
Interaction between [(η6-p-cym)M(H2O)3]2+ (MII = Ru, Os) or [(η5-Cp*)M(H2O)3]2+ (MIII = Rh, Ir) and Phosphonate Derivatives of Iminodiacetic Acid: A Solution Equilibrium and DFT Study


Abstract
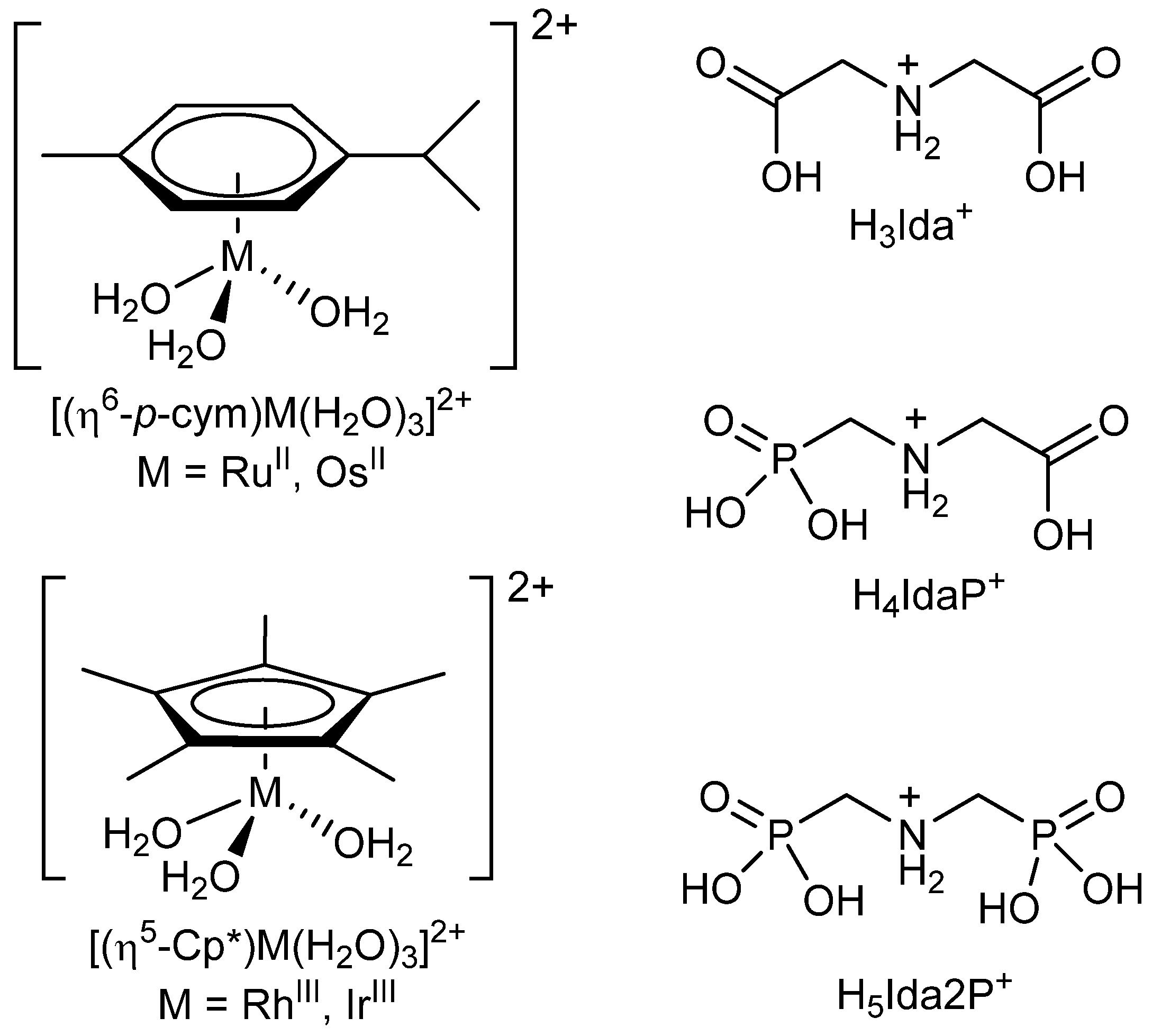
:1. Introduction
