
The Use of Zidovudine Pharmacophore in Multi-Target-Directed Ligands for AIDS Therapy
 , , , , and
, , , , and
Abstract
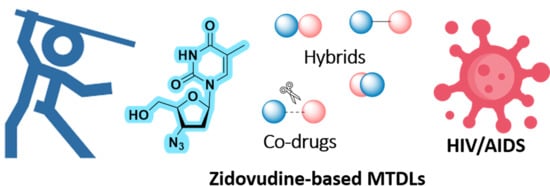
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
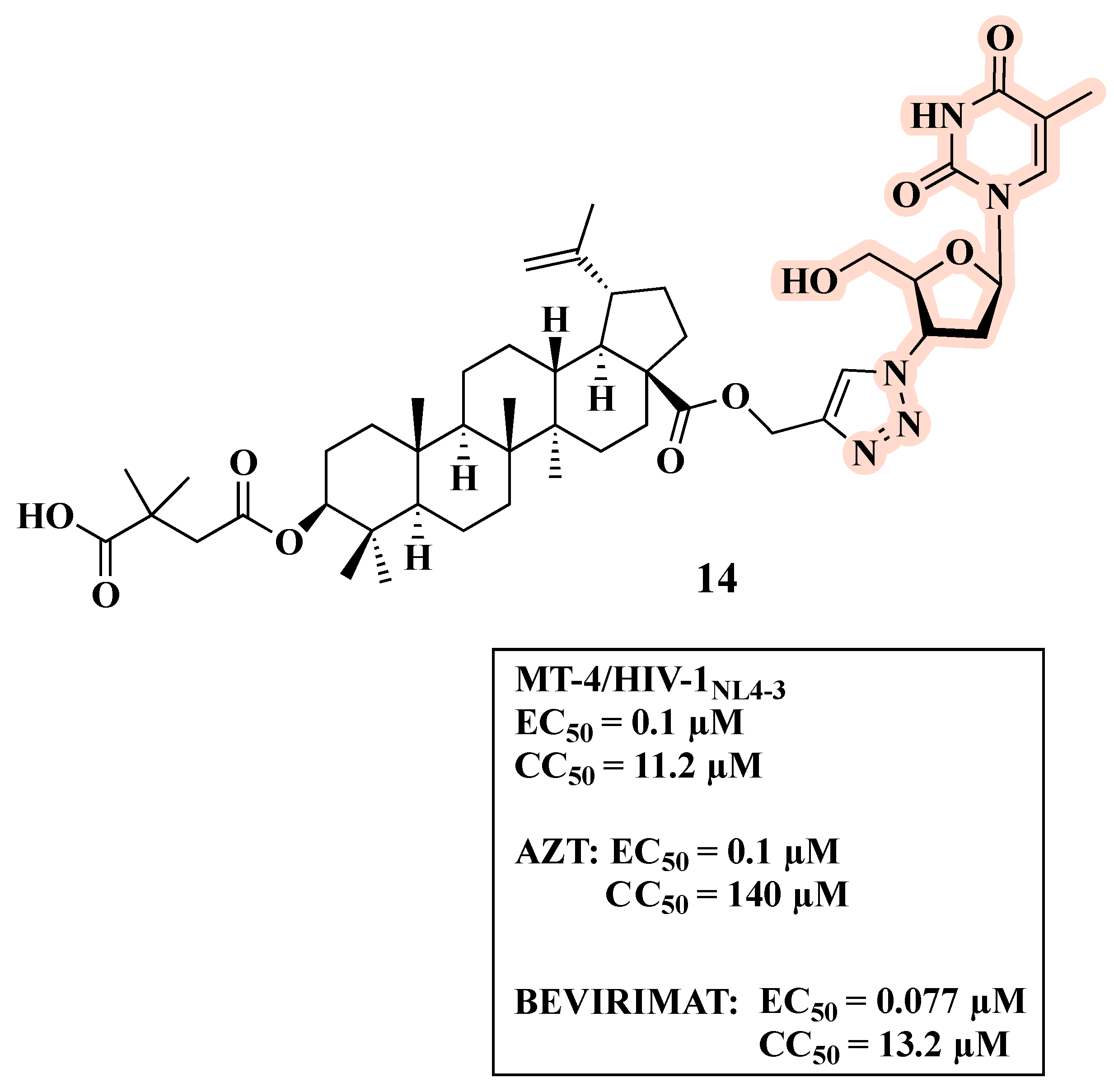{kind=link}
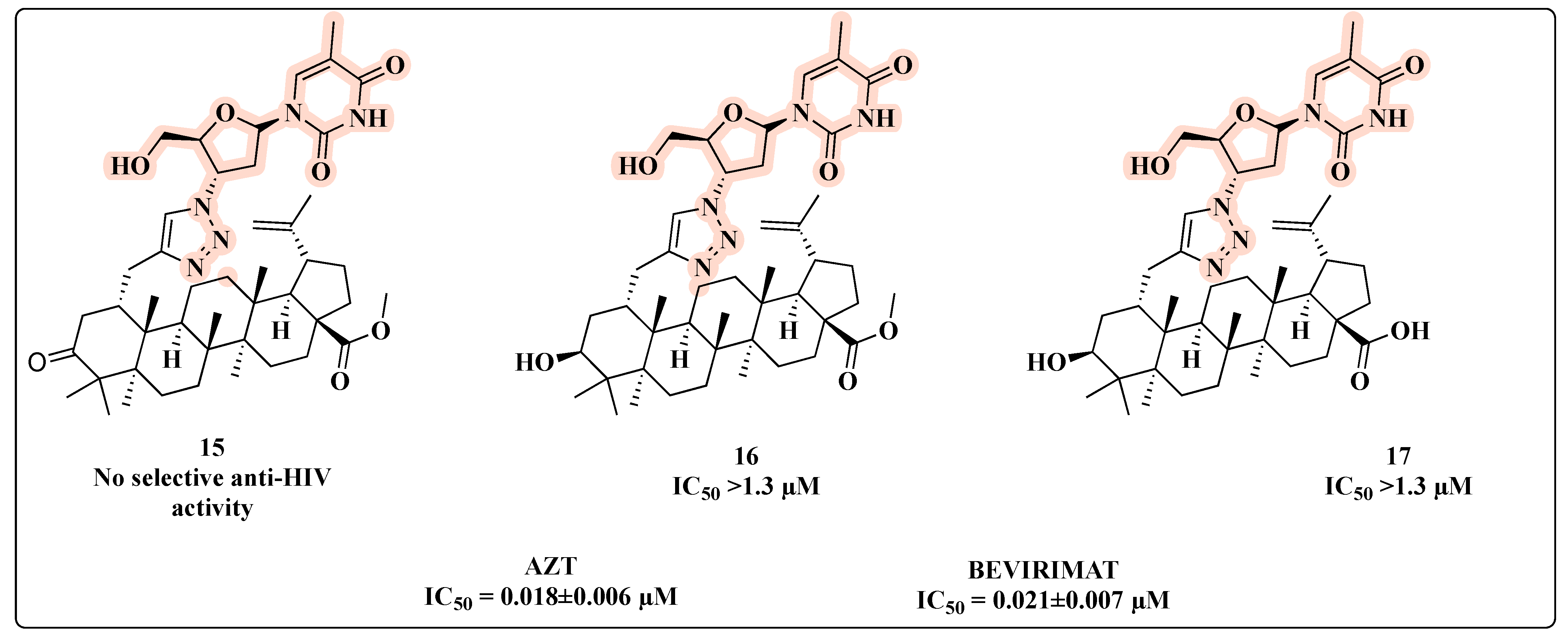{kind=link}
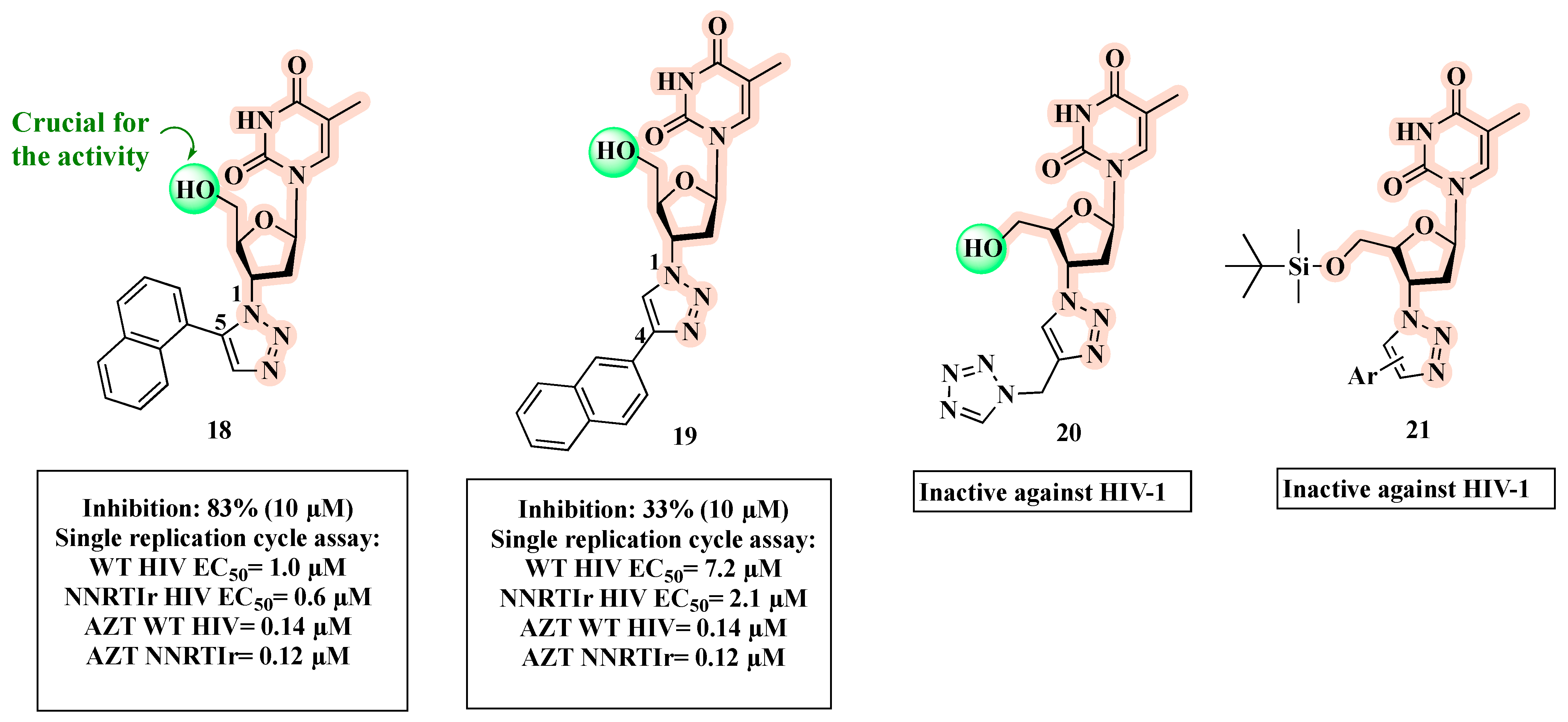{kind=link}
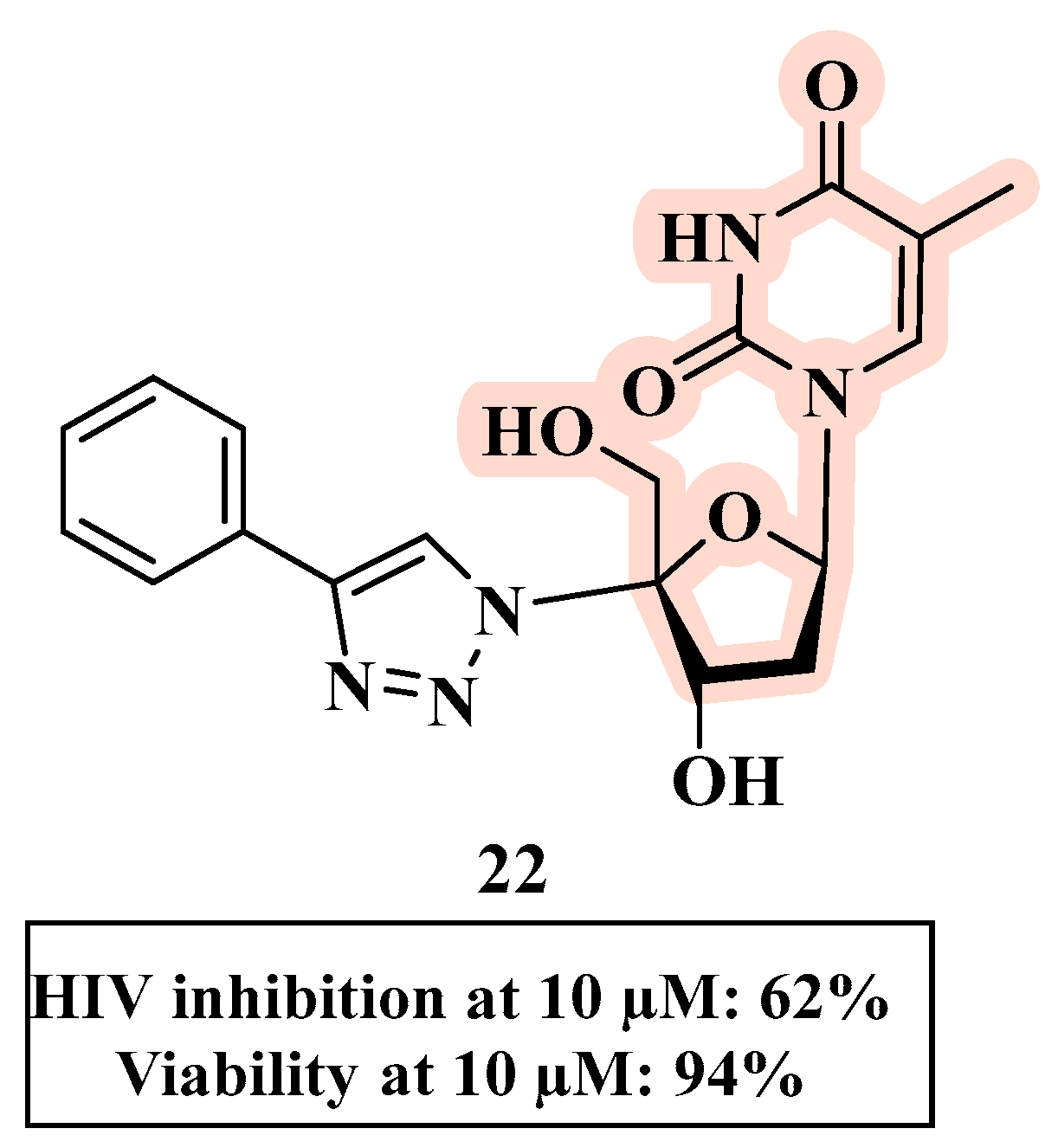{kind=link}
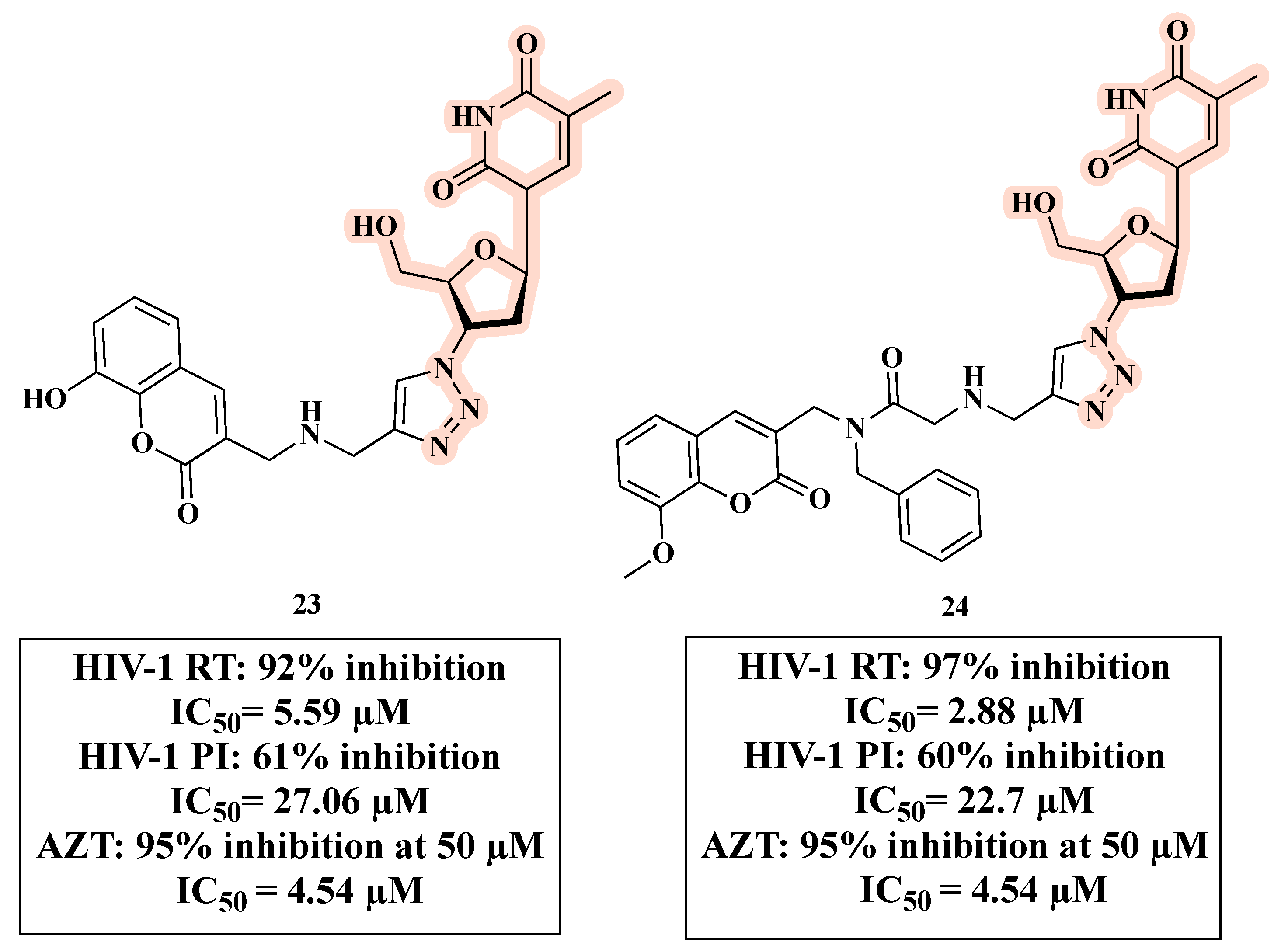{kind=link}
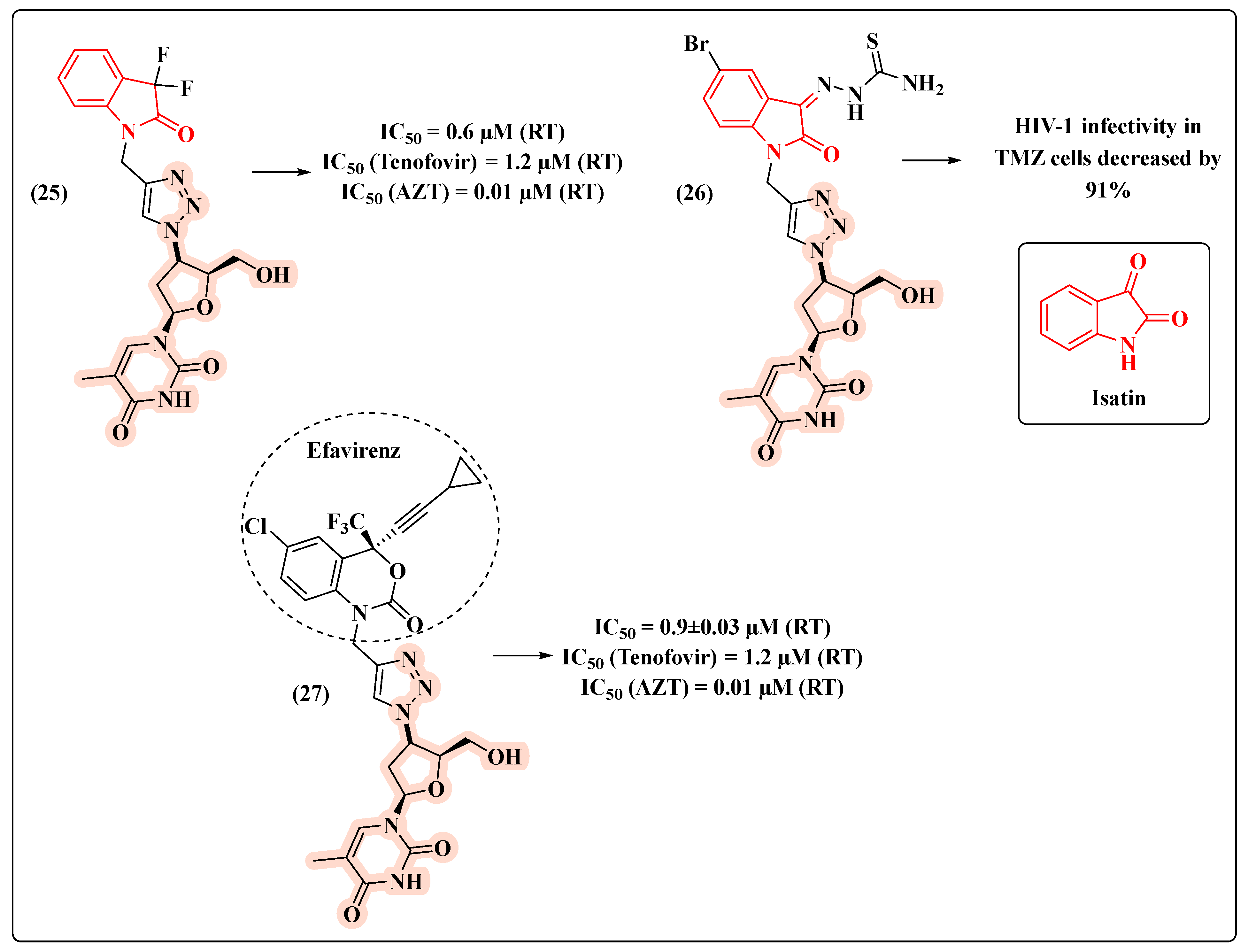{kind=link}
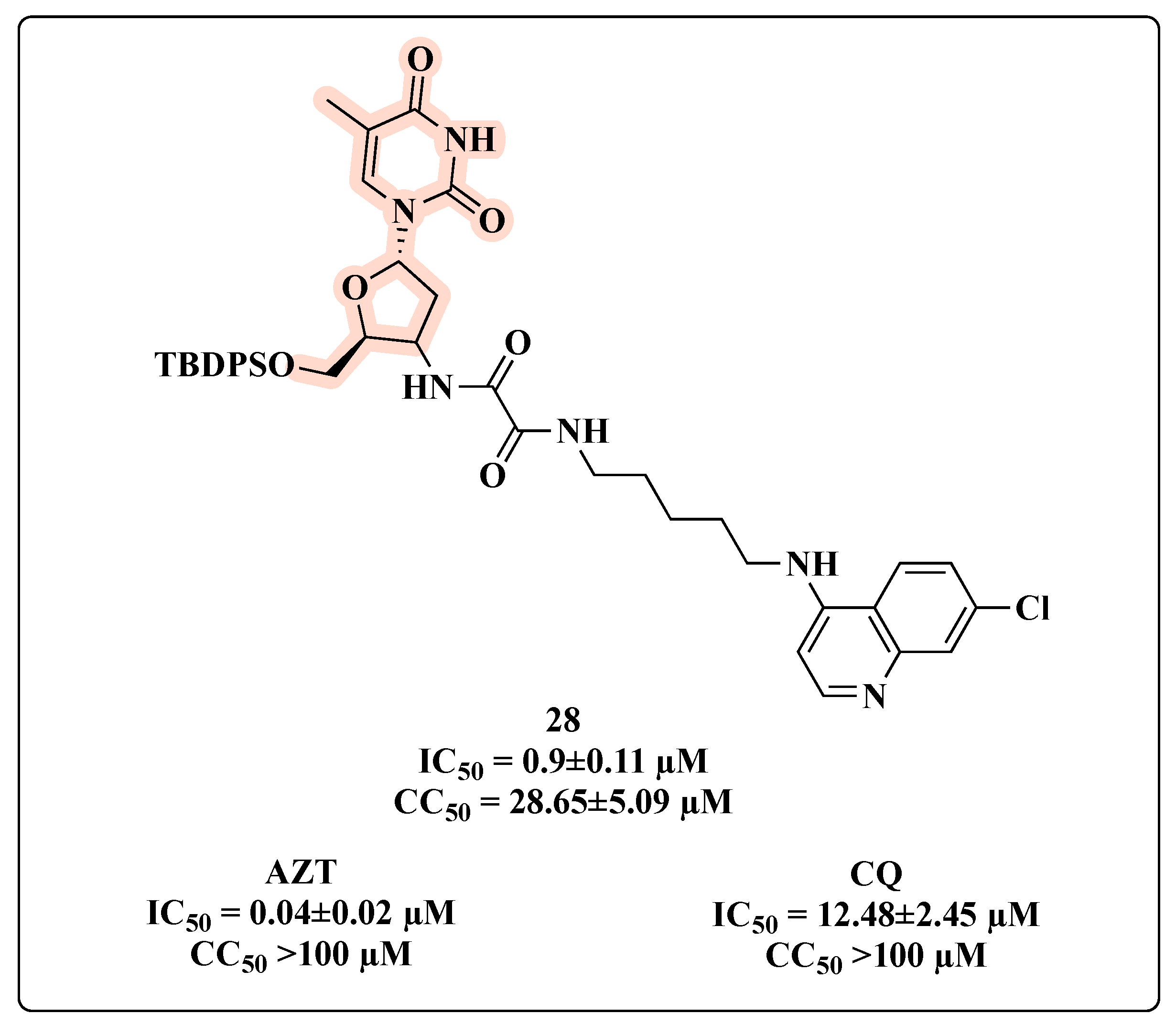{kind=link}
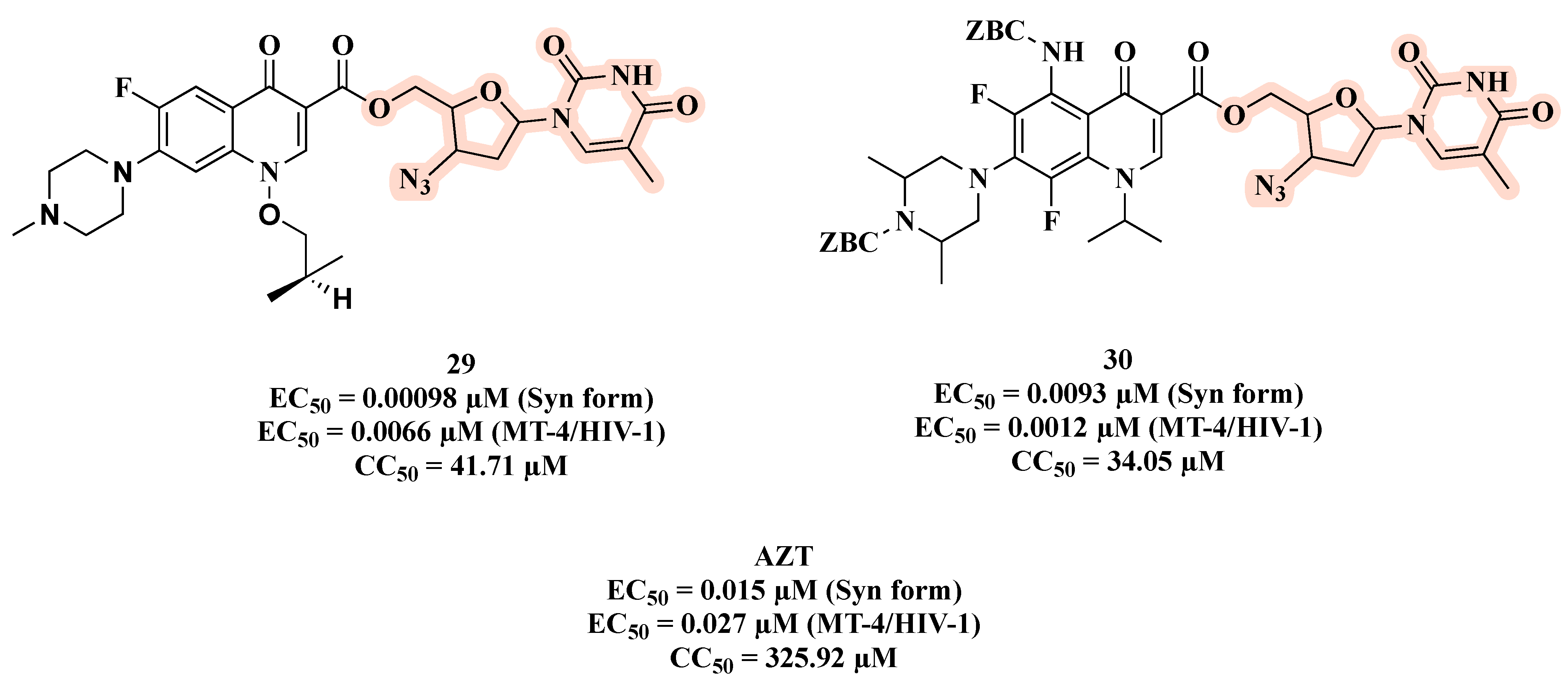{kind=link}
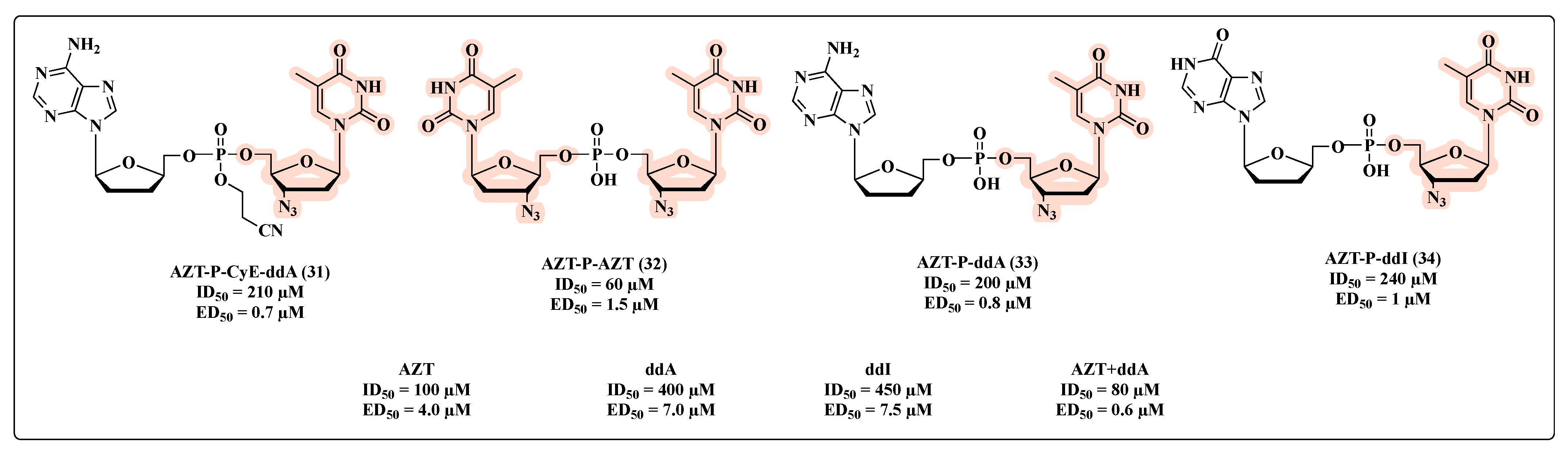{kind=link}
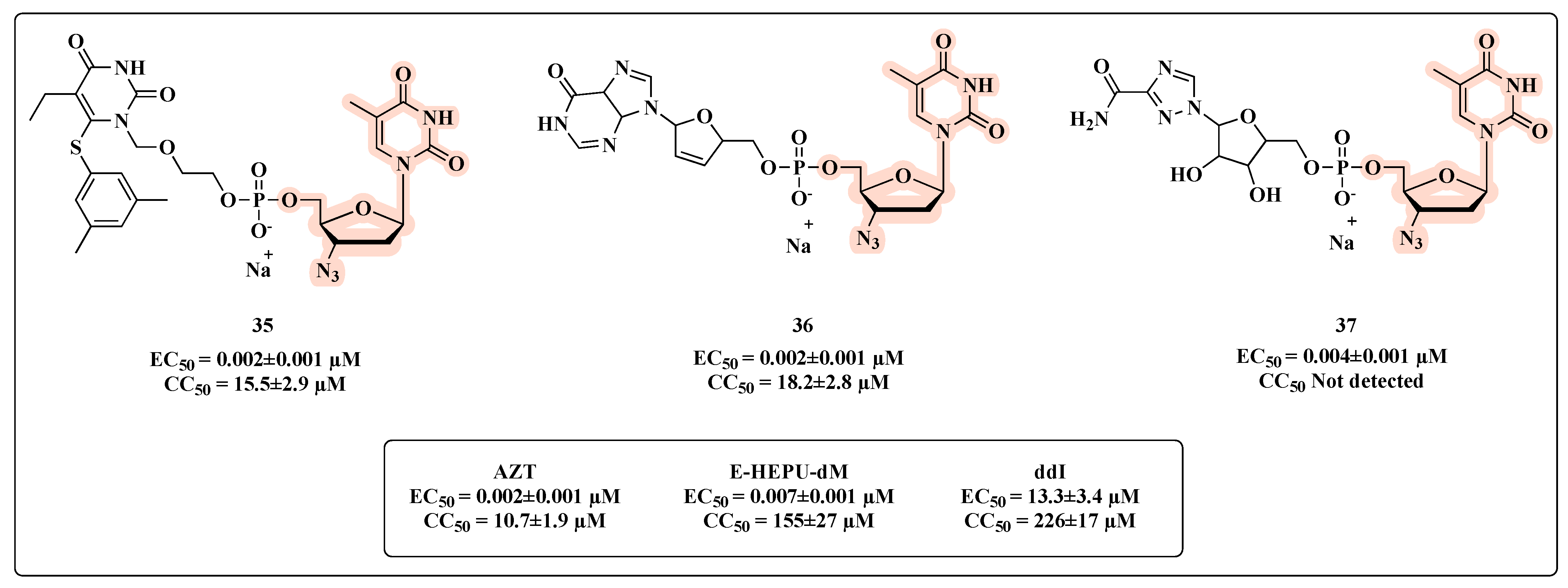{kind=link}
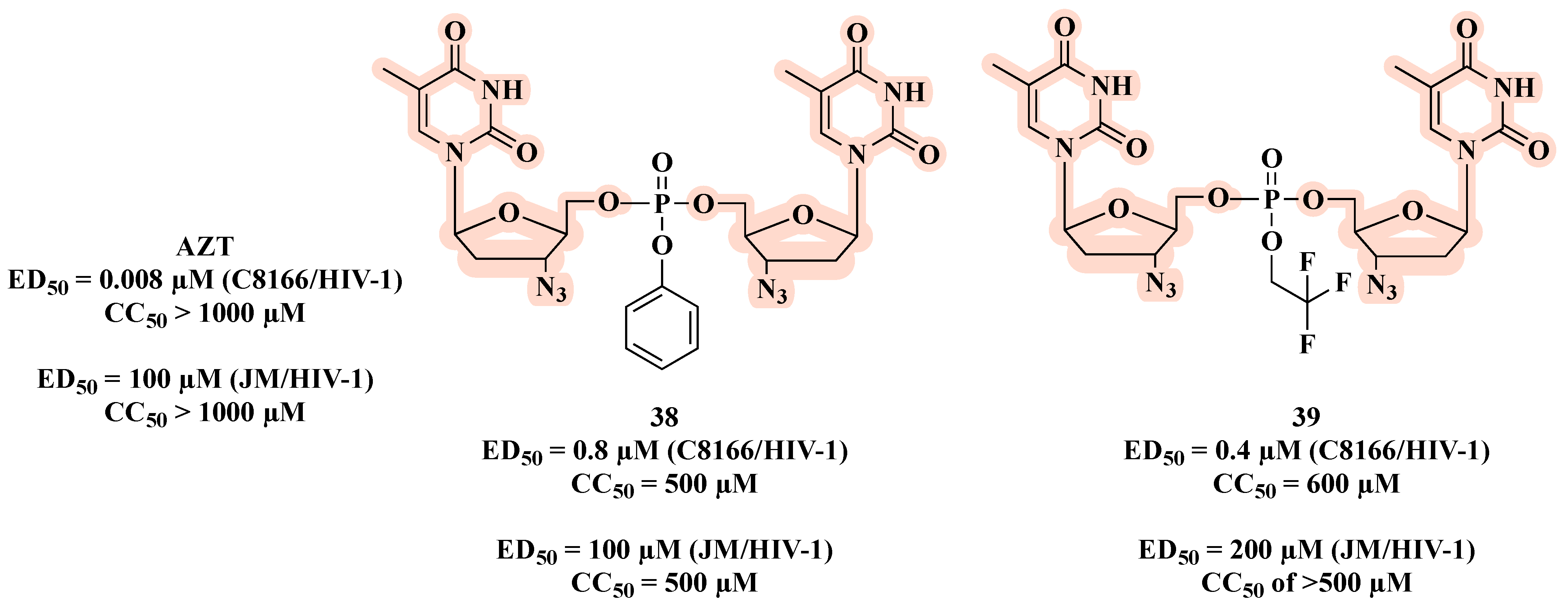{kind=link}
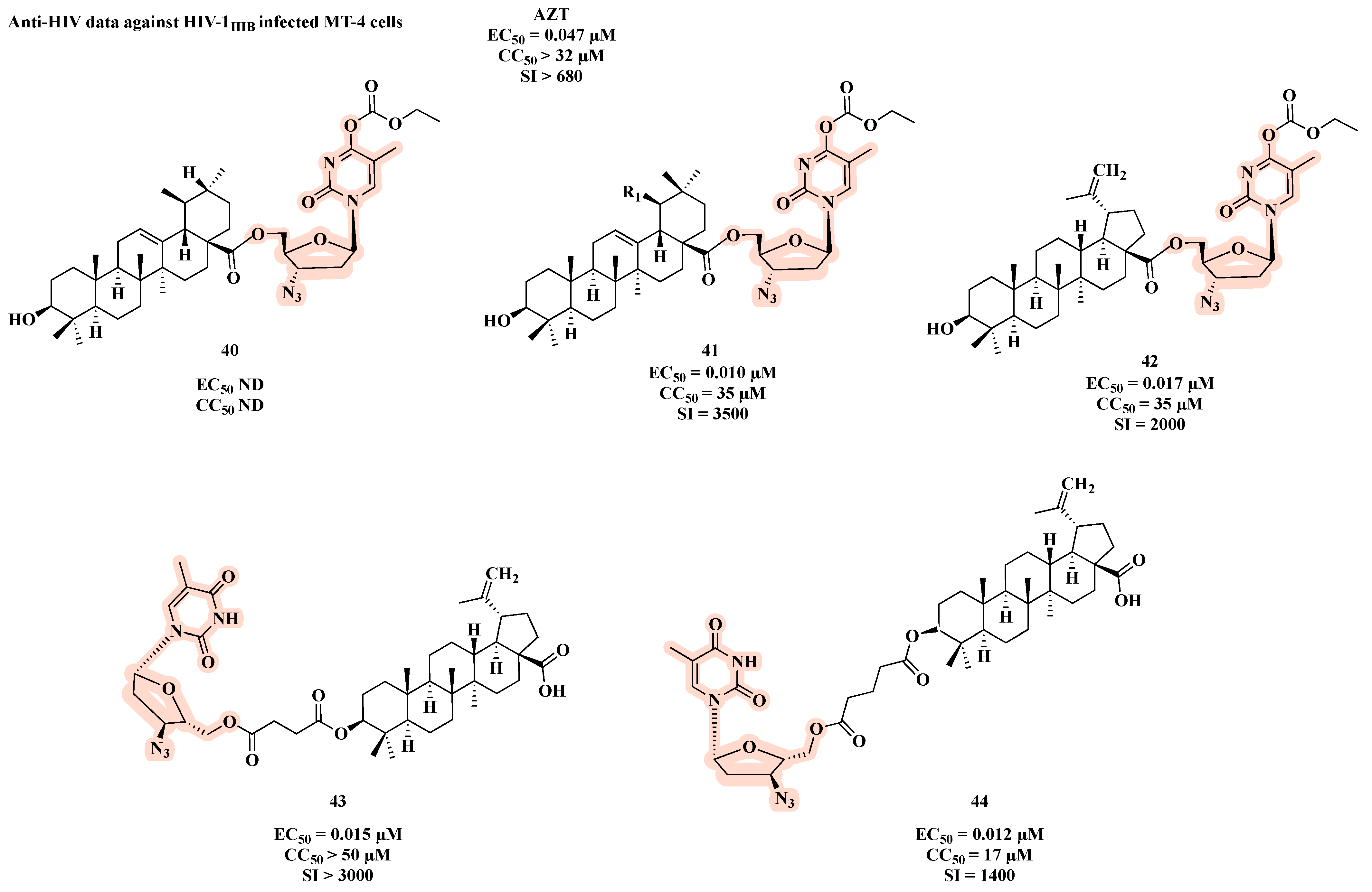{kind=link}
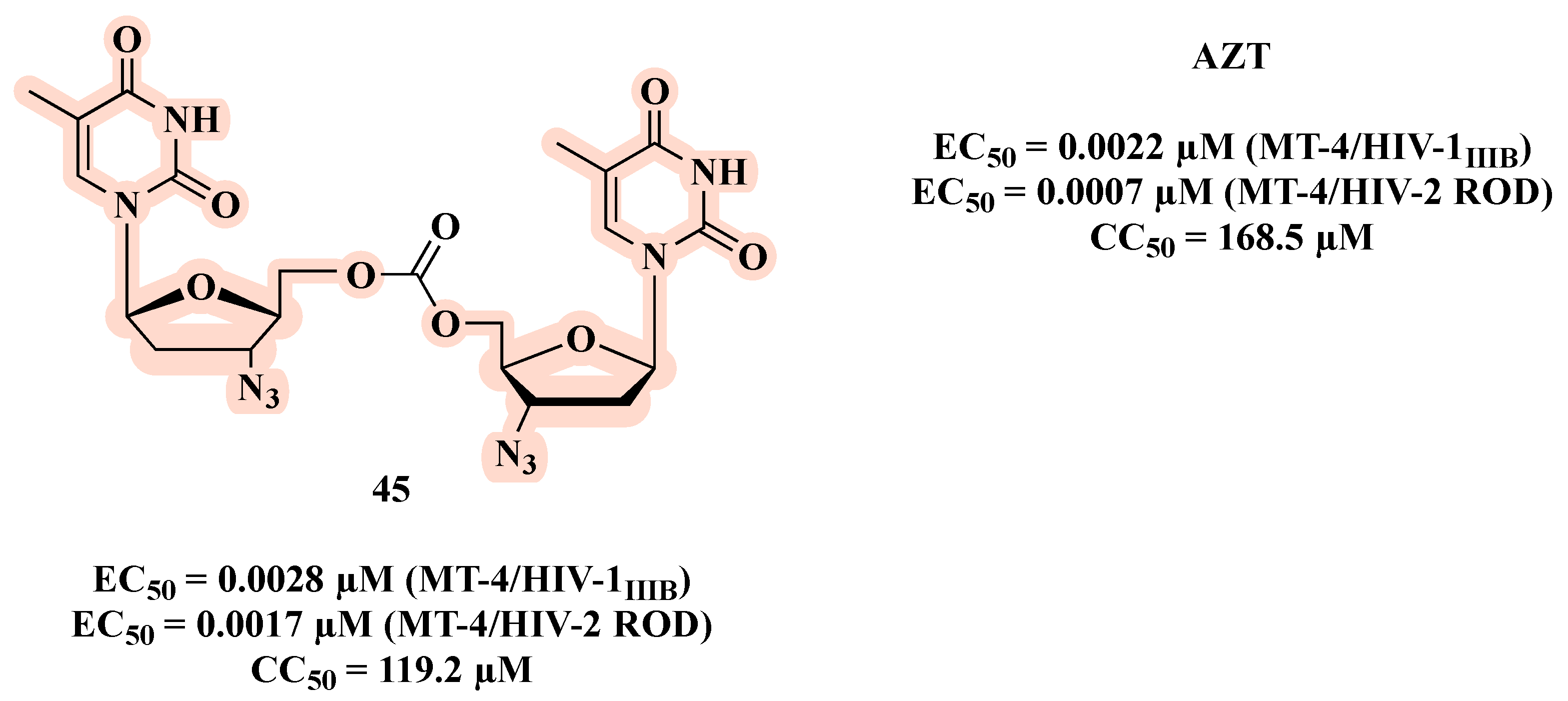{kind=link}
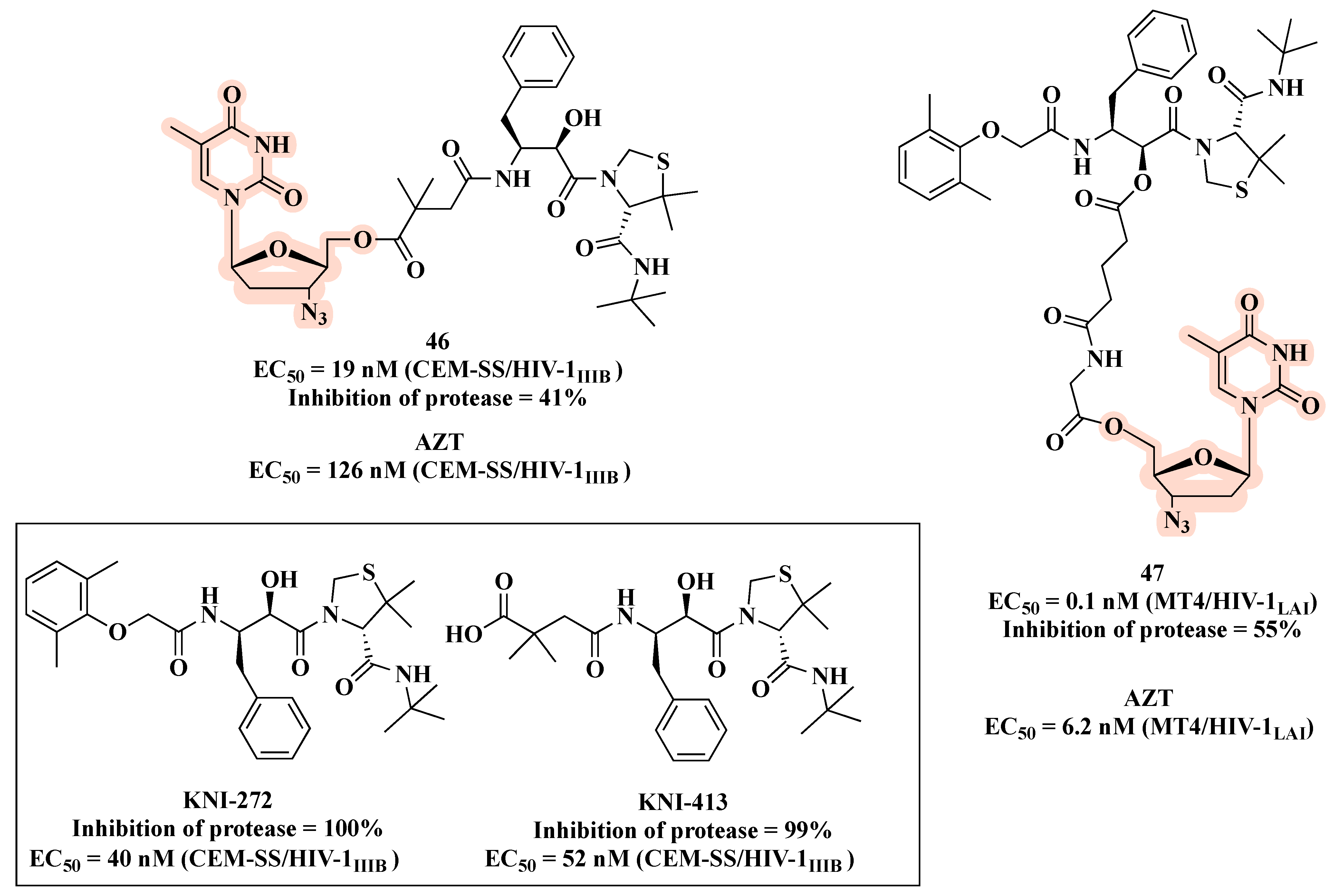{kind=link}
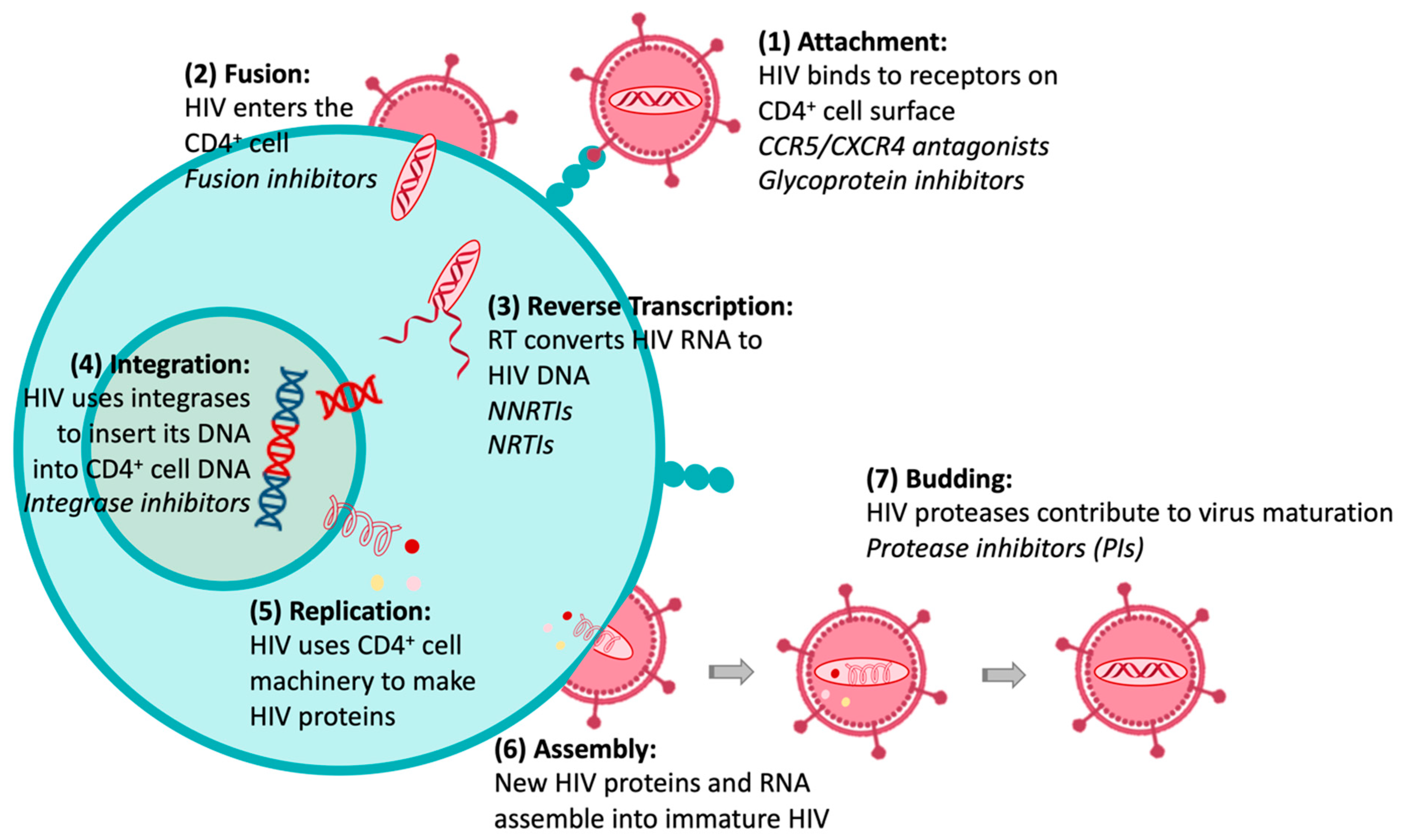
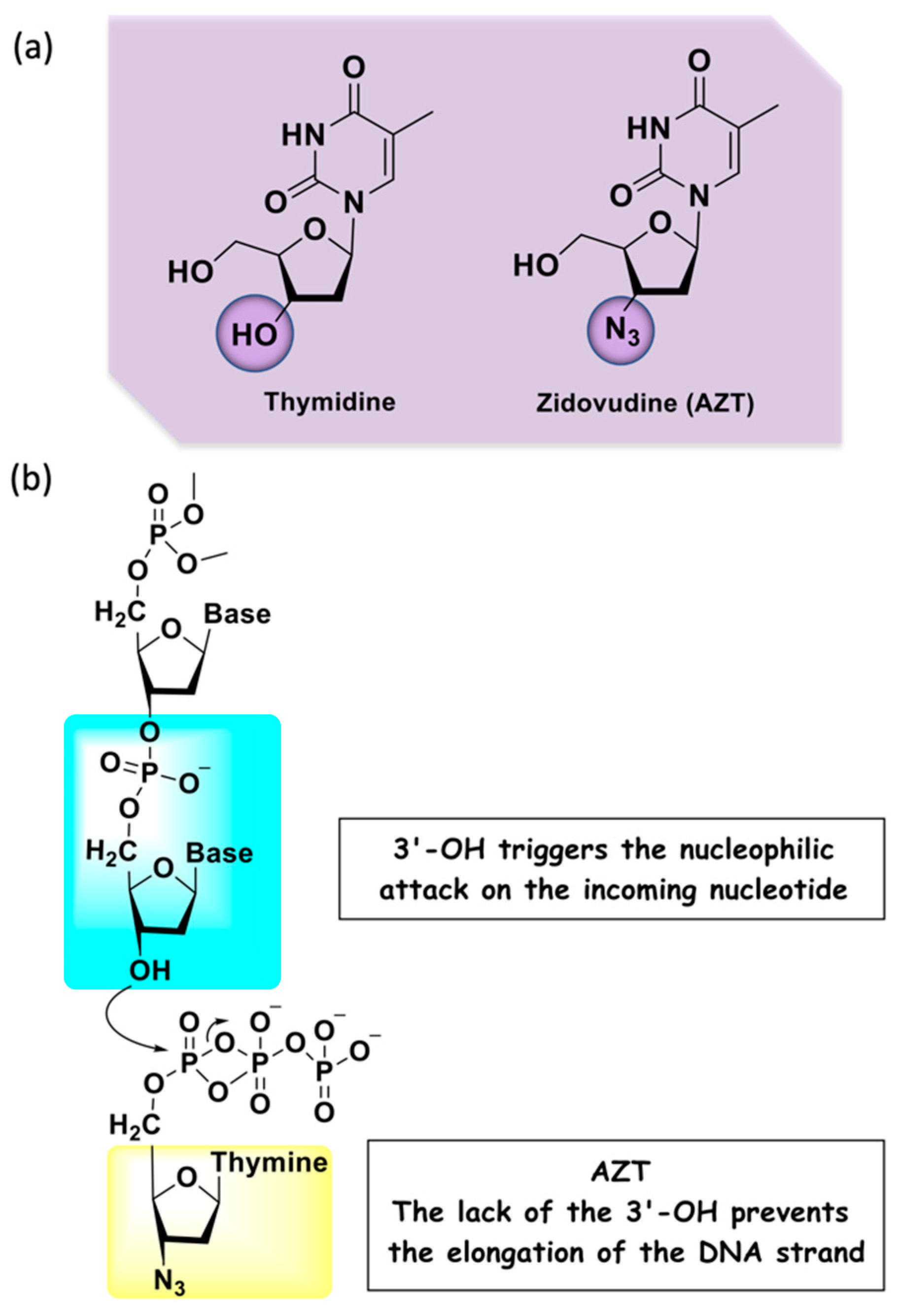
1. Introduction
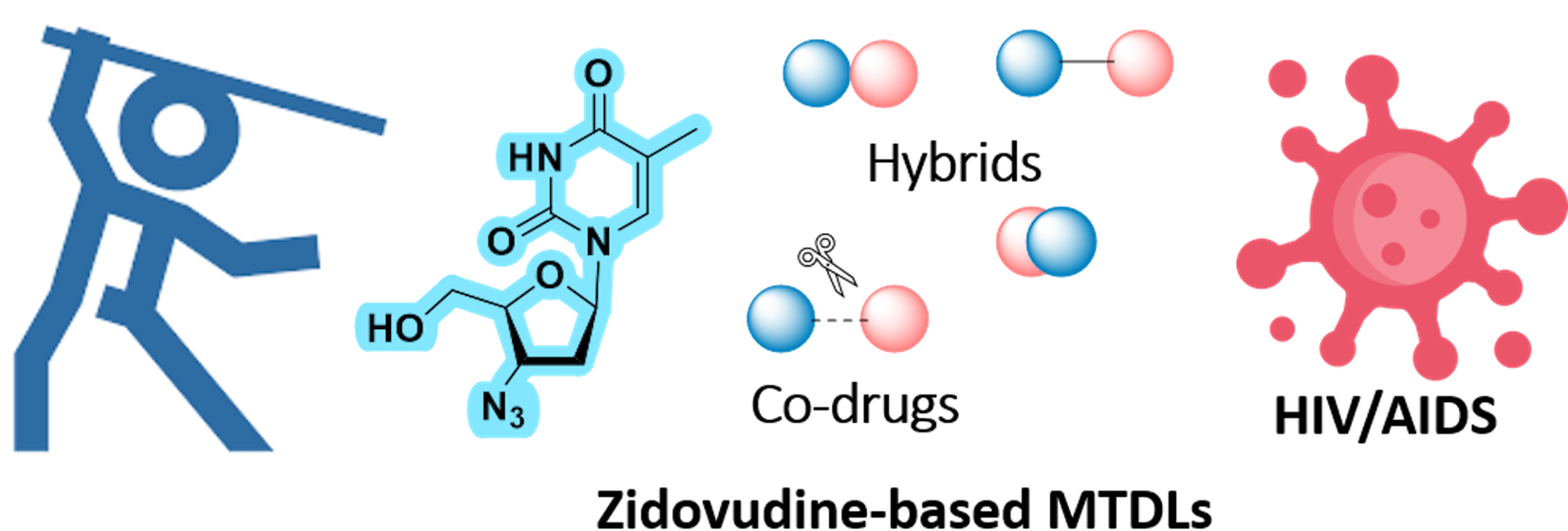
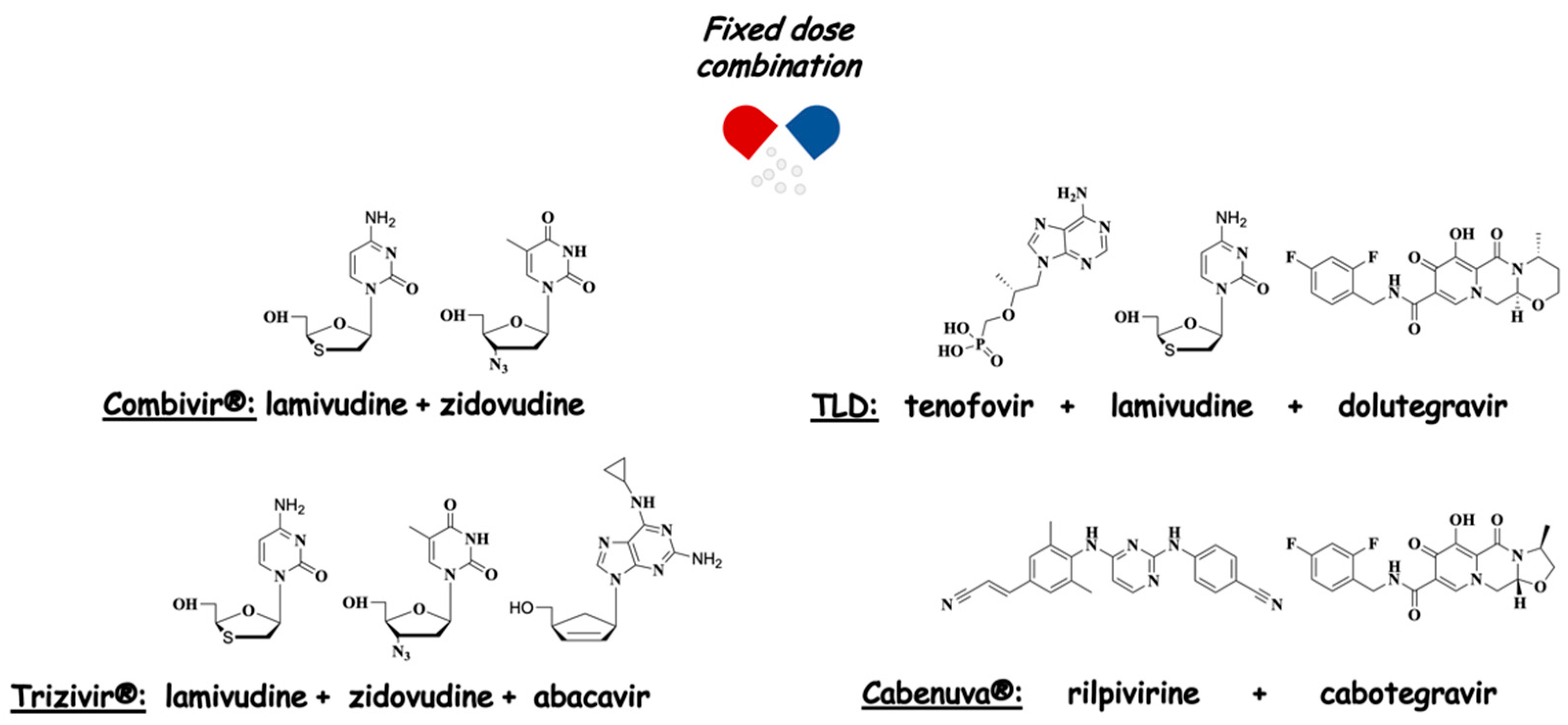
2. Harnessing AZT for Polypharmacology: Drug Cocktails, FDCs, and MTDLs
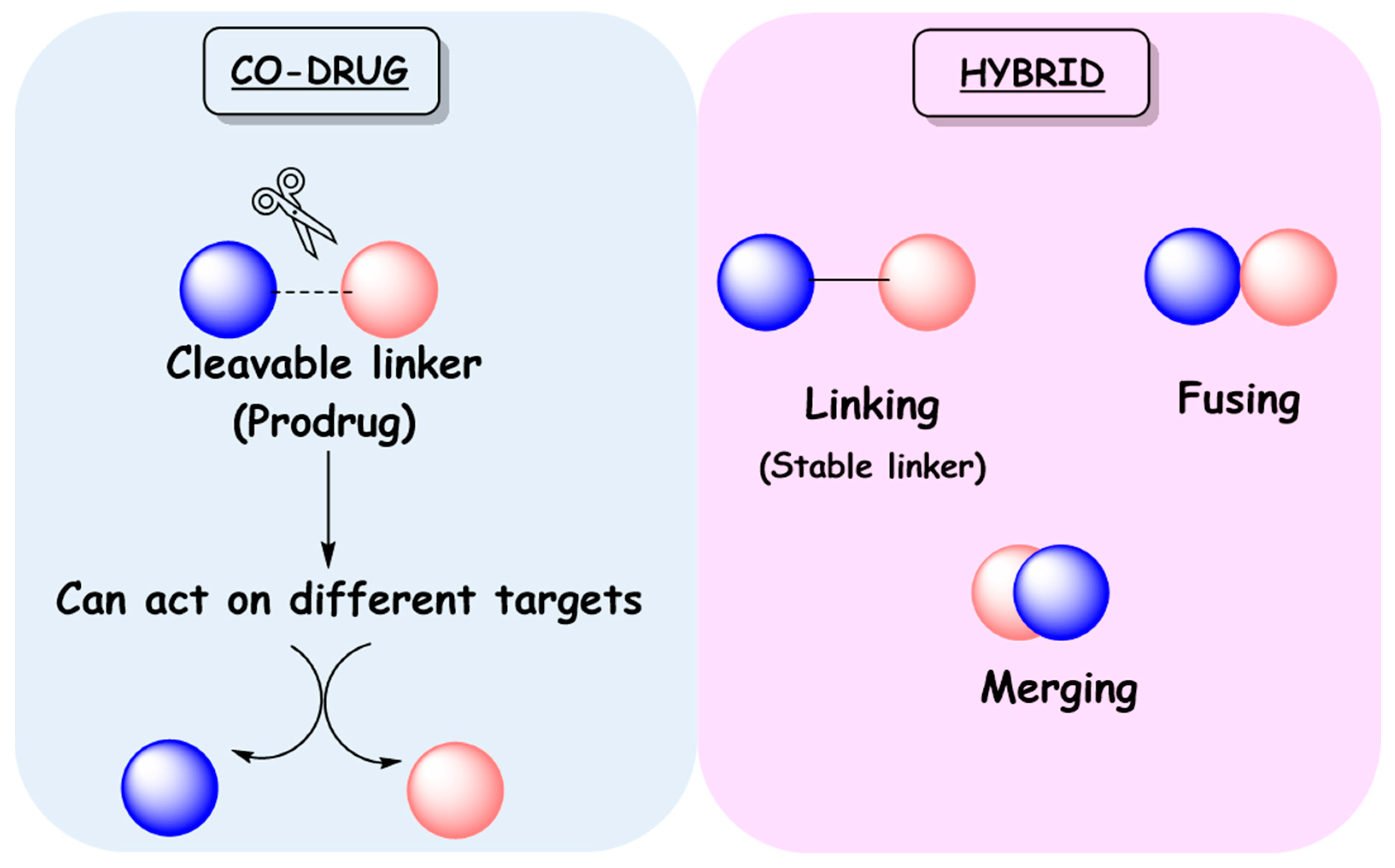
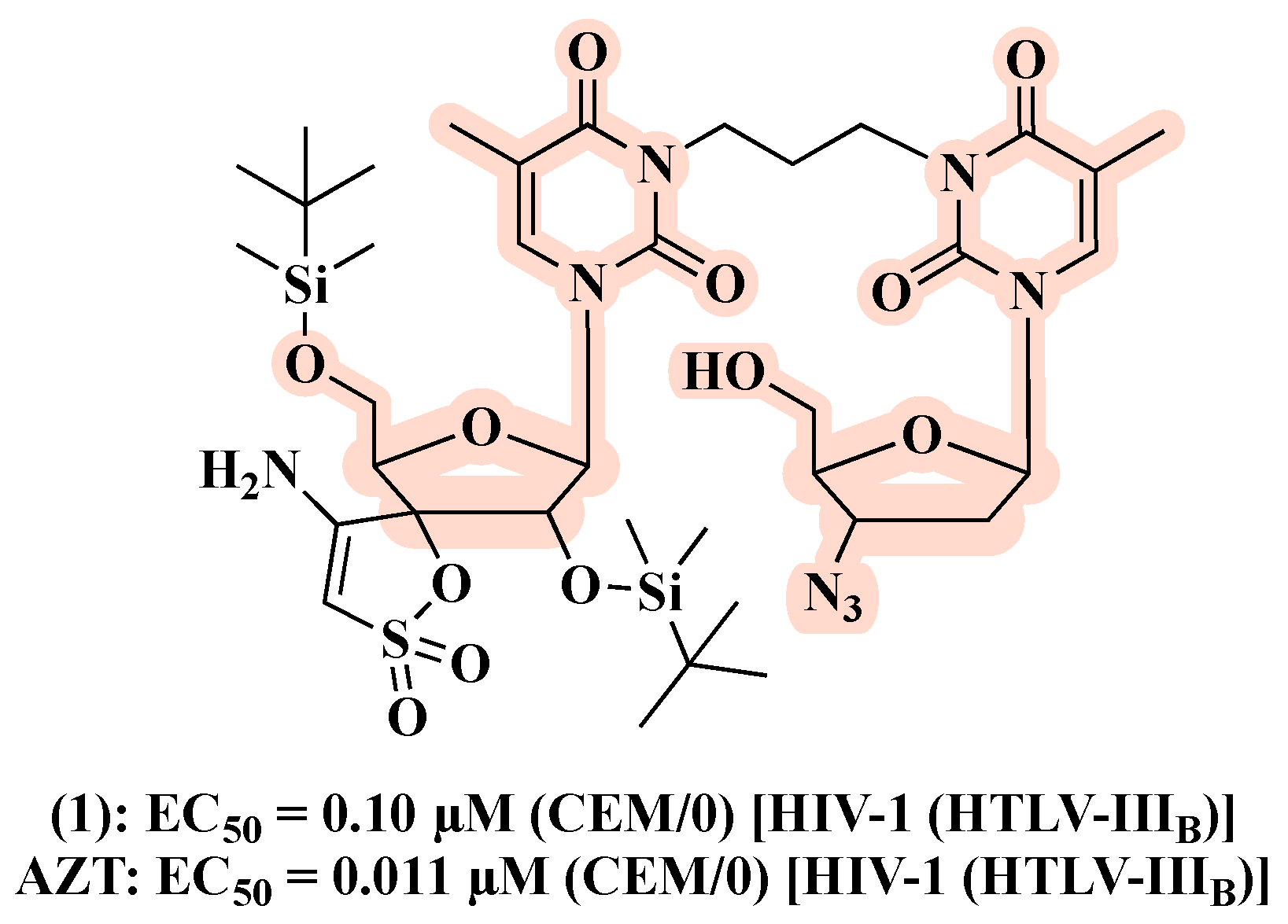
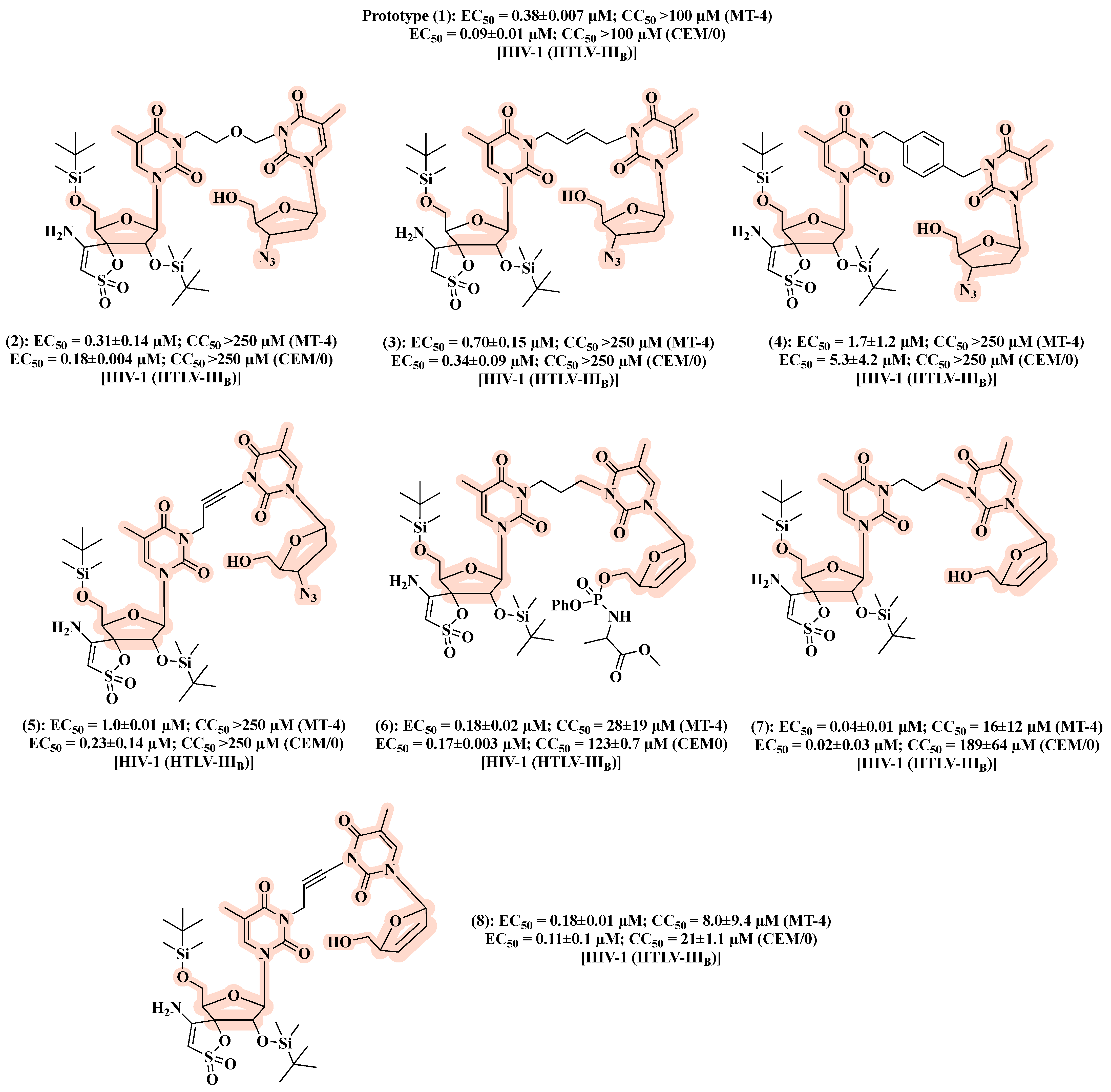
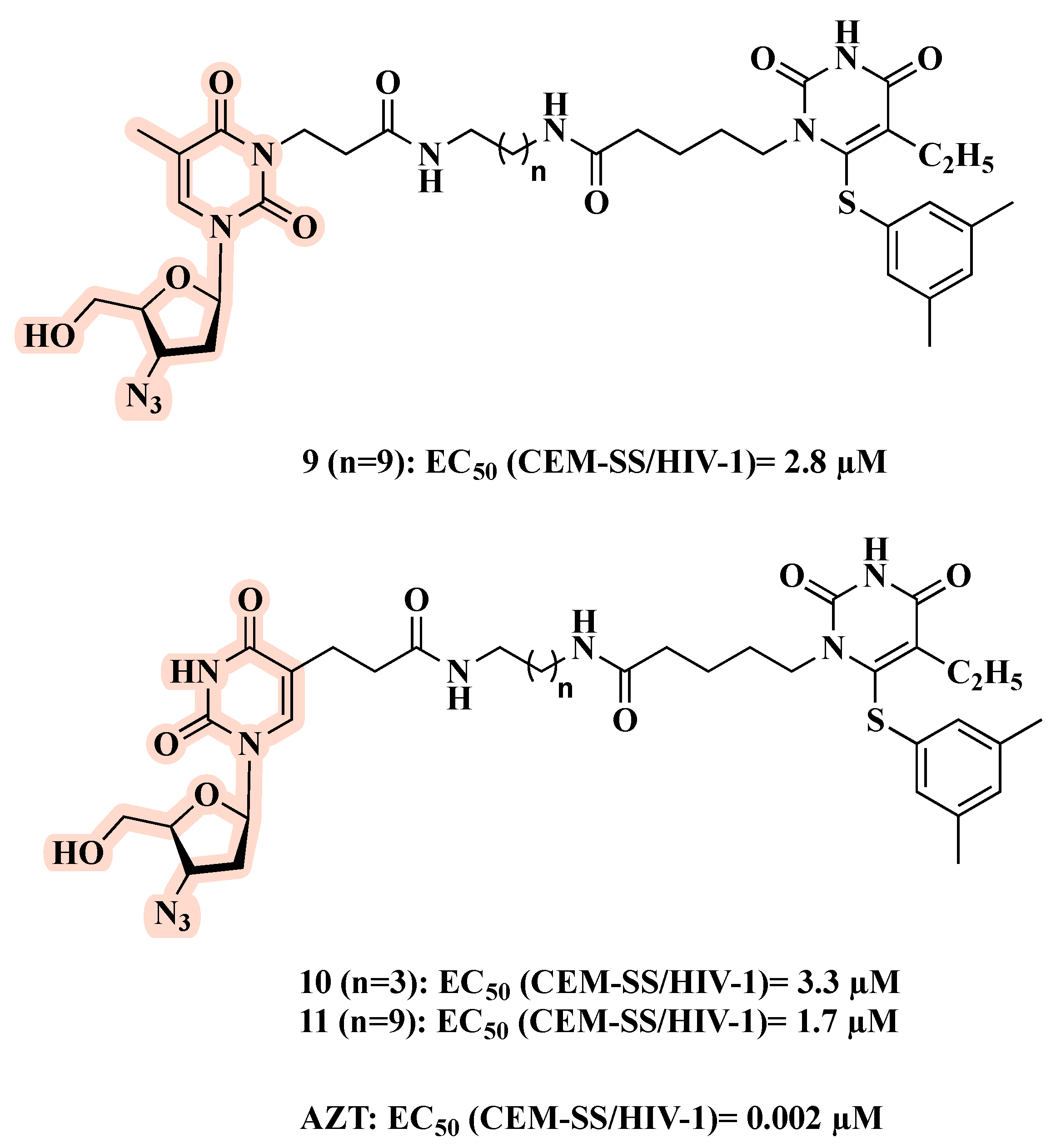
3. Linked Hybrids as Potential HIV-1 Replication Inhibitors
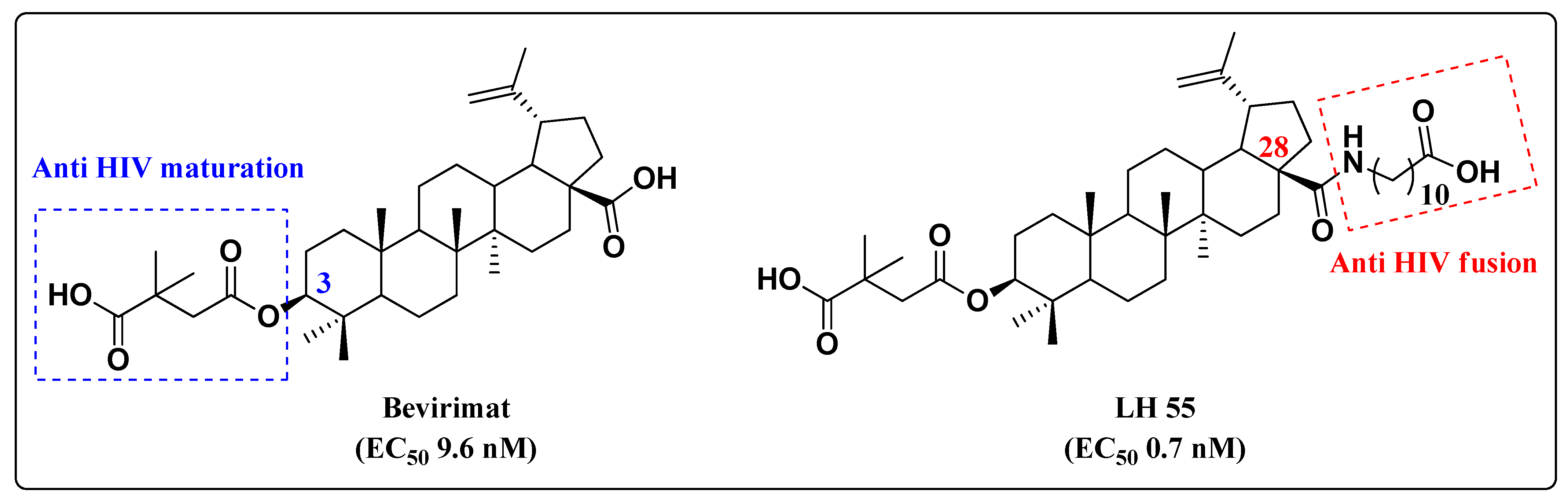
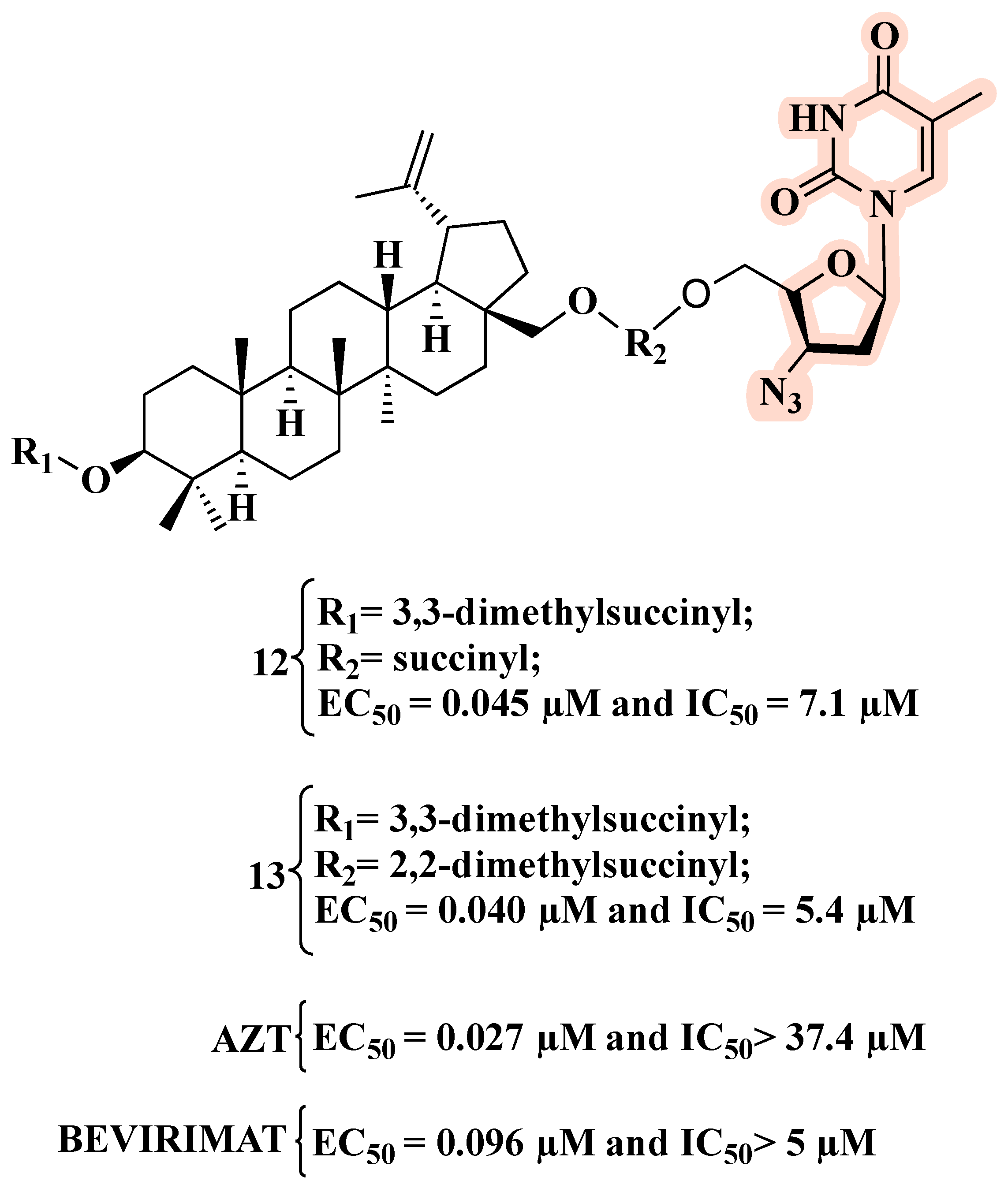
4. Co-Drugs as Potential HIV-1 Inhibitors
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bolognesi, M.L. Harnessing Polypharmacology with Medicinal Chemistry. ACS Med. Chem. Lett. 2019, 10, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.J.; Fraga, C.A.M. Química Medicinal: As Bases Moleculares Da Ação Dos Fármacos, 3rd ed.; Artmed: São Paulo, Brazil, 2015. [Google Scholar]
- Bolognesi, M.L. Polypharmacology in a Single Drug: Multitarget Drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; Uliassi, E.; Bolognesi, M.L. Two diseases, one approach: Multitarget drug discovery in Alzheimer’s and neglected tropical diseases. MedChemComm 2014, 5, 853–861. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Albertini, C.; Salerno, A.; de Sena Murteira Pinheiro, P.; Bolognesi, M.L. From combinations to multitarget-directed ligands: A continuum in Alzheimer’s disease polypharmacology. Med. Res. Rev. 2021, 41, 2606–2633. [Google Scholar] [CrossRef]
