

Rhenium Tricarbonyl Complexes of Azodicarboxylate Ligands



Abstract
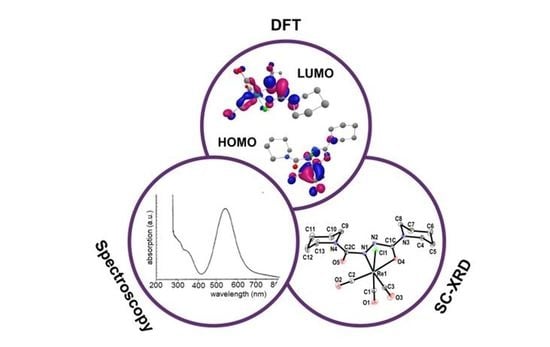
1. Introduction
2. Results and Discussion
2.1. Syntheses
2.2. Exchange Reactions for the Chlorido Coligand
2.3. Structures from X-ray Diffraction and DFT Calculations
2.4. Electrochemistry, EPR and DFT-Calculated Frontier Orbitals
2.5. UV–Vis–NIR Absorption Spectroscopy
3. Conclusions
4. Experimental Section
Syntheses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Usman, M.; Zhang, X.-W.; Wu, D.; Guan, Z.-H.; Liu, W.-B. Application of dialkyl azodicarboxylate frameworks featuring multi-functional properties. Org. Chem. Front. 2019, 6, 1905–1928. [Google Scholar] [CrossRef]
- Beddoe, R.H.; Sneddon, H.F.; Denton, R.M. The catalytic Mitsunobu reaction: A critical analysis of the current state-of-the-art. Org. Biomol. Chem. 2018, 16, 7774–7781. [Google Scholar] [CrossRef] [PubMed]
- Hirose, D.; Gazvoda, M.; Kosmrlj, J.; Taniguchi, T. Advances and mechanistic insight on the catalytic Mitsunobu reaction using recyclable azo reagents. Chem. Sci. 2016, 7, 5148–5159. [Google Scholar] [CrossRef] [PubMed]
- Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P.P. Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, K.; Kainuma, S.; Minakata, S. Electrophilic Amination of Allylic Boranes with Azodicarboxylates: Synthesis of α,α-Disubstituted Allylic Amine Derivatives. Chem. Lett. 2019, 48, 1116–1118. [Google Scholar] [CrossRef]
- Lin, S.; Lin, Z. DFT Studies on Metal-Controlled Regioselective Amination of N-Acylpyrazoles with Azodicarboxylates. J. Org. Chem. 2019, 84, 12399–12407. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J. Aerobic Oxidation of Alkyl 2-Phenylhydrazinecarboxylates Catalyzed by CuCl and DMAP. J. Org. Chem. 2018, 83, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Jang, S.H.; Yim, T.; Kim, J. Oxidation Potential Tunable Organic Molecules and Their Catalytic Application to Aerobic Dehydrogenation of Tetrahydroquinolines. Org. Lett. 2018, 20, 6436–6439. [Google Scholar] [CrossRef]
- Chen, G.J.J.; McDonald, J.W.; Bravard, D.C.; Newton, W.E. Synthetic utility of molybdenum-diazene adducts: Preparation, reactions, and spectral properties of oxo-free and (18O)oxo molybdenum complexes. Inorg. Chem. 1985, 24, 2327–2333. [Google Scholar] [CrossRef]
- McDonald, J.W.; Corbin, J.L.; Newton, W.E. Catalysis by molybdenum complexes. The reaction of diazenes and acetylenes with thiophenol. Inorg. Chem. 1976, 15, 2056–2061. [Google Scholar] [CrossRef]
- Jana, A.; Sen, S.S.; Roesky, H.W.; Schulzke, C.; Dutta, S.; Pati, S.K. End-On Nitrogen Insertion of a Diazo Compound into a Germanium(II) Hydrogen Bond and a Comparable Reaction with Diethyl Azodicarboxylate. Angew. Chem. Int. Ed. 2009, 48, 4246–4248. [Google Scholar] [CrossRef] [PubMed]
- Muniz, K.; Iglesias, A. Phenanthroline Ligands in Aryl Palladium Hydrazinato Complexes: Catalysts for Efficient Coupling of Azo Componds with Aryl Boronic Acids. Angew. Chem. Int. Ed. 2007, 46, 6350–6353. [Google Scholar] [CrossRef]
