid. 1H NMR (CDCl3) δ: 9.16–9.13 (m, 1H), 7.78 (s, 1H), 7.43 (t, J = 7.9 Hz, 1H), 7.06–7.03 (m, 2H), 6.80 (s, br, 1H), 4.93–4.90 (m, 1H), 4.61 (s, 2H), 4.47 (t, J = 4.7 Hz, 2H), 3.79 (t, J = 4.8 Hz, 2H), 3.67 (t, J = 6.0 Hz, 2H), 3.55–3.49 (m, 4H), 2.86–2.65 (m, 3H), 2.55 (t, J = 6.0 Hz, 2H), 2.11–2.05 (m, 1H). 13C NMR (CDCl3) δ: 174.7, 172.0, 169.6, 169.2, 167.6, 146.3, 145.2, 136.3, 132.4, 123.7, 117.6, 112.2, 110.4, 70.3, 70.2, 69.2, 66.5, 50.4, 49.0, 38.4, 34.8, 31.5, 22.8. MS (ESI−): m/z 513 [M−H]−.
The appropriate alkyne 45 or 46 (1 eq) and the azide 38 (1 eq) were dissolved in t-BuOH/H2O 1/1. To the obtained solution, CuSO4 (0.1 eq) and sodium ascorbate (0.2 eq) were successively added. The mixture was stirred at 90 °C for 3 h. After cooling to rt, the mixture was treated with an aqueous solution of EDTA (5 mL). The aqueous phase was extracted with DCM (3 × 20 mL). The organic phase was washed with brine (5 mL), dried, and the solvent removed under reduced pressure. The crude product was purified by flash chromatography (DCM/MeOH 98/2) to afford the desired protected acid as a colorless oil. This intermediate was dissolved in a solution of TFA 10% in DCM and stirred for 12 h. The solvent was evaporated under reduced pressure and the crude product was purified by flash chromatography (DCM/MeOH 95:5).
3-(2-(2-(4-((3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3-oxopropoxy)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)propanoic acid (47).
Compound 38 (0.03 g, 0.08 mmol) was reacted with 45 (0.03 g, 0.08 mmol), CuSO4 10% mmol and sodium ascorbate 20% mmol in t-ButOH/H2O 1/1 (4 mL) using the general procedure described above to obtain, after flash chromatography, a colorless oil. The protected acid was hydrolyzed with a solution of TFA 10% in DCM (10 mL) as described above. The crude product was purified by flash chromatography (DCM/MeOH 95:5) to give 47 (0.05 g; 0.085 mmol, 99%) as a white solid. 1H NMR (CDCl3) δ: 9.95 (s, 1H), 9.47 (s, 1H), 8.79 (d, J = 8.4 Hz, 1H), 7.77 (s, 1H), 7.66 (dd, J = 8.4, 7.5 Hz, 1H), 7.52–7.47 (m, 1H), 4.98–4.96 (m, 1H), 4.80 -4.73 (m, 2H), 4.55–4.43 (m, 2H), 3.89–3.82 (m, 4H), 3.74 -3.72 (m, 2H), 3.69–3.52 (m, 2H), 3.37–3.33 (m, 2H), 2.78–2.70 (m, 3H), 2.61 (t, J = 6.3 Hz, 2H), 2.53 (t, J = 6.0 Hz, 2H), 2.16–2.09 (m, 1H). 13C-NMR (CDCl3) δ: 174.7, 173.8, 173.7, 169.4, 168.8, 167.7, 145.9, 144.7, 136.0, 132.6, 124.2, 116.8, 111.4, 110.3, 70.4, 70.3, 69.7, 69.5, 67.0, 64.9, 50.3, 48.9, 38.4, 34.8, 31.5, 22.8. MS (ESI−): m/z 585 [M−H]−.
3-(2-(2-(4-((2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)propanoic acid (48).
Compound 46 (0.064 g, 0.17 mmol) was reacted with 38 (0.035 g, 0.16 mmol), CuSO4 10% mmol and sodium ascorbate 20% mmol in t-ButOH/H2O 1/1 (5 mL) using the general procedure describe above to obtain, after flash chromatography, a yellow oil. The protected acid was hydrolyzed with a solution of TFA 10% in DCM (10 mL) as described above. follow the general procedure. The crude product was purified by flash chromatography (DCM/MeOH 95:5) to give 48 (0.134 g; 0.222 mmol, 74% yield) as a bright yellow solid. 1H NMR (CDCl3) δ: 8.84 (s, 1H), 7.70 (s, 1H), 7.48–7.44 (m, 1H), 7.04 (d, J =7.0 Hz, 1 H), 6.87 (d, J =8.6 Hz, 1H), 6.45 (t, J = 5.6 Hz, 1H), 4.90–4.87 (m, 1H), 4.65 (s, 2H), 4.48–4.46 (m, 2H), 3.83–3.64 (m, 2H), 3.67–3.63 (m, 8H), 3.53–3.52 (m, 4H), 3.42–3.41 (m, 2H), 2.87–2.67 (m, 3H), 2.44 (t, J = 6.5 Hz, 2H), 2.06 (m, 1H). 13C NMR (CDCl3) δ: 174.6, 170.9, 169.4, 168.8, 167.7, 146.8, 144.8, 136.1, 132.6, 124.0, 116.8, 111.6, 110.3, 70.5, 70.4 (overlapping peaks), 70.2, 69.7, 69.5, 66.9, 64.6, 50.2, 48.9, 42.4, 36.2, 28.2, 22.8. MS (ESI−): m/z 601 [M−H]−.
To a solution of the appropriate acids 30, 32–36 and 43 in dry DMF, HATU (1.2 eq) or HOBt (0.15 eq)/HBTU (1.5 eq) and DIPEA (2 eq) were added and the reaction mixture was stirred at rt. After 2 h, 16 (1 eq) was added and the reaction was stirred at rt for further 16 h. The solvent was evaporated under reduced pressure and the residue treated with an aqueous solution of NaHCO3 10%. The water phase was extracted with DCM (3 × 20 mL) and the organic phase was washed with brine (5 mL). The organic phase was dried and the solvent evaporated under reduced pressure to leave a crude product which was purified by flash chromatography (DCM/MeOH 95/5).