- Broder, S. The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antivir. Res. 2010, 85, 1–18. [Google Scholar] [CrossRef]
- Pennings, P.S. HIV drug resistance: Problems and perspectives. Infect. Dis. Rep. 2013, 5, 21–25. [Google Scholar] [CrossRef]
- World Health Organization. HIV AIDS. Available online: https://www.who.int/data/gho/data/themes/hiv-aids#:~:text=Global%20situation%20and%20trends%3A,people%20have%20died%20of%20HIV (accessed on 23 May 2022).
- Arts, E.J.; Hazuda, D.J. HIV-1 Antiretroviral Drug Therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- World Health Organization. HIV Drug Resistance Report 2021. Available online: https://www.who.int/publications/i/item/9789240038608 (accessed on 31 May 2022).
- World Health Organization. Global Action Plan on HIV Drug Resistance 2017–2021. Available online: https://www.who.int/publications/i/item/978-92-4-151284-8 (accessed on 31 May 2022).
- Lin, J.H.; Ostovic, D.; Vacca, J.P. The Integration of Medicinal Chemistry, Drug Metabolism, and Pharmaceutical Research and Development in Drug Discovery and Development. In Integration of Pharmaceutical Discovery and Development. Pharmaceutical Biotechnology; Borchardt, R.T., Freidinger, R.M., Sawyer, T.K., Smith, P.L., Eds.; Springer: Boston, MA, USA, 1998; Volume 11, pp. 233–255. [Google Scholar] [CrossRef]
- Fischl, M.A.; Richman, D.D.; Grieco, M.H.; Gottlieb, M.S.; Volberding, P.A.; Laskin, O.L.; Leedom, J.M.; Groopman, J.E.; Mildvan, D.; Schooley, R.T.; et al. The efficacy of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N. Engl. J. Med. 1987, 317, 185–191. [Google Scholar] [CrossRef]
- Trivedi, J.; Mohan, M.; Byrareddy, S.N. Drug Repurposing Approaches to Combating Viral Infections. J. Clin. Med. 2020, 9, 3777. [Google Scholar] [CrossRef] [PubMed]
- Rough, K.; Sun, J.W.; Seage, G.R.; Williams, P.L.; Huybrechts, K.F.; Bateman, B.T.; Hernandez-Diaz, S. Zidovudine use in pregnancy and congenital malformations. AIDS 2017, 31, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Veal, G.J.; Back, D.J. Metabolism of zidovudine. Gen. Pharmac. 1995, 26, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K.; Marçais, A.; Nasr, R.; Kato, K.; Fukuda, T.; Hermine, O.; Bazarbachi, A. Diagnostic Approaches and Established Treatments for Adult T Cell Leukemia Lymphoma. Front. Microbiol. 2020, 11, 1207. [Google Scholar] [CrossRef] [PubMed]