- Muniz, K.; Nieger, M. Catalytic Activation of N−N Multiple Bonds: A Homogeneous Palladium Catalyst for Mechanistically Unprecedented Reduction of Azo Compounds. Angew. Chem. Int. Ed. 2006, 45, 2305–2308. [Google Scholar] [CrossRef] [PubMed]
- Cianga, L. Synthesis and characterization of optically active polymers containing azo groups and (L)-α-amino acid moieties. Europ. Polym. J. 2003, 39, 2271–2282. [Google Scholar] [CrossRef]
- Kaim, W. Chelate rings of different sizes with non-innocent ligands. Dalton Trans. 2019, 48, 8521–8529. [Google Scholar] [CrossRef]
- Poornima, S.; Packiaraj, S.; Pushpaveni, A.; Govindarajan, S.; Butcher, R.J.; Jasinski, J.P.; Zeller, M. Neutral and ion-pair silver(I) complexes of Schiff bases derived from methyl and ethyl carbazates with glyoxylic acid: Synthesis, structure, thermal behavior and cytotoxic activity. Inorg. Chim. Acta 2019, 497, 119072. [Google Scholar] [CrossRef]
- Orhan, E.; Garci, A.; Riedel, T.; Soudani, M.; Dyson, P.J.; Therrien, B. Cytotoxic double arene ruthenium metalla-cycles that overcome cisplatin resistance. J. Organomet. Chem. 2016, 803, 39–44. [Google Scholar] [CrossRef]
- Garci, A.; Dobrov, A.A.; Riedel, T.; Orhan, E.; Dyson, P.J.; Arion, V.B.; Therrien, B. Strategy to Optimize the Biological Activity of Arene Ruthenium Metalla-Assemblies. Organometallics 2014, 33, 3813–3822. [Google Scholar] [CrossRef]
- Köthe, C.; Metzinger, R.; Limberg, C. Reduction and Hydrogenation of a Diazene by a (β-Diketiminato)nickel Hydrazide. Eur. J. Inorg. Chem. 2013, 2013, 3937–3942. [Google Scholar] [CrossRef]
- Köthe, C.; Metzinger, R.; Herwig, C.; Limberg, C. Reductive Deprotonation and Dehydrogenation of Phenylhydrazine at a Nickel Center To Give a Nickel Diazenido Complex. Inorg. Chem. 2012, 51, 9740–9747. [Google Scholar] [CrossRef]
- Kaim, W. The Shrinking World of Innocent Ligands: Conventional and Non-Conventional Redox-Active Ligands. Eur. J. Inorg. Chem. 2012, 2012, 343–348. [Google Scholar] [CrossRef]
- Roy, S.; Sarkar, B.; Imrich, H.-G.; Fiedler, J.; Zális, S.; Jimenez-Aparicio, R.; Urbanos, F.A.; Mobin, S.M.; Lahiri, G.K.; Kaim, W. Charged, but Found “Not Guilty”: Innocence of the Suspect Bridging Ligands [RO(O)CNNC(O)OR]2– = L2− in [(acac)2Ru(μ-L)Ru(acac)2]n, n = +,0,−,2−. Inorg. Chem. 2012, 51, 9273–9281. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W. Manifestations of Noninnocent Ligand Behavior. Inorg. Chem. 2011, 50, 9752–9765. [Google Scholar] [CrossRef] [PubMed]
- Azhakar, R.; Sarish, S.P.; Tavcar, G.; Roesky, H.W.; Hey, J.; Stalke, D.; Koley, D. Formation of Silicon Centered Spirocyclic Compounds: Reaction of N-Heterocyclic Stable Silylene with Benzoylpyridine, Diisopropyl Azodicarboxylate, and 1,2-Diphenylhydrazine. Inorg. Chem. 2011, 50, 3028–3036. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, K.; Sugiyama, M.; Miyashita, Y.; Okamoto, K.-i. Structure and chemical properties of a copper(II) hydrazido complex: {Cu(HB(3,5-iPr2pz)3)}2(μ-NCOOEt)2. Inorg. Chem. Commun. 2009, 12, 246–248. [Google Scholar] [CrossRef]
- Roy, S.; Sieger, M.; Singh, P.; Niemeyer, M.; Fiedler, J.; Duboc, C.; Kaim, W. A radical-bridged bis(ferrocenylcopper(I)) complex: Structural identity, multifrequency EPR, and spectroelectrochemistry. Inorg. Chim. Acta 2008, 361, 1699–1704. [Google Scholar] [CrossRef]