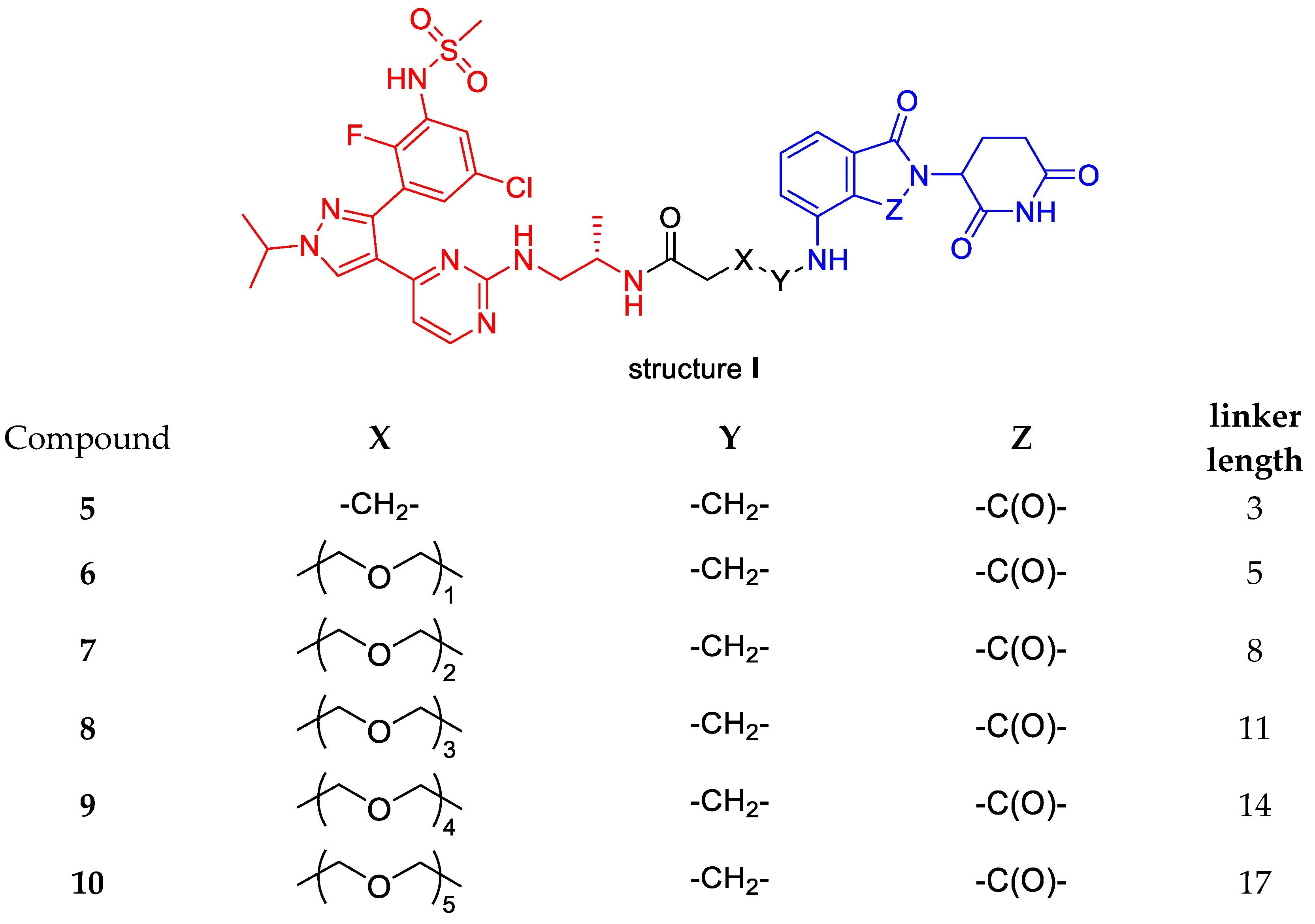
N-((S)-1-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propan-2-yl)-4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)butanamide (5).
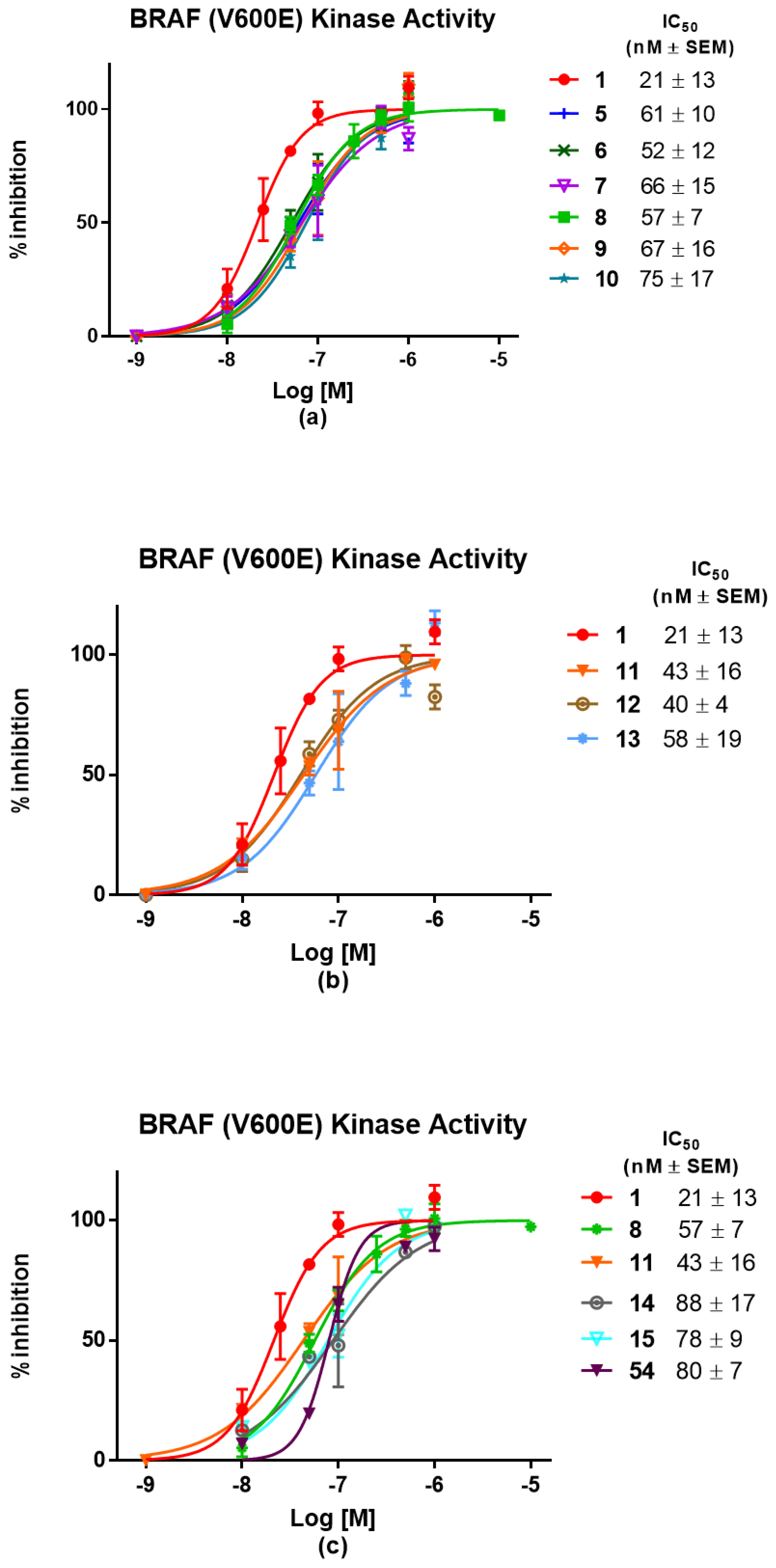
Compound 30 (0.053 g, 0.14 mmol) was reacted with HATU (0.067 g, 0.17 mmol), DIPEA (0.038 g, 0.29 mmol) and 16 (0.085 g, 0.17 mmol) in dry DMF (3 mL) using the general procedure reported above to give 5 (0.075 g, 0.091 mmol, 61%) as a bright yellow solid. 1H NMR (CDCl3) δ: 9.57 (s, 1H), 8.81 (d, J = 16.8 Hz, 1H), 8.20 (s, br, 1H), 8.16 (s, 1H), 7.87 (s, 1H), 7.61 (d, J = 6.1 Hz, 1H), 7.45–7.35 (m, 2H), 7.00 (d, J = 6.7 Hz, 1H), 6.89–6.82 (m, 1H), 6.69 (m, 2H), 4.91 (s, 1H), 4.62–4.55 (m, 1H), 3.95 (m, 1H), 3.41–3.17 (m, 4H), 3.04 (s, 3H), 2.86–2.63 (m, 3H), 2.28 (m, 2H), 2.08 (m, 1H), 1.88 (m, 2H), 1.60 (d, J = 6.6 Hz, 6H), 0.95 (s, 3H). 13C NMR (CDCl3) δ: 174.3, 171.9, 169.5, 169.2, 167.8, 167.0, 162.1, 154.8, 150.6 (d, J = 249 Hz), 146.8, 145.4, 144.3, 136.3, 132.4, 131.2, 130.0, 127.1, 126.4, 124.3, 123.6, 118.2, 116.9, 111.6, 109.9, 105.8, 55.5, 48.9, 45.6, 44.3, 41.9, 40.4, 33.4, 31.4, 24.9, 22.8, 22.7, 17.5. MS (ESI−): m/z 821/823 [M−H]−.