2. Results and Discussion
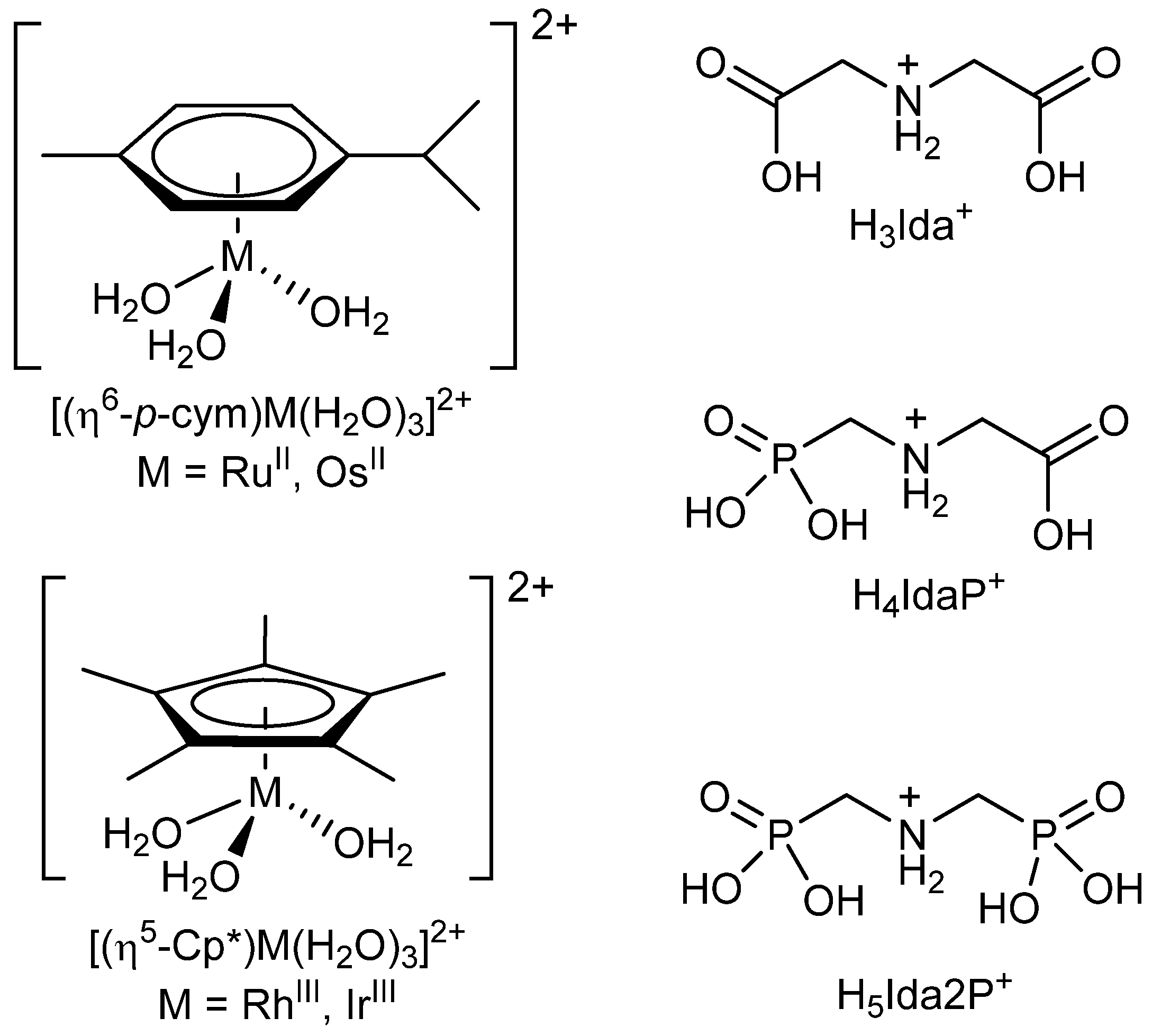