- Roy, S.; Sieger, M.; Sarkar, B.; Schwederski, B.; Lissner, F.; Schleid, T.; Fiedler, J.; Kaim, W. Establishing the Chelating α-Azocarbonyl Function in π-Acceptor Ligands. Angew. Chem. Int. Ed. 2008, 47, 6192–6194. [Google Scholar] [CrossRef]
- Xun, S.; LeClair, G.; Zhang, J.; Chen, X.; Gao, J.P.; Wang, Z.Y. Tuning the Electrical and Optical Properties of Dinuclear Ruthenium Complexes for Near Infrared Optical Sensing. Org. Lett. 2006, 8, 1697–1700. [Google Scholar] [CrossRef]
- Kaim, W.; Klein, A.; Glöckle, M. Exploration of Mixed-Valence Chemistry: Inventing New Analogues of the Creutz-Taube Ion. Acc. Chem. Res. 2000, 33, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.; Cronin, L.; Duckett, S.B.; Hupfield, P.; Perutz, R.N. Synthesis, structure and reactivity of N,O-metallacyclic (dicarbonyldiazene) platinum complexes. New J. Chem. 1998, 22, 511–516. [Google Scholar] [CrossRef]
- Körner, V.; Vogel, S.; Huttner, G.; Zsolnai, L.; Walter, O. Reaktionen des Tripod-Co-Templats CH3C(CH2PPh2)3Co(II) mit funktionalisierten Hydrazinen. Chem. Ber. 1996, 129, 1107–1113. [Google Scholar] [CrossRef]
- Kasack, V.; Kaim, W.; Binder, H.; Jordanov, J.; Roth, E. When Is an Odd-Electron Dinuclear Complex a Mixed-Valent Species? Tuning of Ligand-to-Metal Spin Shifts in Diruthenium(III,II) Complexes of Noninnocent Bridging Ligands OC(R)NNC(R)O. Inorg. Chem. 1995, 34, 1924–1933. [Google Scholar] [CrossRef]
- Dürr, S.; Höhlein, U.; Schobert, R. Neue Synthesen und Reaktionen ungesättigter Heterotitanacyclen. J. Organomet. Chem. 1993, 458, 89–96. [Google Scholar] [CrossRef]
- Moscherosch, M.; Field, J.S.; Kaim, W.; Kohlmann, S.; Krejcik, M. ‘Inverse cryptate’ structure of an exceptionally stable dicopper(I) semiquinonoid intermediate. J. Chem. Soc. Dalton Trans. 1993, 211–216. [Google Scholar] [CrossRef]
- Kaim, W.; Moscherosch, M. Detection of electron paramagnetic resonance signals from three different isotope combinations 63Cu/63Cu, 63Cu/65Cu and 65Cu/65Cu in stable dicopper(I) radical complexes. J. Chem. Soc. Faraday Trans. 1991, 87, 3185–3187. [Google Scholar] [CrossRef]
- Kaim, W.; Kasack, V.; Binder, H.; Roth, E.; Jordanov, J. A Stable Bis(chelate) Analogue of the Creutz-Taube Ion. Angew. Chem. Int. Ed. 1988, 27, 1174–1176. [Google Scholar] [CrossRef]
- Curtis, M.D.; D’Errico, J.J.; Butler, W.M. Metal-metal multiple bonds. Part 22. Addition reactions of organic azides and diethyl azodicarboxylate with Cp2Mo2(CO)4. Molecular structures of Cp2Mo2(CO)2(NAr)(μ-NNN(Ar)CO) (Ar = p-tert-BuC6H4) and Cp’Mo(CO)2.2(Ar-EtO2CN2CO2Et). Organometallics 1987, 6, 2151–2157. [Google Scholar] [CrossRef]
- Avar, G.; Rüsseler, W.; Kisch, H. Transition Metal Complexes of Diazenes, XXIV 1. Complexation and N=N-Cleavage of Diazenes by Dicarbonylbis(cyclopentadienyl)titanium. Z. Naturforsch. B 1987, 42, 1441–1446. [Google Scholar] [CrossRef]
- Einstein, F.W.B.; Nussbaum, S.; Sutton, D.; Willis, A.C. Dimethyl azodicarboxylate derivatives of triosmium carbonyl clusters. Synthesis of Os3(CO)n(CH3OCONNCOOCH3) (n = 10-12) and characterization by spectroscopy and X-ray analysis. Organometallics 1984, 3, 568–574. [Google Scholar] [CrossRef]