N-((S)-1-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propan-2-yl)-3-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)propanamide (6).
Compound 32 (0.14 g; 0.36 mmol) was reacted with HATU (0.164 g; 0.43 mmol), DIPEA (0.139 g; 1.08 mol) and 16 (0.173 g; 0.36 mmol) in dry DMF (3 mL) using the general procedure described above to give 6 (0.128 g; 0.15 mmol; 42% yield) as a bright yellow solid. 1H NMR (CDCl3) δ: 8.12 (d, J = 5.3 Hz, 1H), 8.05 (d, J = 4.6 Hz, 1H), 7.94 (s, 1H), 7.54–7.50 (m, 1H), 7.47–7.41 (m, 1H), 7.38–7.36 (m, 1H), 7.05 (m, 2H), 6.85 (m 2H), 6.49 –6.44 (m, 2H), 6.41 (s, 1H), 4.93–4.83 (m, 1H), 4.54 (m, 1H), 3.61 (m, 5H), 3.35 (m, 2H), 3.12 (m, 1H), 2.94 (s, 3H), 2.82–2.59 (m, 4H), 2.38–2.24 (m, 2H), 2.12–1.99 (m, 1H), 1.65–1.47 (m, 6H), 0.91 (s, 3H). 13C NMR (DMSO-d6) δ: 173.4, 170.5, 170.1, 169.3, 167.9, 162.4, 158.8, 158.6, 151.7 (d, J = 250 Hz), 146.9, 142.1, 136.7, 132.6, 130.5, 130.4, 128.1, 127.4, 125.9, 124.5, 119.8, 117.9, 111.2, 109.8, 106.1, 69.5, 67.4, 54.3, 49.1, 46.0, 44.7, 42.1, 40.8, 36.8, 31.5, 22.9, 22.6, 18.3. MS (ESI−): m/z 852/854 [M−H]−.
N-((S)-1-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propan-2-yl)-3-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)propanamide (7).
Compound 33 (0.066 g; 0.152 mmol) was reacted with HOBt (0.003 g; 0,002 mmol), HBTU (0.086 g, 0.228 mmol), DIPEA (0.039 g, 0.304 mmol) and 16 (0.065 g; 0.136 mmol) in dry DMF (3 mL) using the general procedure described above to give 7 (0.054 g, 0.60 mml, 45%) as a bright yellow solid. 1H NMR (DMSO-d6) δ: 11.08 (s, 1H), 9.86 (s, 1H), 8.57 (s, br, 1H), 8.07 (s, 1H), 7.54–7.49 (m, 2H), 7.45 (s, 1H), 7.29 (s, 1H), 7.07 (d, J = 8.6 Hz, 1H), 6.99 (m, 1H), 6.69 (s, br, 1H), 6.55 (m, 2H), 5.02 (dd, J = 12.8, 5.4 Hz, 1H), 4.54 (dt, J = 13.2, 6.6 Hz, 1H), 3.78 (m, 1H), 3.55–3.39 (m, 12H), 3.01 (s, 3H), 2.84–2.52 (m, 3H), 2.21 (m,2H), 2.03–1.93 (m, 1H), 1.45 (d, J = 6.6 Hz, 6H), 0.83 (s, 3H). 13C NMR (DMSO-d6) δ: 173.4, 170.6, 170.2, 169.4, 167.8, 162.6, 158.8, 158.7, 151.9 (d, J = 249 Hz), 146.9, 142.2, 136.7, 132.6, 130.5, 130.4, 128.2, 127.3, 125.8, 124.6, 119.9, 117.9, 111.2, 109.8, 106.1, 70.2, 70.1, 69.4, 67.4, 54.4, 49.1, 46.1, 44.7, 42.2, 40.8, 36.8, 31.5, 23.0, 22.6, 18.4. MS (ESI−): m/z 895/897 [M−H]−.
N-((S)-1-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propan-2-yl)-3-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)propanamide (8).
Compound 34 (0.043 g, 0.09 mmol) was reacted with HOBt (0.002 g, 0,00135 mmol), HBTU (0.051 g, 0.13 mmol), DIPEA (0.013 g, 0.10 mmol) and 16 (0.043 g, 0.09 mmol) in dry DMF (3 mL) using the general procedure described above to give 8 (0.04 g, 0.042 mmol, 47%) as a bright yellow solid. 1H NMR (DMSO-d6) δ: 11.07 (s, 1H), 9.89 (s, 1H), 8.83 (s, br 1H), 8.20 (s, br, 1H), 7.60 (s, br 1H), 7.55–7.51 (m, 2H), 7.48 (d, J = 6.1 Hz, 1H), 7.32 (d, J = 4.5 Hz, 1H), 7.09 (d, J = 8.6 Hz, 1H), 6.99 (d, J = 7.0 Hz, 1H), 6.91 (s, br, 1H), 6.55 (s, 1H), 5.01 (dd, J = 12.9, 5.4 Hz, 1H), 4.64–4.50 (m, 1H), 3.76 (s, 1H), 3.57–3.38 (m, 16H), 3.04 (s, 3H), 2.84–2.52 (m, 3H), 2.22 (m,, 2H), 2.01–1.95 (m, 1H), 1.47 (d, J = 6.5 Hz, 7H), 0.81 (s, 3H). 13C NMR (CD3CN) δ: 172.2, 171.8, 169.8, 169.5, 167.6, 166.6, 154.6, 151.2 (d, J = 255 Hz), 146.8, 145.7, 143.4, 136.2, 132.7, 132.6, 128.9, 127.0, 126.9, 124.4, 124.3, 117.2, 115.4, 111.0, 109.9, 105.7, 70.1–69.0 (three signals, multiple overlapping peaks), 66.8, 55.1, 48.9, 45.8, 43.8, 42.0, 40.1, 36.4, 31.50, 22.4, 21.8, 17.0. MS (ESI−): m/z 939/941 [M−H]−.