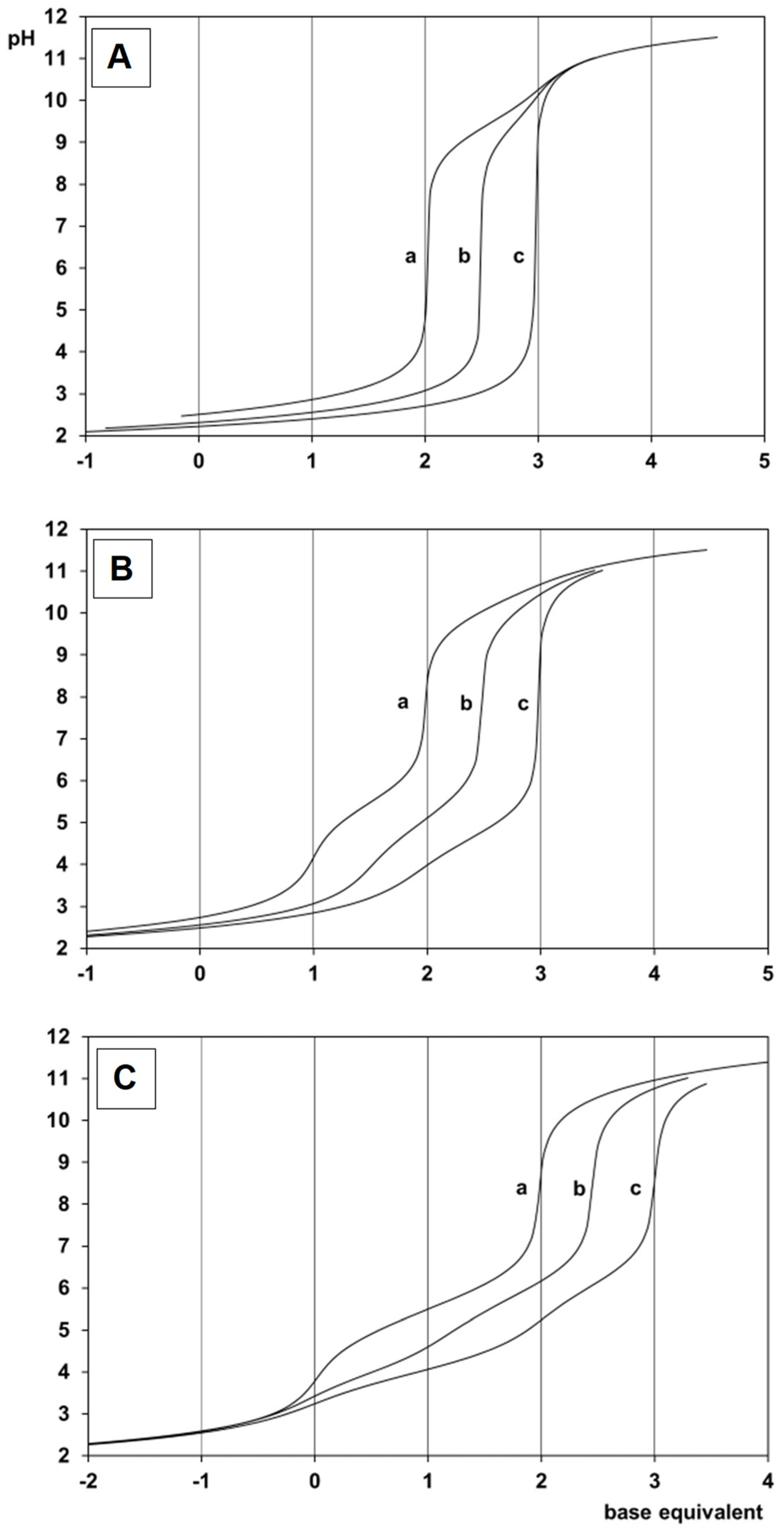
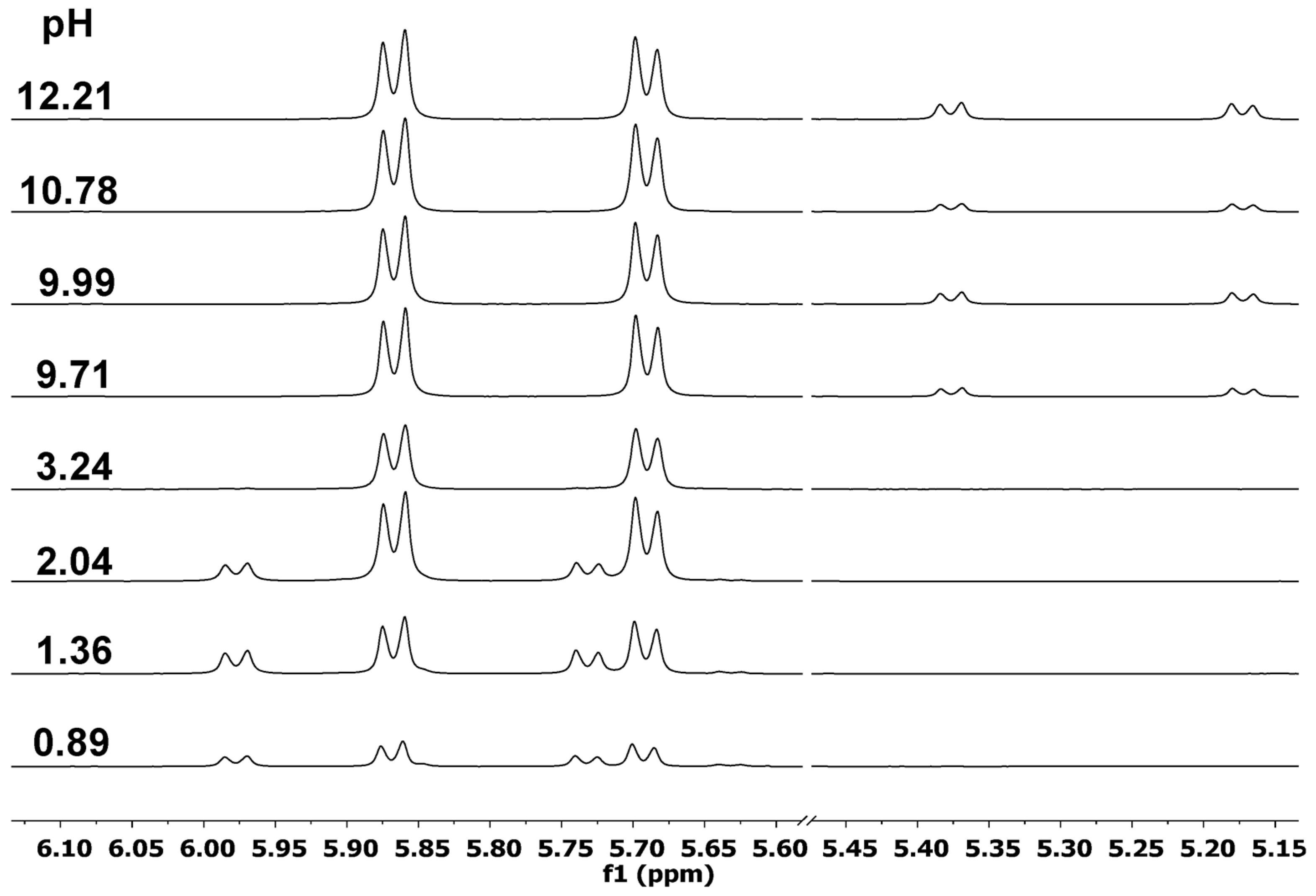