- Einstein, F.W.B.; Nussbaum, S.; Sutton, D.; Willis, A.C. Syntheses and x-ray structure analyses of Os3(CO)n(MeO2CNNCO2Me) (n = 10-12). A triangular Os3(CO)12X2 structure. Organometallics 1983, 2, 1259–1261. [Google Scholar] [CrossRef]
- Creber, K.A.M.; Ho, T.-I.; Depew, M.C.; Weir, D.; Wan, J.K.S. Some physical and chemical aspects of spin trapping of organometallic radicals by quinones: Optically active radical complexes. Can. J. Chem. 1982, 60, 1504–1511. [Google Scholar] [CrossRef]
- Chen, K.S.; Wan, J.K.S. Electron spin resonance studies of alkali radical ion pairs and triple ions of 1,2- and 1,4-dicarbonyl compounds. J. Am. Chem. Soc. 1978, 100, 6051–6056. [Google Scholar] [CrossRef]
- Henke, W.C.; Kerr, T.A.; Sheridan, T.R.; Henling, L.M.; Takase, M.K.; Day, V.W.; Gray, H.B.; Blakemore, J.D. Synthesis, structural studies, and redox chemistry of bimetallic Mn(CO)3 and Re(CO)3 complexes. Dalton Trans. 2021, 50, 2746–2756. [Google Scholar] [CrossRef]
- Frantz, S.; Sieger, M.; Hartenbach, I.; Lissner, F.; Schleid, T.; Fiedler, J.; Duboc, C.; Kaim, W. Structure, electrochemistry, spectroscopy, and magnetic resonance, including high-field EPR, of {(μ-abpy)Re(CO)3X.2}o/•−, where abpy = 2,2′-azobispyridine and X = F, Cl, Br, I.J. Organomet. Chem. 2009, 694, 1122–1133. [Google Scholar] [CrossRef]
- Alsindi, W.Z.; Easun, T.L.; Sun, X.Z.; Ronayne, K.L.; Towrie, M.; Herrera, J.-M.; George, M.W.; Ward, M.D. Probing the Excited States of d6 Metal Complexes Containing the 2,2‘-Bipyrimidine Ligand Using Time-Resolved Infrared Spectroscopy. 1. Mononuclear and Homodinuclear Systems. Inorg. Chem. 2007, 46, 3696–3704. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, B.; Frantz, S.; Kaim, W.; Duboc, C. High-frequency EPR study of reduced diruthenium and dirhenium polypyridine complexes based on the 1,2,4,5-tetrazine radical bridge. Dalton Trans. 2004, 3727–3731. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Fiedler, J.; Hartenbach, I.; Schleid, T.; Kaim, W. A complete series of tricarbonylhalidorhenium(I) complexes (abpy)Re(CO)3(Hal), Hal = F, Cl, Br, I; abpy = 2,2′-azobispyridine: Structures, spectroelectrochemistry and EPR of reduced forms. J. Organomet. Chem. 2004, 689, 3031–3039. [Google Scholar] [CrossRef]
- Frantz, S.; Hartmann, H.; Doslik, N.; Wanner, M.; Kaim, W.; Kümmerer, H.-J.; Denninger, G.; Barra, A.-L.; Duboc-Toia, C.; Fiedler, J.; et al. Multifrequency EPR Study and Density Functional g-Tensor Calculations of Persistent Organorhenium Radical Complexes. J. Am. Chem. Soc. 2002, 124, 10563–10571. [Google Scholar] [CrossRef]
- Hartmann, H.; Scheiring, T.; Fiedler, J.; Kaim, W. Structures and spectroelectrochemistry (UV–vis, IR, EPR) of complexes (OC)3ClRe.n(abpy), n = 1, 2; abpy = 2,2′-azobispyridine. J. Organomet. Chem. 2000, 604, 267–272. [Google Scholar] [CrossRef]