N-((S)-1-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propan-2-yl)-1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12-tetraoxapentadecan-15-amide (9).
Compound 35 (0.071 g, 0.13 mmol) was reacted with HOBt (0.001 g, 0,00186 mmol), HBTU (0.07 g, 0.186 mmol), DIPEA (0.032 g, 0.24 mmol) and 16 (0.06 g, 0.12 mmol) in dry DMF (3 mL) using the general procedure described above to give 9 (0.05 g, 0.05 mmol, 41%) as a bright yellow solid. 1H NMR (CD3CN) δ: 9.15 (s, br, 1H), 9.05 (s, br, 1H), 8.47 (s, 1H), 8.16 (s, 1H), 7.98–7.87 (m, 1H), 7.58 (m, 1H), 7.51 (m, 1H), 7.36 (m, 1H), 7.01 (m, 3H), 6.87–6.85 (m, 1H), 6.64–6.83 (m, 1H), 4.92 (m, 1H), 4.61 (m, 1H), 3.88 (m, 1H), 3.66–3.42 (m, 21H), 3.12–3.09 (m, 2 H), 3.03 (s, 3H), 2.89–2.63 (m, 3H), 2.30 (m, 1H), 2.03–1.93 (m, 1H), 1.54 (d, J = 6.5 Hz, 6H), 0.93 (s, 3H). 13C NMR (CD3CN) δ: 173.3, 172.6, 170.8, 170.5, 168.7, 168.6, 167.3, 161.4, 156.0, 152.2 (d, J = 247 Hz), 147.8, 147.3, 144.8, 138.8, 137.2, 133.5, 129.9, 128.0, 125.3, 122.5, 119.0, 111.9, 110.9, 106.7, 71.1–70.9 (four signals, multiple overlapping peaks), 70.7, 70.1, 67.8, 56.1, 50.0, 45.9, 44.7, 42.4, 40.9, 36.8, 31.5, 22.9, 22.5, 18.3. MS (ESI−): m/z 983/985 [M−H]−.
N-((S)-1-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propan-2-yl)-1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15-pentaoxaoctadecan-18-amide (10).
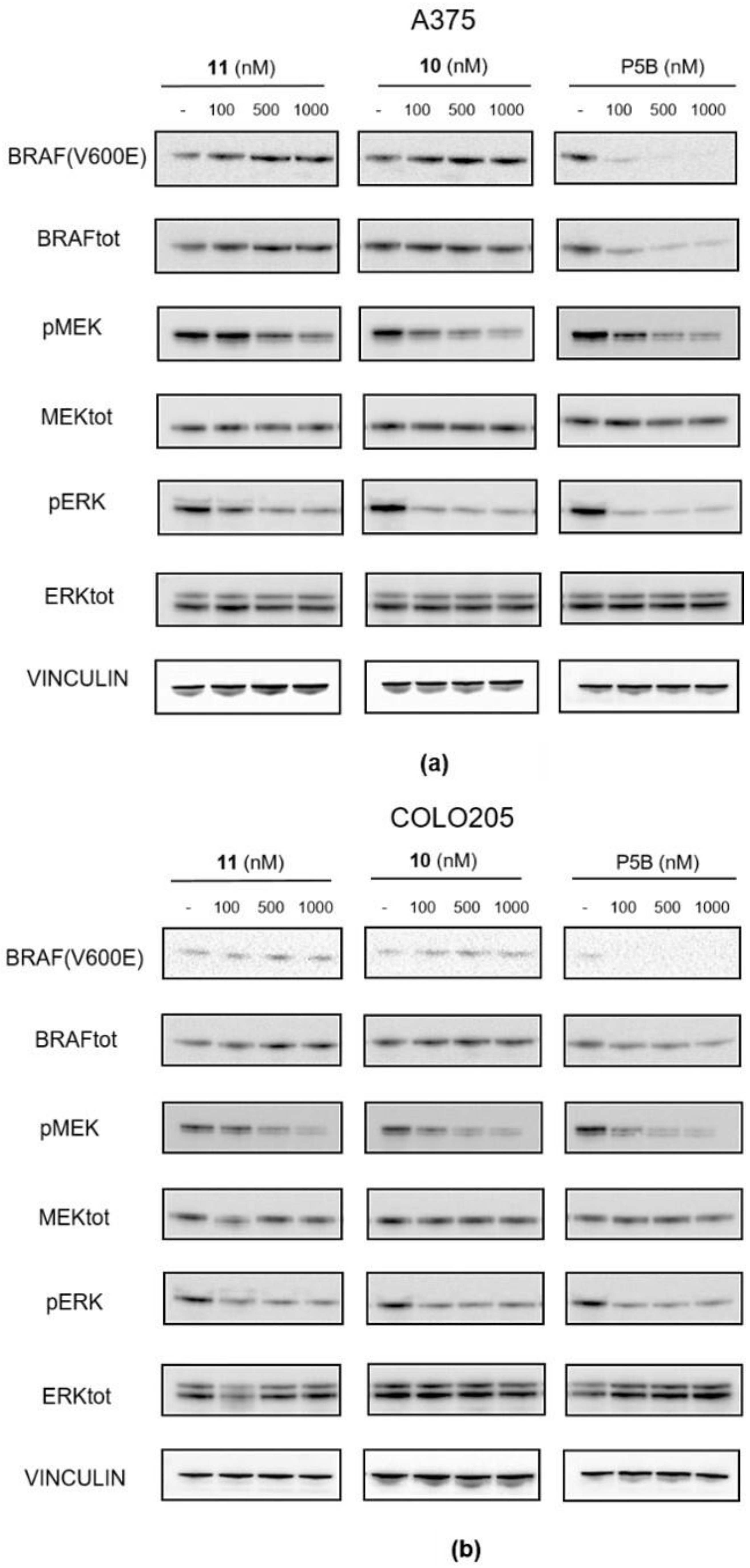
Compound 36 (0.03 g, 0.05 mmol) was reacted with HATU (0.024 g, 0.06 mmol), DIPEA (0.14 g, 0.10 mmol) and 16 (0.035 g, 0.07 mmol) in dry DMF (3 mL) using the general procedure reported above to give 10 (0.027 g, 0.026 mmol, 40%) as a bright yellow solid. 1H NMR (CD3CN) δ: 8.19 (s, br, 1H), 8.10–8.09 (m, 1H), 7.52–7.49 (m, 2H), 7.37 (m, 1H), 7.01–7.00 (m, 2H), 6.64–6.63 (s, br, 1H), 6.45 (m, 1H), 4.90 (m, 1H), 4.55 (m, 1H), 3.64–3.63 (m, 1H), 3.53- 3.40 (m, 23H), 2.95 (s, 3H), 2.71–2.62 (m, 4H), 2.07–2.04 (m, 2H), 1.51 (d, J = 6.5 Hz, 6H), 0.86 (s, 3H). 13C NMR (DMSO-d6) δ: 174.0, 173.3, 171.4, 170.5, 168.7, 165.9, 161.4, 157.9, 154.8, 152.6 (d, J = 230 Hz), 147.9, 143.4, 137.2, 133.6, 130.8 (two overlapping peaks), 129.5, 128.3, 127.6, 125.4, 125.2, 122.2, 111.9, 111.0, 106.5, 71.2–71.1 (four overlapping signals), 71.0, 70.9, 70.1, 67.8, 55.6, 50.0, 43.0, 40.9, 37.6, 32.1, 23.4, 23.0 (two overlapping signals), 18.4. MS (ESI+): m/z 1030/1032 [MH+].