- Styrt, B.A.; Piazza-Hepp, T.D.; Chikami, G.K. Clinical toxicity of antiretroviral nucleoside analogs. Antivir. Res. 1996, 31, 121–135. [Google Scholar] [CrossRef]
- Enomoto, L.; Anderson, P.L.; Li, S.; Edelstein, C.L.; Weinberg, A. Effect of Nucleoside and Nucleotide Analog Reverse Transcriptase Inhibitors on Cell-Mediated Immune Functions. AIDS Res. Hum. Retrovir. 2011, 27, 47–55. [Google Scholar] [CrossRef]
- Santos, C.; Morais, J.; Gouveia, L.; de Clercq, E.; Pannecouque, C.; Nielsen, C.U.; Steffansen, B.; Moreira, R.; Gomes, P. Dipeptide Derivatives of AZT: Synthesis, Chemical Stability, Activation in Human Plasma, hPEPT1 Affinity, and Antiviral Activity. ChemMedChem 2008, 3, 970–978. [Google Scholar] [CrossRef]
- Phanuphak, N.; Gulick, R.M. HIV treatment and prevention 2019: Current standards of care. Curr. Opin. HIV AIDS 2020, 15, 4–12. [Google Scholar] [CrossRef]
- Weichseldorfer, M.; Reitz, M.; Latinovic, O.S. Past HIV-1 Medications and the Current Status of Combined Antiretroviral Therapy Options for HIV-1 Patients. Pharmaceutics 2021, 13, 1798. [Google Scholar] [CrossRef]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]
- Cheng, F.; Kovacs, I.A.; Barabási, A.-L. Network-based prediction of drug combinations. Nat. Commun. 2019, 10, 1197. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Ivasiv, V.; Albertini, C.; Gonçalves, A.E.; Rossi, M.; Bolognesi, M.L. Molecular Hybridization as a Tool for Designing Multitarget Drug Candidates for Complex Diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, S.; Alvarez, R.; San-Felix, A.; Jimeno, M.L.; De Clercq, E.; Balzarini, J.; Camarasa, M.J. Synthesis and Anti-HIV Activity of [AZT]-[TSAO-T] and [AZT]-[HEPT] Dimers as Potential Multifunctional Inhibitors of HIV-1 Reverse Transcriptase. J. Med. Chem. 1995, 38, 1641–1649. [Google Scholar] [CrossRef]
- Velázquez, S.; Tuñón, V.; Jimeno, M.L.; Chamorro, C.; De Clercq, E.; Balzarini, J.; Camarasa, M.J. Potential Multifunctional Inhibitors of HIV-1 Reverse Transcriptase. Novel [AZT]-[TSAO-T] and [d4T]-[TSAO-T] Heterodimers Modified in the Linker and in the Dideoxynucleoside Region. J. Med. Chem. 1999, 42, 5188–5196. [Google Scholar] [CrossRef]