- Klein, A.; Kasack, V.; Reinhardt, R.; Sixt, T.; Scheiring, T.; Zalis, S.; Fiedler, J.; Kaim, W. 2,5-Bis(1-phenyliminoethyl)pyrazine (bpip): A conjugated metal–metal bridging acceptor ligand and its homodinuclear complexes with low-valent metal centres. J. Chem. Soc. Dalton Trans. 1999, 575–582. [Google Scholar] [CrossRef]
- Braterman, P.S.; Song, J.-I.; Kohlmann, S.; Vogler, C.; Kaim, W. Spectroelectrochemistry of aromatic ligands and their derivatives: III. Binuclear transition metal complexes of CuI, Mo0, and ReI with 2,2′-bipyrimidine. J. Organomet. Chem. 1991, 411, 207–213. [Google Scholar] [CrossRef]
- Matheis, W.; Kaim, W. Electronic structure of heterodinuclear complexes (bpy)2RuII-(μ-bpym)MLn; bpy = 2,2′-bipyridine; bpym = 2,2′-bipyrimidine; MLn = +Cu(PPh3)2, Mo(CO)4, Re(CO)3Cl. Inorg. Chim. Acta 1991, 181, 15–21. [Google Scholar] [CrossRef]
- Kaim, W.; Kohlmann, S. The nature of reduced and excited states of π-electron-deficient complexes between tricarbonylhalorhenium and diimine ligands. Inorg. Chem. 1990, 29, 2909–2914. [Google Scholar] [CrossRef]
- Gaviraghi, G.; Pinza, M.; Pifferi, G. A Mild and Convenient Synthesis of Ethyl 2-Phenyl-and 2-(3-Pyridazine)-diazenecarboxylates (Azocarboxylates). Synthesis 1981, 1981, 608–610. [Google Scholar] [CrossRef]
- Zeng, H.; Ju, J.; Hua, R. ReCl(CO)5-catalyzed cyclocondensation of phenols with 2-methyl-3-butyn-2-ol to afford 2,2-dimethyl-2H-chromenes. Tetrahedron Lett. 2011, 52, 3926–3928. [Google Scholar] [CrossRef]
- Bichler, P.; Sun, A.D.; Patrick, B.O.; Love, J.A. Design, synthesis and coordination chemistry of sidearm substituted bisoxazoline ligands. Inorg. Chim. Acta 2009, 362, 4546–4552. [Google Scholar] [CrossRef]
- Orgel, L.E. Carbonyl Stretching Bands of Tetracarbonyl Halide Dimers of Manganese, Technetium, and Rhenium. Inorg. Chem. 1964, 3, 303. [Google Scholar] [CrossRef]
- El-Sayed, M.A.; Kaesz, H.D. Infrared Spectra and Structure of the Tetracarbonyl Halide Dimers of Manganese, Technetium, and Rhenium. Inorg. Chem. 1963, 2, 158–162. [Google Scholar] [CrossRef]
- Klein, A.; Vogler, C.; Kaim, W. The δ in 18+δ Electron Complexes: Importance of the Metal/Ligand Interface for the Substitutional Reactivity of “Re(0)” Complexes (α-diimine-)ReI(CO)3(X). Organometallics 1996, 15, 236–244. [Google Scholar] [CrossRef]
- Brown, T.L.; Darensbourg, D.J. Intensities of CO stretching modes in the infrared spectra of adsorbed CO and metal carbonyls. Inorg. Chem. 1967, 6, 971–977. [Google Scholar] [CrossRef]
- Gerschel, P.; Cordes, A.L.; Bimmermann, S.; Siegmund, D.; Metzler-Nolte, N.; Apfel, U.-P. Investigation of Cyclam Based Re-Complexes as Potential Electrocatalysts for the CO2 Reduction Reaction. Z. Anorg. Allgem. Chem. 2021, 647, 968–977. [Google Scholar] [CrossRef]
- Maser, L.; Vogt, M.; Langer, R. Facial vs. Meridional Coordination Modes in ReI Tricarbonyl Complexes with a Carbodiphosphorane-based Tridentate Ligand. Z. Anorg. Allgem. Chem. 2021, 647, 1518–1523. [Google Scholar] [CrossRef]