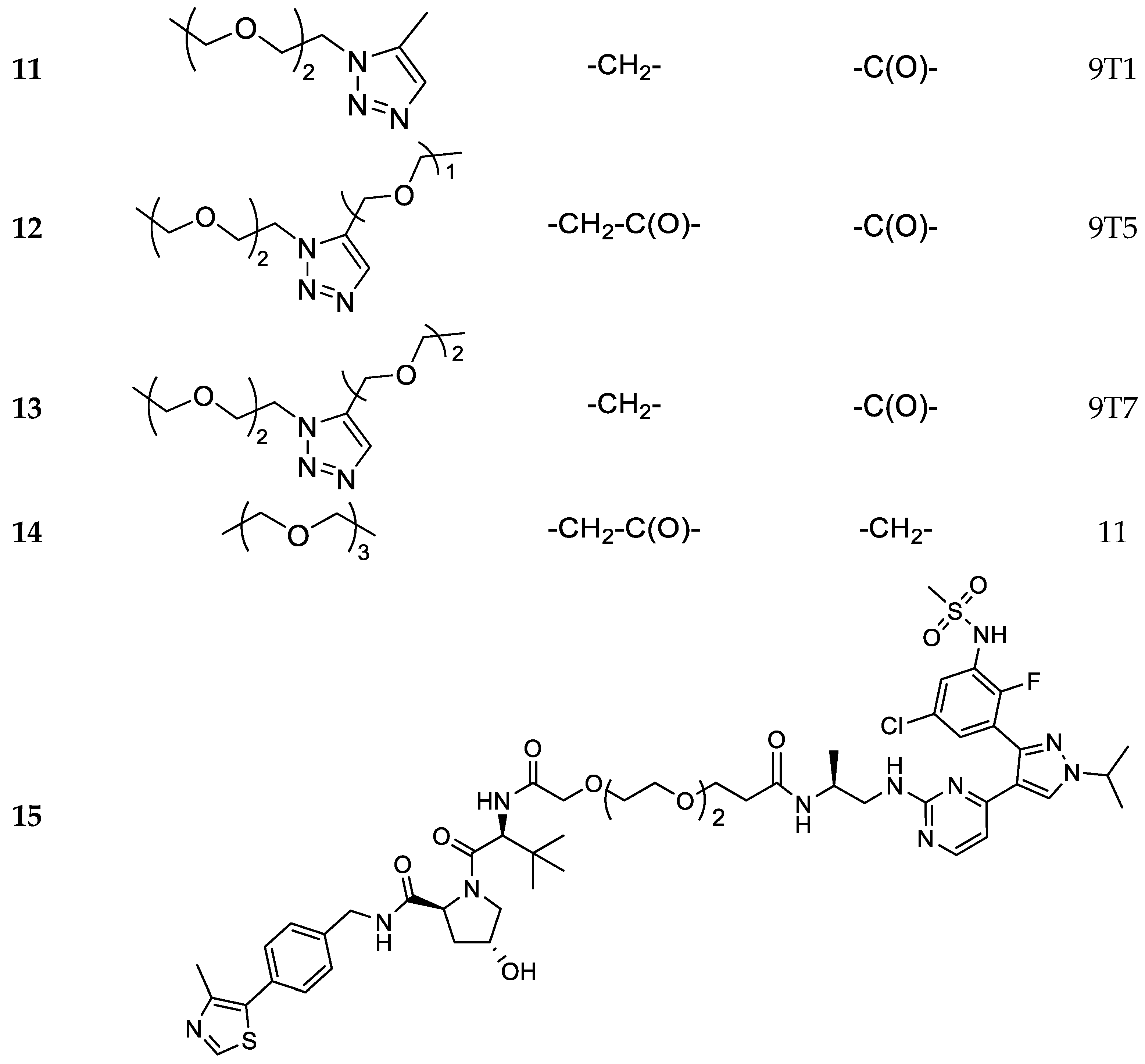
N-((S)-1-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propan-2-yl)-3-(2-(2-(4-(((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)propanamide (11).
Compound 43 (0.05 g, 0.09 mmol) was reacted with HATU (0.04 g, 0.11 mmol), DIPEA (0.025 g, 0.19 mmol) and 16 (0.046 g, 0.09 mmol) in dry DMF (3 mL) using the general procedure described above to give 11 (0.04 g, 0.04 mmol, 42%) as a bright yellow solid. 1H NMR (CD3CN) δ: 8.19 (s, br 1H), 8.10 (m, 1H), 7.79–7.78 (m, 1H), 7.59–7.53 (m, 2H), 7.38 (m, 1H), 7.10 (dd, J = 8.6, 1.4 Hz, 1H), 7.04 (d, J = 7.2 Hz, 1H), 6.80 (m, 1H), 6.63 (s, br, 1H), 4.93- 4.90 (m, 1H), 4.59–4.55 (m, 3H), 4.43 (t, J = 5.0 Hz, 2H), 3.77 (m, 3H), 3.44–3.42 (m, 4H), 3.44–3.42 (m, 3H), 3.36 (m, 2h), 2,97 (m, 3H), 2.75–2.63 (m, 4H), 2.17–2.15 (m, 2H), 2.10–2.04 (m, 2H), 1.52–1.51 (d, J = 6.1 Hz, 6H), 1.30–1.32 (m, 1H), 0.90 (s, 3H). 13C NMR (CD3CN) δ: 173.4, 170.4, 168.7, 161.2, 158.6, 152.1 (d, J = 231 Hz), 147.3, 145.9, 143.3, 137.2, 133.6, 130.7, 128.4, 127.6, 127.5, 124.2, 120.5, 119.0, 112.3, 111,5, 107.5, 71.0, 70.9, 70.8, 69.9 (two overlapping peaks), 67.8, 55.6, 51.0, 50.0, 40.9, 38.9, 37.7, 32.1, 23.4, 23.0 (two overlapping peaks), 18.4. MS (ESI-): m/z 976/978 [M−H]−.