- Pontikis, R.; Dollé, V.; Guillaumel, J.; Dechaux, E.; Note, R.; Nguyen, C.H.; Legraverend, M.; Bisagni, E.; Aubertin, A.-M.; Grierson, D.S.; et al. Synthesis and Evaluation of “AZT-HEPT”, “AZT-Pyridinone”, and “ddC-HEPT” Conjugates as Inhibitors of HIV Reverse Transcriptase. J. Med. Chem. 2000, 43, 1927–1939. [Google Scholar] [CrossRef]
- Hashimoto, F.; Kashiwada, Y.; Cosentino, L.; Chen, C.-H.; Garrett, P.E.; Lee, K.-H. Anti-AIDS agents—XXVII. Synthesis and anti-HIV activity of betulinic acid and dihydrobetulinic acid derivatives. Bioorg. Med. Chem. 1997, 5, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zoumplis, D.; Matallana, C.; Kilgore, N.R.; Reddick, M.; Yunus, A.S.; Adamson, C.S.; Salzwedel, K.; Martin, D.E.; Allaway, G.P.; et al. Determinants of activity of the HIV-1 maturation inhibitor PA-457. Virology 2006, 356, 217–224. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soler, F.; Poujade, C.; Evers, M.; Carry, J.-C.; Hénin, Y.; Bousseau, A.; Huet, T.; Pauwels, R.; De Clercq, E.; Mayaux, J.-F.; et al. Betulinic Acid Derivatives: A New Class of Specific Inhibitors of Human Immunodeficiency Virus Type 1 Entry. J. Med. Chem. 1996, 39, 1069–1083. [Google Scholar] [CrossRef]
- Lai, W.; Huang, L.; Ho, P.; Li, Z.; Montefiori, D.; Chen, C.-H. Betulinic Acid Derivatives That Target gp120 and Inhibit Multiple Genetic Subtypes of Human Immunodeficiency Virus Type 1. Antimicrob. Agents Chemother. 2008, 52, 128–136. [Google Scholar] [CrossRef]
- Xiong, J.; Kashiwada, Y.; Chen, C.-H.; Qian, K.; Morris-Natschke, S.L.; Lee, K.-H.; Takaishi, Y. Conjugates of betulin derivatives with AZT as potent anti-HIV agents. Bioorg. Med. Chem. 2010, 18, 6451–6469. [Google Scholar] [CrossRef] [PubMed]
- Bori, I.D.; Hung, H.-Y.; Qian, K.; Chen, C.-H.; Morris-Natschke, S.L.; Lee, K.-H. Anti-AIDS agents 88. Anti-HIV conjugates of betulin and betulinic acid with AZT prepared via click chemistry. Tetrahedron Lett. 2012, 53, 1987–1989. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.T.A.; Nguyen, T.K.T.; Pham, T.C.; Nguyen, H.T.; Ba, T.C.; Doan, D.T.; D’hooghe, M.; Nguyen, V.T. Synthesis and cytotoxic evaluation of novel ester-triazole-linked triterpenoid–AZT conjugates. Bioorg. Med. Chem. Lett. 2014, 24, 5190–5194. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.T.A.; Nguyen, T.K.T.; Pham, T.C.; Nguyen, H.T.; Ba, T.C.; Phuong, H.T.; Luu, V.B.; Nguyen, V.T.; D’hooghe, M. Synthesis and cytotoxic evaluation of novel amide–triazole-linked triterpenoid–AZT conjugates. Tetrahedron Lett. 2015, 56, 218–224. [Google Scholar] [CrossRef]