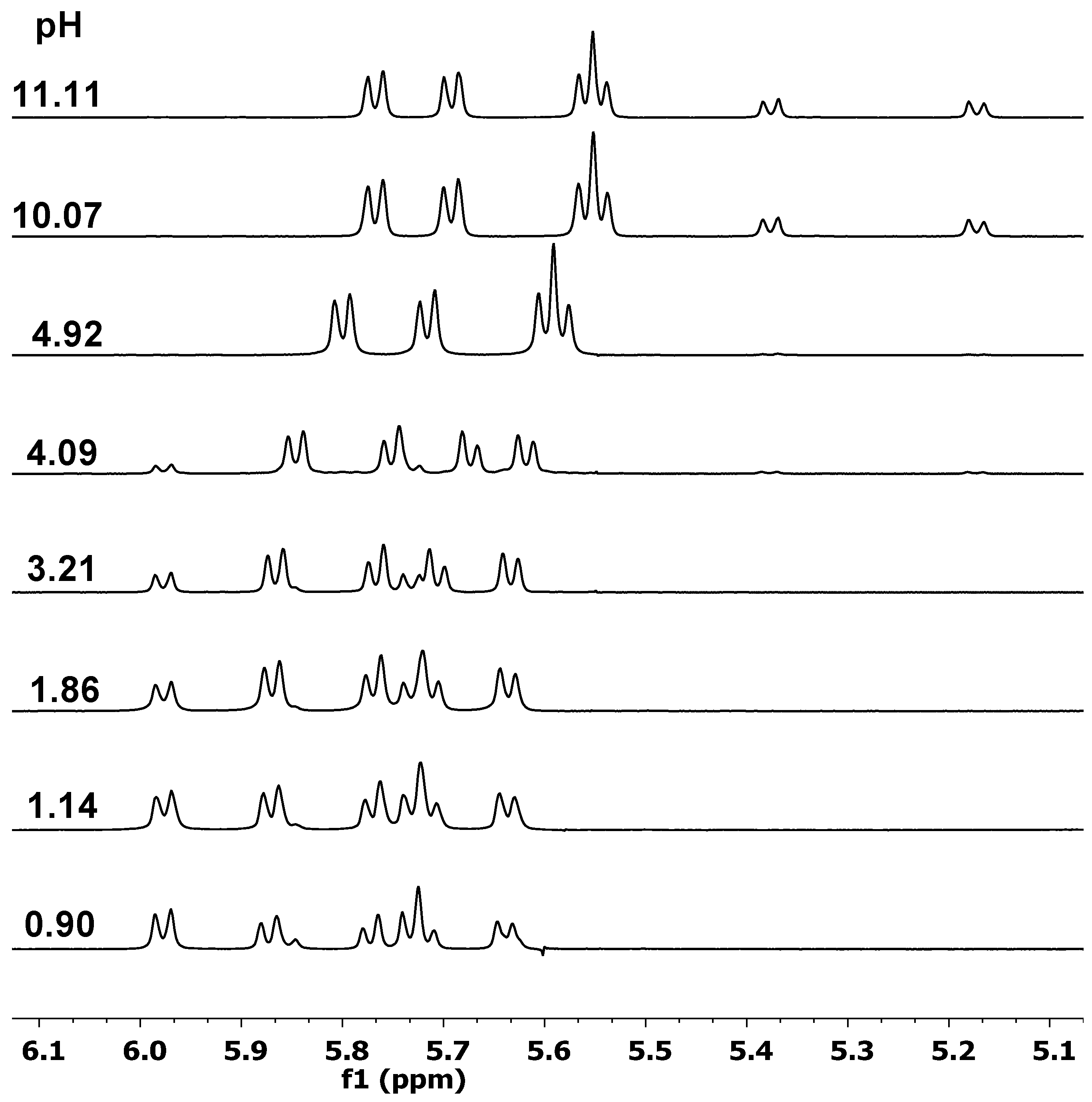
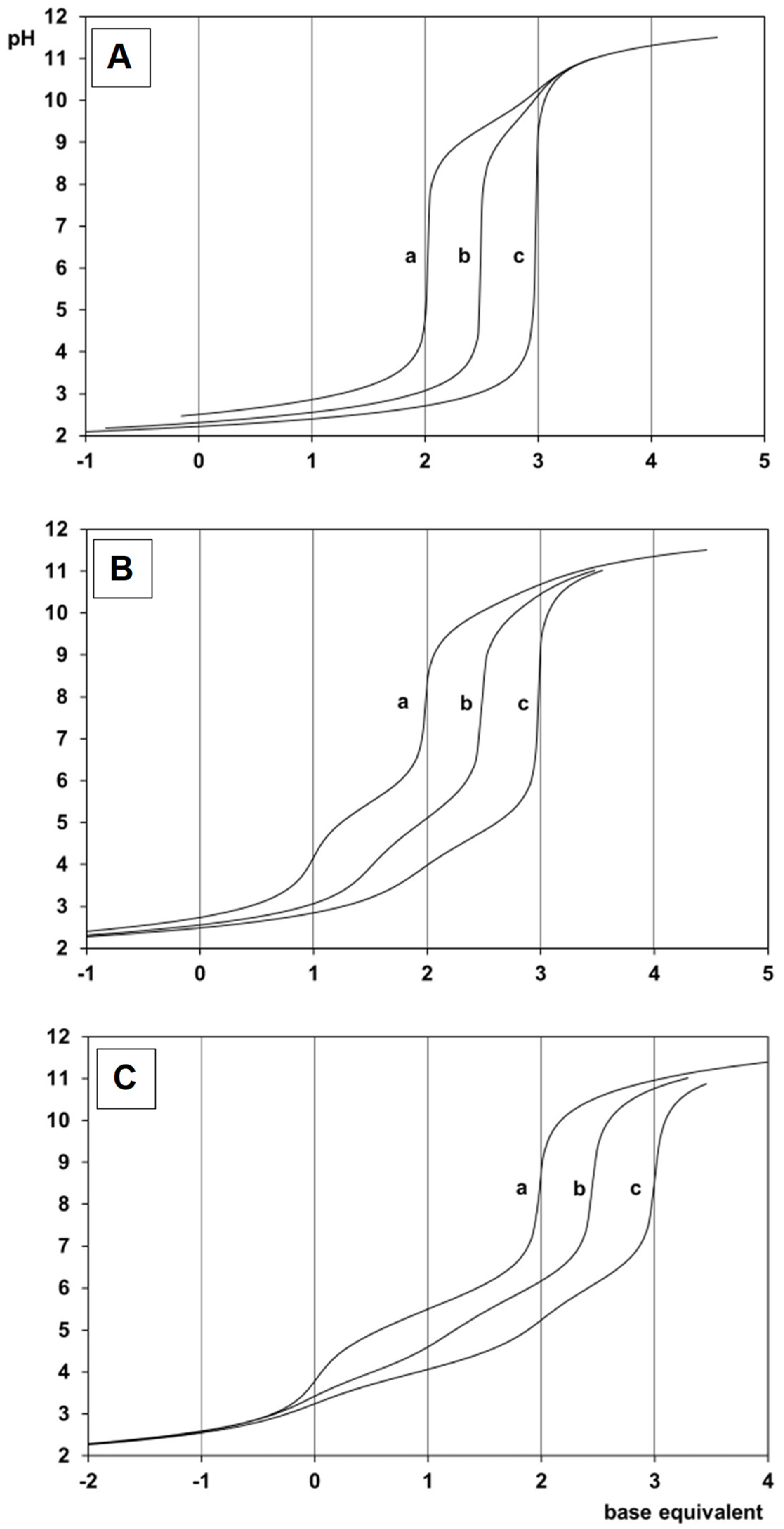