To a stirred solution of the appropriate acid 47 or 48 (1.0 eq) in dry DMF, HATU (1.2 eq) or HOBt (0.15 eq)/HBTU (1.5 eq) and DIPEA (2 eq) were added and the mixture was stirred at rt. After 2 h, 16 (1 eq) was added and the reaction was stirred at rt for further 16 h. The solvent was evaporated under reduced pressure and the residue treated with an aqueous solution of NaHCO3 10%. The water phase was extracted with DCM (3 × 20 mL) and the organic phase was washed with brine (5 mL). The organic phase was dried and the solvent evaporated under reduced pressure to leave a crude product which was purified by flash chromatography (DCM/MeOH 95/5).
N-((S)-1-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propan-2-yl)-3-(2-(2-(4-((3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3-oxopropoxy)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)propanamide (12).
Compound 12 was obtained by starting from 16 (0.035 g, 0.085 mmol), 47 (0.05 g, 0.085 mmol), HATU (0.038 g, 0.0102 mmol) and DIPEA (0.033 g, 0.25 mmol) in DMF (2 mL), using the general procedure described above to give a yellow solid (0.30 g, 0.028 mmol, 33%). 1H NMR (600 MHz, CDCl3) δ 10.2 (s, br, 1H), 10.04 (s, 1H), 8.83 (m, 1H), 8.11 (dd, J = 5.2, 1.4 Hz, 1H), 7.91 (d, J = 3.0 Hz, 1H), 7.74–7.69 (m, 2H), 7.57–7.53 (m, 2H), 7.43 (s, 1H), 6.49–6.42 (m, 2H), 4.96 (dd, J = 12.3, 6.0 Hz, 1H), 4.79–4.72 (m, 2H), 4.56 (dt, J = 13.4, 6.7 Hz, 1H), 4.54–4.46 (m, 2H), 3.87–3.81 (m, 4H), 3.64–3.42 (m, 7H), 3.24 (m, 1H), 2.96 (s, 3H), 2.88–2.74 (m, 5H), 2.30–2.28 (m, 2H), 2.19–2.09 (m, 1H), 1.57 (d, J = 6.7 Hz, 6H), 0.97 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 172.4, 171.0, 169.5, 168.7, 166.9, 162.0, 160.3, 157.3, 150.9 (d, J = 232 Hz), 144.8, 142.3. 137.6, 136.3, 131.4, 129.5, 128.6, 128.0, 126.0, 125.9, 124.7, 124.6, 120.0, 118.5, 115.8, 111,0, 109.9, 106.7, 70.2, 70.1, 69.4, 67.1, 65.7, 64.7, 55.5, 54.8, 50.1, 49.3, 43.5, 40.2, 38.6, 37.0, 31.5, 22.9, 22.8, 18.2. MS (ESI−): m/z 1049/1051 [M−H]−.
N-((S)-1-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propan-2-yl)-2-(2-(2-(4-((2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)acetamide (13).
Compound 48 (0.135 g, 0.22 mmol) was reacted with HATU (0.102 g, 0.26 mmol), DIPEA (0.034 g, 0.26 mmol) and 16 (0.135 g, 0.22 mmol) in dry DMF (3 mL) using the general procedure reported above to give 13 (0.16 g, 0.15 mmol, 67%) as a bright yellow solid. 1H NMR (DMSO-d6) δ: 11.06 (s, 1H), 9.86 (s, 1H), 8.56 (s, br, 1H), 8.08 (s, 1H), 7.97 (s, 1H), 7.53–7.51 (m, 2H), 7.45 (m, 1H), 7.29 (s, 1H), 7.09 (d, J = 8.6 Hz, 1H), 6.99 (d, J = 7.0 Hz, 1H), 6.67 (s, 1H), 6.56 (s, 2H), 5.01 (dd, J = 12.8, 5.3 Hz, 1H), 4.58–4.51 (m, 1H), 4.47–4.42 (m, 4H), 3.73 (m, 3H), 3.62–3.37 (m, 15H), 3.00 (s, 3H), 2.89–2.52 (m, 3H), 2.20 (m, 2H), 2.02–1.94 (m, 1H), 1.45 (d, J = 6.5 Hz, 6H), 0.83 (s, 3H). 13C NMR (CDCl3) δ: 172.6, 172.2, 170.0, 169.4, 167.8, 162.2, 160.3, 157.4, 151.0 (d, J = 246 Hz), 146.8, 144.8, 142.7, 136.2, 132.5, 129.4, 128.6, 128.0, 125.9 (d, J = 14.7 Hz), 125.3, 124.8, 124.2, 119.9, 117.0, 111.7, 110.2, 106.7, 70.8–68.9 (six partially overlapping signals), 67.2, 64.6, 54.8, 50.0, 48.9, 45.7, 43.8, 42.4, 40.2, 36.8, 31.5, 22.9, 22.8, 18.1. MS (ESI+): m/z 1067/1069 [MH+].
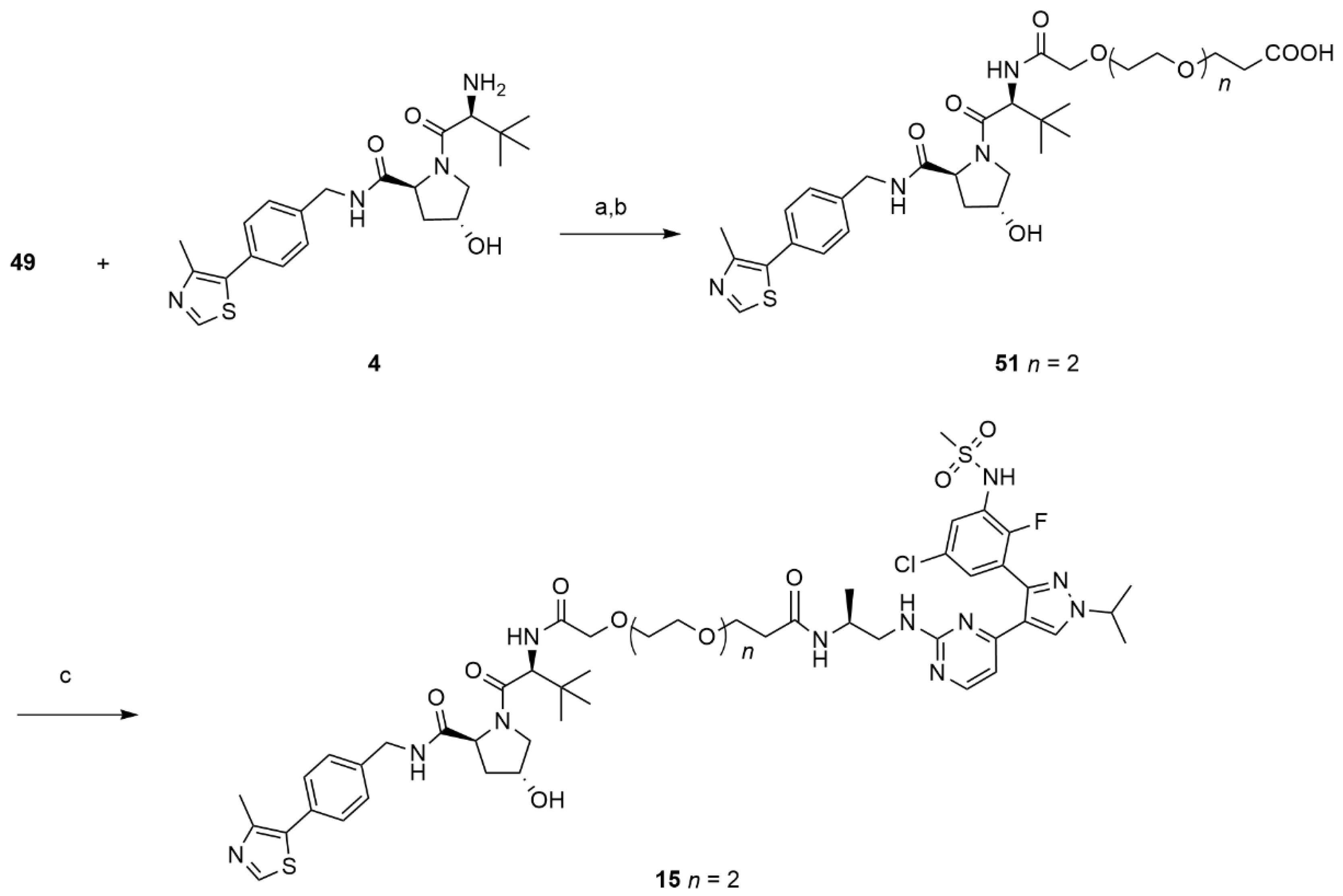
>14,14-dimethyl-12-oxo-3,6,9,13-tetraoxapentadecanoic acid (49).