- Vasilevsky, S.F.; Govdi, A.I.; Sorokina, I.V.; Tolstikova, T.G.; Baev, D.S.; Tolstikov, G.A.; Mamatuyk, V.I.; Alabugin, I.V. Rapid access to new bioconjugates of betulonic acid via click chemistry. Bioorg. Med. Chem. Lett. 2011, 21, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Majeed, R.; Sangwan, P.L.; Chinthakindi, P.K.; Khan, I.; Dangroo, N.A.; Thota, N.; Hamid, A.; Sharma, P.R.; Saxena, A.K.; Koul, S. Synthesis of 3-Opropargylated betulinic acid and its 1,2,3-triazoles as potential apoptotic agents. Eur. J. Med. Chem. 2013, 63, 782–792. [Google Scholar] [CrossRef]
- Pertino, M.W.; Lopez, C.; Theoduloz, C.; Schmeda-Hirschmann, G. 1,2,3-Triazole-Substituted Oleanolic Acid Derivatives: Synthesis and Antiproliferative Activity. Molecules 2013, 18, 7661–7674. [Google Scholar] [CrossRef]
- Cheng, K.; Liu, J.; Liu, X.; Li, H.; Sun, H.; Xie, J. Synthesis of glucoconjugates of oleanolic acid as inhibitors of glycogen phosphorylase. Carbohydr. Res. 2009, 344, 841–850. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, S.-J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef]
- Oramas-Royo, S.; López-Rojas, P.; Amesty, Á.; Gutiérrez, D.; Flores, N.; Martín-Rodríguez, P.; Fernández-Pérez, L.; Estévez-Braun, A. Synthesis and Antiplasmodial Activity of 1,2,3-Triazole-Naphthoquinone Conjugates. Molecules 2019, 24, 3917. [Google Scholar] [CrossRef]
- Xu, Z. 1,2,3-Triazole-containing hybrids with potential antibacterial activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Med. Chem. 2020, 206, 112686. [Google Scholar] [CrossRef] [PubMed]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef]
- Zhou, L.; Amer, A.; Korn, M.; Burda, R.; Balzarini, J.; De Clercq, E.; Kern, E.R.; Torrence, P.F. Synthesis and Antiviral Activities of 1,2,3-triazole Functionalized Thymidines: 1,3-dipolar Cycloaddition for Efficient Regioselective Diversity Generation. Antivir. Chem. Chemother. 2005, 16, 375–383. [Google Scholar] [CrossRef]