- Suntrup, L.; Klenk, S.; Klein, J.; Sobottka, S.; Sarkar, B. Gauging Donor/Acceptor Properties and Redox Stability of Chelating Click-Derived Triazoles and Triazolylidenes: A Case Study with Rhenium(I) Complexes. Inorg. Chem. 2017, 56, 5771–5783. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Al-Balushi, R.; Al-Busaidi, I.J.; Al-Rasbi, N.K.; Al-Bahri, S.; Al-Suti, M.K.; Khan, M.S.; Abou-Zied, O.K.; Skelton, J.M.; Raithby, P.R. Two Is Better than One? Investigating the Effect of Incorporating Re(CO)3Cl Side Chains into Pt(II) Diynes and Polyynes. Inorg. Chem. 2021, 60, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, B.; Kaim, W.; Schleid, T.; Hartenbach, I.; Fiedler, J. 3,6-Bis(2-pyrazinyl)-1,2,4,5-tetrazine: A New Multifunctional Ligand and its syn,fac-configurated Bis(tricarbonylchlororhenium) Complex. Z. Anorg. Allgem. Chem. 2003, 629, 1353–1357. [Google Scholar] [CrossRef]
- Scheiring, T.; Klein, A.; Kaim, W. EPR study of paramagnetic rhenium(I) complexes (bpy•−)Re(CO)3X relevant to the mechanism of electrocatalytic CO2 reduction. J. Chem. Soc. Perkin Trans. 1997, 2569–2572. [Google Scholar] [CrossRef]
- Kaim, W.; Scheiring, T.; Weber, M.; Fiedler, J. The Conjugative Bridging of Organometallic Reaction Centers in Heterodinuclear Complexes (OC)3ClRe(μ-L)MCl(C5Me5)., M = Rh or Ir − Spectroscopic Consequences of Reductive Activation. Z. Anorg. Allgem. Chem. 2004, 630, 1883–1893. [Google Scholar] [CrossRef]
- Berger, S.; Klein, A.; Kaim, W.; Fiedler, J. Variable Reduction Sequences for Axial (L) and Chelate Ligands (N∧N) in Rhenium(I) Complexes (N∧N)Re(CO)3(L)n. Inorg. Chem. 1998, 37, 5664–5671. [Google Scholar] [CrossRef]
- Matheis, W.; Kaim, W. Homo- and Heterodinuclear Complexes of the D2h-symmetric Bis(chelate) Ligand 2,2’-Bipyrimidine with Electron-Rich Metal Fragments Mo(CO)4, Re(CO)3Cl, Cu(PPh3)2+, and Ru(bpy)22+. Z. Anorg. Allgem. Chem. 1991, 593, 147–159. [Google Scholar] [CrossRef]
- Herrmann, W.A. On a Simple Synthesis of the Halfsandwich Rhenium Complexes (η-C,H,X)Re(CO)3 X = CI, Br, I. Chem. Ber. 1978, 111, 2458–2460. [Google Scholar] [CrossRef]
- Schmidt, S.P.; Trogler, W.C.; Basolo, F. Mechanism of halogenation of dimanganese, manganese-rhenium, and dirhenium decacarbonyls. J. Am. Chem. Soc. 1984, 106, 1308–1313. [Google Scholar] [CrossRef]
- Schmidt, S.P.; Trogler, W.C.; Basolo, F.; Urbancic, M.A.; Shapley, J.R. Pentacarbonylrhenium Halides. In Inorganic Syntheses; John Wiley&Sons: Hoboken, NY, USA, 1985; pp. 41–46. [Google Scholar] [CrossRef]
- Kaim, W.; Fiedler, J. Spectroelectrochemistry: The best of two worlds. Chem. Soc. Rev. 2009, 38, 3373–3382. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W.; Klein, A. Spectroelectrochemistry; RSC Publishing: Cambridge, UK, 2008; ISBN 978-0-85404-550-1. [Google Scholar]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P.; Yue, W. Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation. Phys. Rev. B 1986, 33, 8800–8802. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 1993, 5, 799–805. [Google Scholar] [CrossRef]
- Petrenko, T.; Kossmann, S.; Neese, F. Efficient time-dependent density functional theory approximations for hybrid density functionals: Analytical gradients and parallelization. J. Chem. Phys. 2011, 134, 054116. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Stavoverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 8 November 2020).