BAIB (1.74 g, 5.422 mmol) and TEMPO (0.084 g, 0.54 mmol) were added to a solution of ACN/H2O 1:1 (5 mL) containing compound 19 (0.68 g, 2.46 mmol) The resulting mixture was stirred at room temperature until complete conversion of the starting material was observed by TLC (DCM/MeOH 9:1). The crude was purified using flash chromatography (DCM/MeOH 98:2) to give 49 (0.26 g, 0.89 mmol, 36%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ: 4.16–4.05 (m, 2H), 3.75–3.53 (m, 10H), 2.50–2.40 (m, 2H), 1.40 (dd, J = 3.4, 2.3 Hz, 9H). 13C NMR (151 MHz, CDCl3) δ: 173.9, 171.2, 80.8, 71.1, 70.6, 70.4, 70.3, 68.7, 66.9, 36.6, 27.9. MS (ESI−): m/z 291 [M−H]−.
Compound 49 (0.078 g, 0.103 mmol) was reacted with HATU (0.12 g, 0.32 mmol), DIPEA (0.041 g, 0.32 mmol) and lenalidomide (0.07 g, 0.27 mmol) in dry DMF (2 mL) using the general procedure reported above to afford the desired protected acid as a colorless oil. This intermediate was dissolved in a solution of TFA 10% in DCM and stirred for 12 h. The solvent was evaporated under reduced pressure and the crude product was purified by flash chromatography (DCM/MeOH 95:5) to give 50 (0.095 g, 0.198 mmol, 80%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 9.29 (s, br, 1H), 9.12 (s, 1H), 7.87–7.55 (m, br 1H), 7.69 (d, J = 7.4 Hz, 1H), 7.55 (d, J = 7.9 Hz, 1H), 7.44 (m, 1H), 5.16 (dd, J = 13.2, 4.9 Hz, 1H), 4.42 (s, 2H), 4.17 (m, 2H), 3.82–3.46 (m, 11H), 2.79–2.67 (m, 2H), 2.39–2.37 (m, 2H), 2.30–2.29 (m, 1H), 2.12–2.09 (m, 1H). MS (ESI−): m/z 476 [M−H]−.
Compound 50 (0.075 g, 0.16 mmol) was reacted with HATU (0.071 g, 0.17 mmol), DIPEA (0.061 g, 0.47 mmol) and 16 (0.075 g, 0.16 mmol) in dry DMF (2 mL) using the general procedure described for compounds 5–11 to give 14 (0.065g, 0.069 mmol, 45%) as a bright white solid. 1H NMR (600 MHz, CDCl3) δ 11.02 (s, 1H), 9.89 (s, br, 1H), 9.69 (s, 1H), 8.60 (s, br, 1H), 8.12 (d, J = 5.5 Hz, 1H), 7.73 (d, J = 7.6 Hz, 1H), 7.56–7.48 (m, 4H), 7.34 (s, 1H), 6.42–6.81 (m, 2H), 5.14 (dd, J = 13.4, 5.2 Hz, 1H), 4.58 (m, 1H), 4.37 (m, 2H), 4.12 (s, 2H), 3.74–3.92 (m, 1H), 3.67–3.44 (m, 10H), 3.13 (m, 1H), 3.05 (s, 3H), 2.89 (m, 2H), 2.66–2.55 (m, 1H), 2.43–2.30 (m, 1H), 2.24–2.23 (m, 2H), 2.04–1.95 (m, 1H), 1.49 (d, J = 6.9 Hz, 6H), 1.26–1.23 (m, 3H). 13C-NMR (151 MHz, DMSO-D6) δ 173.4, 171.6, 170.2, 168.9, 168.3, 162.7, 158.7, 151.1, 142.2, 135.4, 133.4, 133.3, 130.5, 130.4, 129.2, 128.2, 127.3 (two overlapping peaks), 126.9, 120.3, 119.9, 11.7, 70.9, 70.5, 70.2, 70.1, 70.0, 67.3, 54.4, 54.1, 52.1, 47.0, 42.3, 41.0, 36.8, 31.7, 23.1, 23.0, 18.4. MS (ESI+): m/z 941/943 [MH+].
(S)-3-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-2,2-dimethyl-5-oxo-7,10,13-trioxa-4-azahexadecan-16-oic acid (51).
Compound 49 (0.030 g, 0.103 mmol) was reacted with HATU (0.046 g, 0.12 mmol), DIPEA (0.039 g, 0.31 mmol) and 4 (0.044 g, 0.103 mmol) in dry DMF (2 mL) using the same procedure described for 50 to afford the desired protected acid as a colorless oil. This intermediate was dissolved in a solution of TFA 10% in DCM and stirred for 12 h. The solvent was evaporated under reduced pressure and the crude product was purified by flash chromatography (DCM/MeOH 95:5) to give 51 (0.041 g, 0.063 mmol, 45%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 8.92 (s, 1H), 7.48 (s, br, 1H), 7.35–7.32 (m, 4H), 4.74–4.41 (m, 3H), 4.32 (m, 1H), 4.11–3.97 (m, 3H), 3.76–3.50 (m, 12H), 2.53–2.52 (m, 5H), 2.33 (m, 1H), 2.18 (m, 1H), 0.98 (s, 9H). MS (ESI−): m/z 647 [M−H]−.
(2S,4R)-1-((2S,17S)-2-(tert-butyl)-18-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)-17-methyl-4,15-dioxo-6,9,12-trioxa-3,16-diazaoctadecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (15).