- Feng, L.-S.; Zheng, M.-J.; Zhao, F.; Liu, D. 1,2,3-Triazole hybrids with anti-HIV-1 activity. Arch. Pharm. 2021, 354, e2000163. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Y.; Zheng, L.; Huang, X.; Wang, Y.; Chen, C.-H.; Cheng, Y.-Y.; Morris-Natschke, S.L.; Lee, K.-H. Novel Betulinic Acid–Nucleoside Hybrids with Potent Anti-HIV Activity. ACS Med. Chem. Lett. 2020, 11, 2290–2293. [Google Scholar] [CrossRef] [PubMed]
- Sirivolu, V.R.; Vernekar, S.K.V.; Ilina, T.; Myshakina, N.S.; Parniak, M.A.; Wang, Z. Clicking 3′-Azidothymidine into Novel Potent Inhibitors of Human Immunodeficiency Virus. J. Med. Chem. 2013, 56, 8765–8780. [Google Scholar] [CrossRef] [PubMed]
- Popova, E.A.; Ovsepyan, G.K.; Protas, A.V.; Erkhitueva, E.B.; Kukhanova, M.K.; Yesaulkova, Y.L.; Zarubaev, V.V.; Starova, G.L.; Suezov, R.V.; Eremin, A.V.; et al. Synthesis and in vitro Biological Evaluation of Novel Thymidine Analogs Containing 1H-1,2,3-Triazolyl, 1H-Tetrazolyl, and 2H-Tetrazolyl Fragments. Nucleosides Nucleotides Nucleic Acids 2019, 38, 713–731. [Google Scholar] [CrossRef]
- Vernekar, S.K.V.; Qiu, L.; Zhang, J.; Kankanala, J.; Li, H.; Geraghty, R.J.; Wang, Z. 5′-Silylated 3′-1,2,3-triazolyl Thymidine Analogues as Inhibitors of West Nile Virus and Dengue Virus. J. Med. Chem. 2015, 58, 4016–4028. [Google Scholar] [CrossRef]
- Vernekar, S.K.V.; Qiu, L.; Zacharias, J.; Geraghty, R.J.; Wang, Z. Synthesis and antiviral evaluation of 4′-(1,2,3-triazol-1-yl)thymidines. MedChemComm 2014, 5, 603–608. [Google Scholar] [CrossRef]
- Olomola, T.O.; Klein, R.; Mautsa, N.; Sayed, Y.; Kaye, P.T. Synthesis and evaluation of coumarin derivatives as potential dual-action HIV-1 protease and reverse transcriptase inhibitors. Bioorg. Med. Chem. 2013, 21, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Boechat, N.; Macedo, M.B.; Souza, T.M.L.; Leite, D.I.; Bernardino, A.M.R. Isatin-Derived Compounds, Use of the Compounds for the Treatment of AIDS and Method of Treatment Using These Compounds. U.S. Patent 10538515B2, 10 May 2016. [Google Scholar]
- Costa, C.C.P. Novos Derivados de Isatina Como Potenciais Inibidores da Coinfecção HIV-TB. Ph.D. Thesis, Universidade Federal Fluminense, Niterói, Brazil, February 2019. [Google Scholar]