- OriginLab Corporation. OriginPro, Version 2021b; OriginLab Corporation: Northampton, MA, USA, 2021. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bruker AXS, Inc. APEX4—Software Suite for Crystallographic Programs; Bruker AXS, Inc.: Madison, WI, USA, 2021. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A: Found. Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT – Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crytsallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Exp. | Calc. (THF) | |||||
|---|---|---|---|---|---|---|---|
| νCO | (cm−1) | Solvent | |||||
| Band | I | II | III | I | II | III | |
| [Re(CO)3Cl(adcpip)] | 2040 | 1960 | 1920 | DCE | 2007 b | 1927 b | 1894 b |
| [Re(CO)3(PPh3)(adcpip)]Cl | 2010 | 1910 | 1876 | DCE | |||
| [Re(CO)3Cl(pacOEt)] | 2022 | 1962 | 1928 | DCE | |||
| syn-[{Re(CO)3Cl}2(µ-adcpip)] | 2015 | 1913 | c,d | DCE | 1978 b | 1932, 1929 b | 1903, 1895 b |
| anti-[{Re(CO)3Cl}2(µ-adcpip)] | 1981 b | 1938, 1927 b | 1908, 1895 b | ||||
| [{Re(CO)3Cl}2(µ-adcOEt)] | 2019 | 1916 | DCE | ||||
| [{Re(CO)3Cl}2(µ-adcOiPr)] | 2021 | 1919 | DCE | ||||
| [Re(CO)3Cl(bpy)] e | 2024 | 1917 | 1900 | MeCN | |||
| [Re(CO)3Cl(bpip)] f | 2027 | 1934 | 1905 | THF | |||
| [{Re(CO)3Cl}2(µ-apy)] g | 2005 | 1952 | 1938 | THF | |||
| [{Re(CO)3Cl}2(µ-bptz)] h | 2010 | 1950 | 1921 | Acetone | |||
| [{Re(CO)3Cl}2(µ-bpip)] f | 2020 | 1940 | 1910 | THF | |||
| [{Re(CO)3Cl}2(µ-bpym)] i | 2025 | 1938 | 1900 | Acetone | |||
| Bond Lengths (Å) | Angles (°) | ||||
|---|---|---|---|---|---|
| Exp. | Calc. | Exp. | Calc. | ||
| Re1–Cl1 | 2.457(1) | 2.473 | Cl1–Re1–C1 | 174.5(1) | 177.4 |
| Re1–C1 | 1.924(2) | 1.933 | Cl1–Re1–C2 | 95.6(1) | 93.6 |
| Re1–C2 | 1.897(2) | 1.912 | Cl1–Re1–C3 | 87.0(1) | 88.6 |
| Re1–C3 | 1.942(2) | 1.969 | Cl1–Re1–O4 | 81.6(1) | 81.9 |
| Re1–O4 | 2.151(1) | 2.169 | Cl1–Re1–N1 | 89.6(1) | 90.0 |
| Re1–N1 | 2.117(2) | 2.078 | O4-Re1-N1 | 71.6(1) | 72.1 |
| N1–N2 | 1.261(2) | 1.283 | O4–Re1–C1 | 95.2(1) | 96.9 |
| C1C–O4 | 1.257(3) | 1.275 | O4–Re1–C2 | 170.2(1) | 169.4 |
| C2C–O5 | 1.211(3) | 1.227 | O4–Re1–C3 | 100.9(1) | 98.5 |
| N1–C2C | 1.489(2) | 1.484 | C1–Re1–C2 | 88.3(1) | 87.9 |
| N2–C1C | 1.443(2) | 1.414 | C1–Re1–C3 | 89.2(1) | 89.3 |
| C1–O1 | 1.148(3) | 1.164 | C1–Re1–N1 | 93.7(1) | 91.8 |
| C2–O2 | 1.156(3) | 1.167 | C2–Re1–C3 | 88.3(1) | 91.0 |
| C3–O3 | 1.145(3) | 1.157 | C2–Re1–N1 | 99.0(1) | 98.3 |
| C3–Re1–N1 | 172.2(1) | 170.7 | |||
| N2–N1–Re1 | 123.1(1) | 122.7 | |||
| Dihedral angles (°) | |||||
| Re1–N1–N2–C1C | 0.9(2) | 2.9 | N1–N2–C1C–N3 | 176.5(2) | 173.5 |
| C2C–N1–N2–C1C | 172.9(2) | 168.2 | N2–N1–C2C–N4 | 99.8(2) | 102.8 |