Compound 51 (0.041 g, 0.631 mmol, 1 eq) was reacted with HATU (0.030 g, 0.076 mmol, 1.2 eq), DIPEA (0.024 g, 0.19 mmol, 3 eq) and 16 (0.041 g, 0.063 mmol, 1 eq) in dry DMF (2 mL) using the general procedure described for compounds 5–11 to give 15 (0.031 g, 0.028 mmol, 44%) as a bright white solid. 1H-NMR (600 MHz, DMSO-D6) δ 9.9 (s, br, 1H), 8.98 (m, 1H), 8.60 (t, J = 5.9 Hz, 2H), 8.11 (d, J = 4.8 Hz, 1H), 7.49–7.4.7 (s, br, 1H), 7.45–7.43 (m, 1H), 7.42 -7.33 (m, 6H), 6.89–6.44 (m, 2H), 5.16 (d, J = 3.4 Hz, 1H), 4.59–4.55 (m, 2H), 4.48–4.35 (m, 3H), 4.27–4.23 (m, 1H), 3.96–3.94 (m, 2H), 3.67–3.44 (m, 15H), 3.04 (s, 3H), 2.45–2.44 (m, 3H), 2.27–2.22 (m, 2H), 1.49 (d, J = 6.9 Hz, 6H), 1.26–1.23 (m, 3H), 0.94–0.92 (s, 9H). 13C-NMR (CD3CN) δ 175.2, 173.0, 172.7, 171.4, 170.6, 160.9, 159.1, 151.8, 149.3, 143.2, 140.3, 132.5, 131.4, 130.5, 130.1, 129.5, 129.0, 128.8, 128.4, 127.5, 120.6, 107.5, 71.8, 71.7, 71.0–70.8 (four overlapping peaks), 67.8, 67.6, 60.3, 57.8, 57.4, 55.4, 43.3, 40.9, 37.7, 36.9, 36.7, 28.8, 23.0, 18.4, 16.4, 12.7. MS (ESI+): m/z 1112/1114 [MH+].
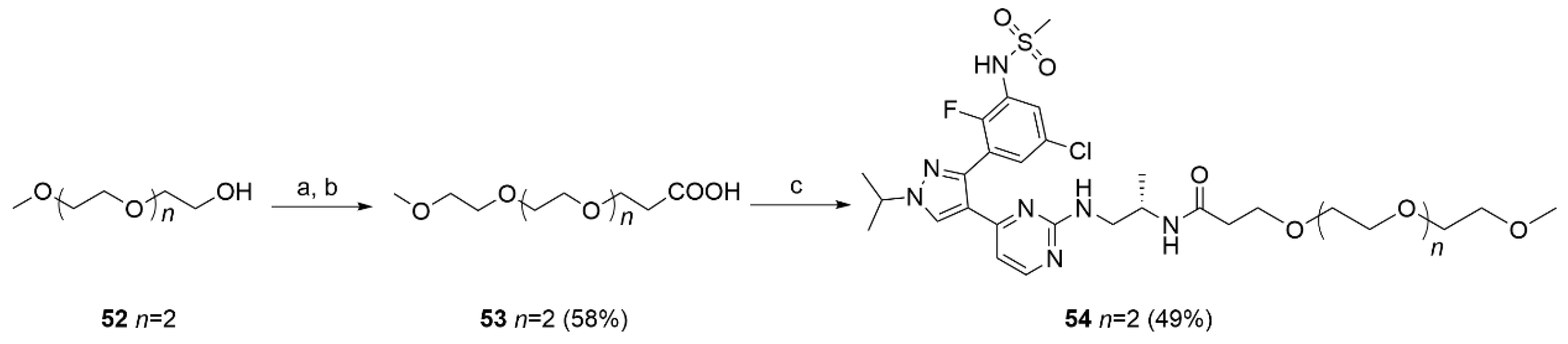
To a stirred solution of 52 (1.14 g, 6.94 mmol) in dry THF (20 mL), NaH 60% in mineral oil (0.03 g, 0.07 mmol) was added at 0 °C. The suspension was stirred for 10 min at 50 °C. The reaction mixture was cooled to rt and tert-butyl acrylate (0.89 g, 6.94 mmol, 1.01 mL) was added dropwise. After 18 h of stirring at rt, the reaction was quenched by the addition of few drops of acetic acid, and the solvent evaporated under reduced pressure. The residue was treated with H2O (5 mL) and extracted with EtOAc (3 × 20 mL). The combined organic phases were dried, the solvent evaporated and the crude product purified by flash chromatography PE/EtOAc 1:1 to give the protected acid as a colorless oil. This intermediate was dissolved in a solution of TFA 10% in DCM (10mL) and left stirring at rt for 12 h. The solvent was evaporated and the crude product purified by flash chromatography (DCM/MeOH 95:5) to afford 53 (0.254 g; 1.07 mmol; 92%) as a bright yellow solid. 1H NMR (CDCl3) δ: 9.2 (s, br, 1H), 3.76–3.74 (m, 2H), 3.66–3.62 (m, 10H), 3.57–3.56 (m, 2H), 3.37 (dd, J = 4.0, 1.5 Hz, 3H), 2.62–2.60 (m, 2H). 13C-NMR (151 MHz, CDCl3) δ: 175.8, 71.9, 70.6, 70.6, 70.5, 70.3 (two overlapping peaks), 66.5, 58.9, 35.0. MS (ESI−): m/z 235 [M−H]−.
(S)-N-(1-((4-(3-(5-chloro-2-fluoro-3-(methylsulfonamido)phenyl)-1-isopropyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)propan-2-yl)-2,5,8,11-tetraoxatetradecan-14-amide (54).
To a stirred solution of 53 (0.02 g, 0.08 mmol) in dry DMF (2 mL), HATU (0.04 g, 0.10 mmol) and DIPEA (0.02 g, 0.17 mmol) were added and the mixture left stirring at rt. After 2 h, 16 (0.05 g, 0.10 mmol) was added, and the reaction stirred at rt for a further 16 h. The solvent was evaporated under reduced pressure, the residue was treated with a 10% NaHCO3 solution (5 mL) and extracted with DCM (3 × 20 mL). The organic layer was washed with brine (5 mL), dried and evaporated. The crude product was purified by flash chromatography (DCM/MeOH 95/5) to give 54 (0.029 g, 0.041 mmol, 49%) as a bright yellow solid. 1H NMR (CDCl3) δ: 8.08 (s, 1H), 7.92 (s, 1H), 7.68 (s, 1H), 7.55 (dd, J = 6.3, 2.7 Hz, 1H), 7.45 (d, J = 2.4 Hz, 1H), 6.64 (s, 1H), 6.50 (d, J = 5.2 Hz, 1H), 4.55 (dt, J = 13.4, 6.7 Hz, 1H), 3.68–3.46 (m, 23H), 3.37–3.29 (m, 5H), 2.94 (s, 4H), 2.45–2.26 (m, 3H), 1.57 (dd, J = 9.4, 6.8 Hz, 8H), 1.17–0.88 (m, 4H). MS (ESI−): m/z 699/701 [M−H]−.





 ,
, 

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}