- Costa, C.C.P.; Boechat, N.; Bastos, M.M.; Silva, F.D.C.D.; Marttorelli, A.; Souza, T.; Baptista, M.S.; Hoelz, L.V.; Cafffarena, E.R. New Efavirenz Derivatives and 1,2,3-Triazolyl-phosphonates as Inhibitors of Reverse Transcriptase of HIV-1. Curr. Top. Med. Chem. 2018, 18, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Aminake, M.N.; Mahajan, A.; Kumar, V.; Hans, R.; Wiesner, L.; Taylor, D.; de Kock, C.; Grobler, A.; Smith, P.J.; Kirschner, M.; et al. Synthesis and evaluation of hybrid drugs for a potential HIV/AIDS-malaria combination therapy. Bioorg. Med. Chem. 2012, 20, 5277–5289. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, P.; Long, J.; Swetha, R.; Shruthi, V.; Wang, R.-R.; Preethi, S.; Yogeeswari, P.; Zheng, Y.-T.; Sriram, D. Synthesis of Zidovudine Derivatives with Anti-HIV-1 and Antibacterial Activities. Nucleosides Nucleotides Nucleic Acids 2009, 28, 89–102. [Google Scholar] [CrossRef]
- Castro, S.; Caramasa, M.J. Polypharmacology in HIV inhibition: Can a drug with simultaneous action against two relevant targets be an alternative to combination therapy? Eur. J. Med. Chem. 2018, 150, 206–227. [Google Scholar] [CrossRef]
- Rossi, M.; Petralla, S.; Protti, M.; Baiula, M.; Kobrlova, T.; Soukup, O.; Spampinato, S.M.; Mercolini, L.; Monti, B.; Bolognesi, M.L. α-Linolenic Acid–Valproic Acid Conjugates: Toward Single-Molecule Polypharmacology for Multiple Sclerosis. ACS Med. Chem. Lett. 2020, 11, 2406–2413. [Google Scholar] [CrossRef]
- Albertini, C.; Naldi, M.; Petralla, S.; Strocchi, S.; Grifoni, D.; Monti, B.; Bartolini, M.; Bolognesi, M.L. From Combinations to Single-Molecule Polypharmacology—Cromolyn-Ibuprofen Conjugates for Alzheimer’s Disease. Molecules 2021, 26, 1112. [Google Scholar] [CrossRef]
- Busso, M.; Mian, A.M.; Hahn, E.F.; Resnick, L. Nucleotide Dimers Suppress HIV Expression In Vitro. AIDS Res. Hum. Retrovir. 1988, 4, 449–455. [Google Scholar] [CrossRef]
- Ijichi, K.; Fujiwara, M.; Mori, K.; Morozumi, M.; Machida, H.; Shigeta, S.; Konno, K.; Yokota, T.; Baba, M. Antiviral activities of nucleotide heterodimers against human immunodeficiency virus type 1 in vitro. Antivir. Res. 1996, 31, 115–120. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Pathirana, R.N.; Balzarini, J.; De Clercq, E. Intracellular delivery of bioactive AZT nucleotides by aryl phosphate derivatives of AZT. J. Med. Chem. 1993, 36, 1048–1052. [Google Scholar] [CrossRef]
- Dang, A.T.T.; Pham, C.T.; Le, T.A.; Truong, H.H.; Vu, H.T.T.; Soldatenkov, A.T.; Van Nguyen, T. New hybrids between triterpenoid acids and nucleoside HIV-RT inhibitors. Mendeleev Commun. 2015, 25, 96–98. [Google Scholar] [CrossRef]