| Compound | E1/2 Red1 | Epc Red2 | Epa Ox1 | Ref. |
|---|---|---|---|---|
| [Re(CO)3Cl(adcpip)] | −0.50 | −1.18 | >1.2 | this work |
| [{Re(CO)3Cl}2(µ-adcpip)] | −0.20 | −1.48 | 1.12 | this work |
| syn-[{Re(CO)3Cl}2(µ-adcOEt)] b | −0.05 | −0.94 | 1.17 | this work |
| anti-[{Re(CO)3Cl}2(µ-adcOEt)] b | −0.16 | −0.95 | 1.17 | this work |
| [{Re(CO)3Cl}2(µ-adcOiPr)] | −0.17 | −0.99 | 1.13 | this work |
| [Re(CO)3Cl(pacOEt)] | −0.77 | −1.58 | 1.11 | this work |
| [Re2(µ-Cl)2(CO)8] | −2.31 | −2.68 | 1.32 | this work |
| [Re(CO)3Cl(bpy)] b | −1.74 | −2.20 | [59] | |
| [Re(CO)3Cl(bpip)] c | −1.17 | −1.89 | [50] | |
| [Re(CO)3Cl(apy)] d | −0.78 | −1.54 | [47] | |
| anti-[{Re(CO)3Cl}2(µ-apy)] d | 0.00 | −0.81 | [44] | |
| syn-[{Re(CO)3Cl}2(µ-apy)] d | 0.53 | −0.63 | [44] | |
| [{Re(CO)3Cl}2(µ-bptz)] | −0.16 | −1.49 | [53] | |
| [{Re(CO)3Cl}2(µ-bpip)] | −0.54 | −1.30 | 0.76 | [50] |
| [{Re(CO)3Cl}2(µ-bpym)] d | 0.02. | −0.71 | 0.73 | [53] |
| Compound | λ1 | λ2 | λ3 | Eopt Max | Solvent | Ref. |
|---|---|---|---|---|---|---|
| [Re(CO)3Cl(adcpip)] | 538 | 2.30 | CH2Cl2 | |||
| [Re(CO)3Cl(pacOEt)] | 502 | 2.47 | CH2Cl2 | |||
| [{Re(CO)3Cl}2(µ-adcpip)] | 452 | 510 | 718 | 1.73 | CH2Cl2 | |
| [{Re(CO)3Cl}2(µ-adcOEt)] | 853 | 1.45 | CH2Cl2 | |||
| [{Re(CO)3Cl}2(µ-adcOiPr)] | 690 | 837 | 1.48 | CH2Cl2 | ||
| [{Re(CO)3Cl}2(µ-apy)] | 705 | 784 | 1.58 | CH2Cl2 | [44] | |
| [{Re(CO)3Cl}2(µ-apy)] | 588 | 747 | 1.66 | DMF | [44] | |
| [{Re(CO)3Cl}2(µ-bptz)] | 510 | 697 | 1.78 | acetone | [53] | |
| [{Re(CO)3Cl}2(µ-bptz)] | 550 | 732 | 1.69 | DCE | [53] | |
| [{Re(CO)3Cl}2(µ-bpip)] | 366 | 584 | 2.12 | acetone | ||
| [{Re(CO)3Cl}2(µ-bpip)] | 378 | 636 | 1.95 | DCE | [50] | |
| [{Re(CO)3Cl}2(µ-bpym) | 357 | 469 | 2.64 | acetone | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordan, R.; Niazi, M.; Schäfer, S.; Kaim, W.; Klein, A. Rhenium Tricarbonyl Complexes of Azodicarboxylate Ligands. Molecules 2022, 27, 8159. https://doi.org/10.3390/molecules27238159
Jordan R, Niazi M, Schäfer S, Kaim W, Klein A. Rhenium Tricarbonyl Complexes of Azodicarboxylate Ligands. Molecules. 2022; 27(23):8159. https://doi.org/10.3390/molecules27238159
Chicago/Turabian StyleJordan, Rose, Maryam Niazi, Sascha Schäfer, Wolfgang Kaim, and Axel Klein. 2022. "Rhenium Tricarbonyl Complexes of Azodicarboxylate Ligands" Molecules 27, no. 23: 8159. https://doi.org/10.3390/molecules27238159
APA StyleJordan, R., Niazi, M., Schäfer, S., Kaim, W., & Klein, A. (2022). Rhenium Tricarbonyl Complexes of Azodicarboxylate Ligands. Molecules, 27(23), 8159. https://doi.org/10.3390/molecules27238159