- Taourirte, M.; Mohamed, L.A.; Rochdi, A.; Vasseur, J.; Fernández, S.; Ferrero, M.; Gotor, V.; Pannecouque, C.; De Clercq, E.; Lazrek, H.B. Chemoenzymatic Syntheses of Homo- and Heterodimers of AZT and d4T, and Evaluation of Their Anti-HIV Activity. Nucleosides Nucleotides Nucleic Acids 2004, 23, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Matsumoto, H.; Matsuda, T.; Hamawaki, T.; Akaji, K.; Kiso, Y. A new class of anti-HIV agents: Synthesis and activity of conjugates of HIV protease inhibitors with a reverse transcriptase inhibitor. Bioorg. Med. Chem. Lett. 1999, 9, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Matsuda, T.; Nakata, S.; Mitoguchi, T.; Kimura, T.; Hayashi, Y.; Kiso, Y. Synthesis and biological evaluation of prodrug-type anti-HIV agents: Ester conjugates of carboxylic acid-containing dipeptide HIV protease inhibitors and a reverse transcriptase inhibitor. Bioorg. Med. Chem. 2001, 9, 417–430. [Google Scholar] [CrossRef] [PubMed]
























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianco, M.d.C.A.D.; Inacio Leite, D.; Silva Castelo Branco, F.; Boechat, N.; Uliassi, E.; Bolognesi, M.L.; Bastos, M.M. The Use of Zidovudine Pharmacophore in Multi-Target-Directed Ligands for AIDS Therapy. Molecules 2022, 27, 8502. https://doi.org/10.3390/molecules27238502
Bianco MdCAD, Inacio Leite D, Silva Castelo Branco F, Boechat N, Uliassi E, Bolognesi ML, Bastos MM. The Use of Zidovudine Pharmacophore in Multi-Target-Directed Ligands for AIDS Therapy. Molecules. 2022; 27(23):8502. https://doi.org/10.3390/molecules27238502
Chicago/Turabian StyleBianco, Maria da Conceição Avelino Dias, Debora Inacio Leite, Frederico Silva Castelo Branco, Nubia Boechat, Elisa Uliassi, Maria Laura Bolognesi, and Monica Macedo Bastos. 2022. "The Use of Zidovudine Pharmacophore in Multi-Target-Directed Ligands for AIDS Therapy" Molecules 27, no. 23: 8502. https://doi.org/10.3390/molecules27238502
APA StyleBianco, M. d. C. A. D., Inacio Leite, D., Silva Castelo Branco, F., Boechat, N., Uliassi, E., Bolognesi, M. L., & Bastos, M. M. (2022). The Use of Zidovudine Pharmacophore in Multi-Target-Directed Ligands for AIDS Therapy. Molecules, 27(23), 8502. https://doi.org/10.3390/molecules27238502