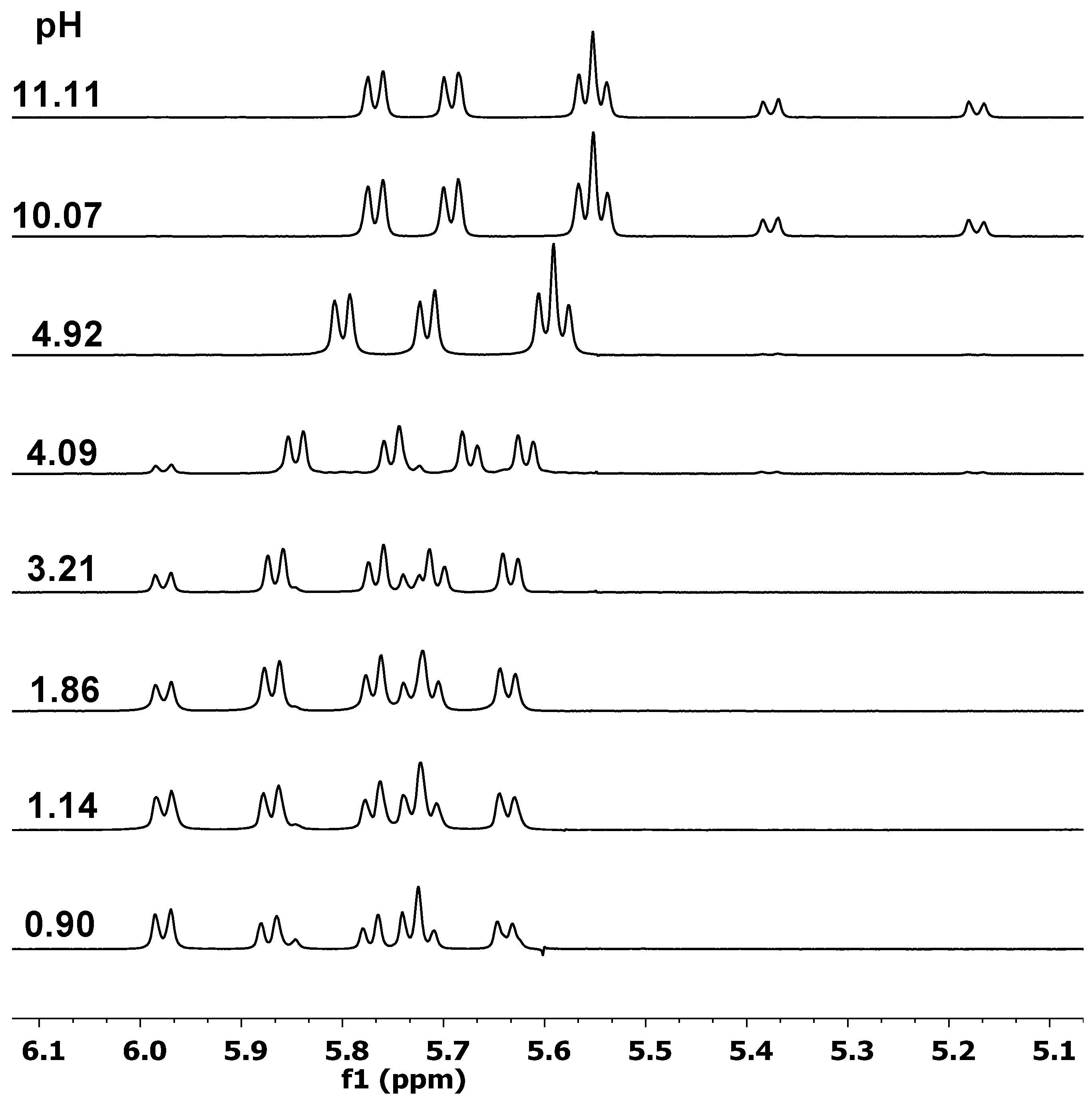
2.1. Proton Dissociation Processes of the Ligands
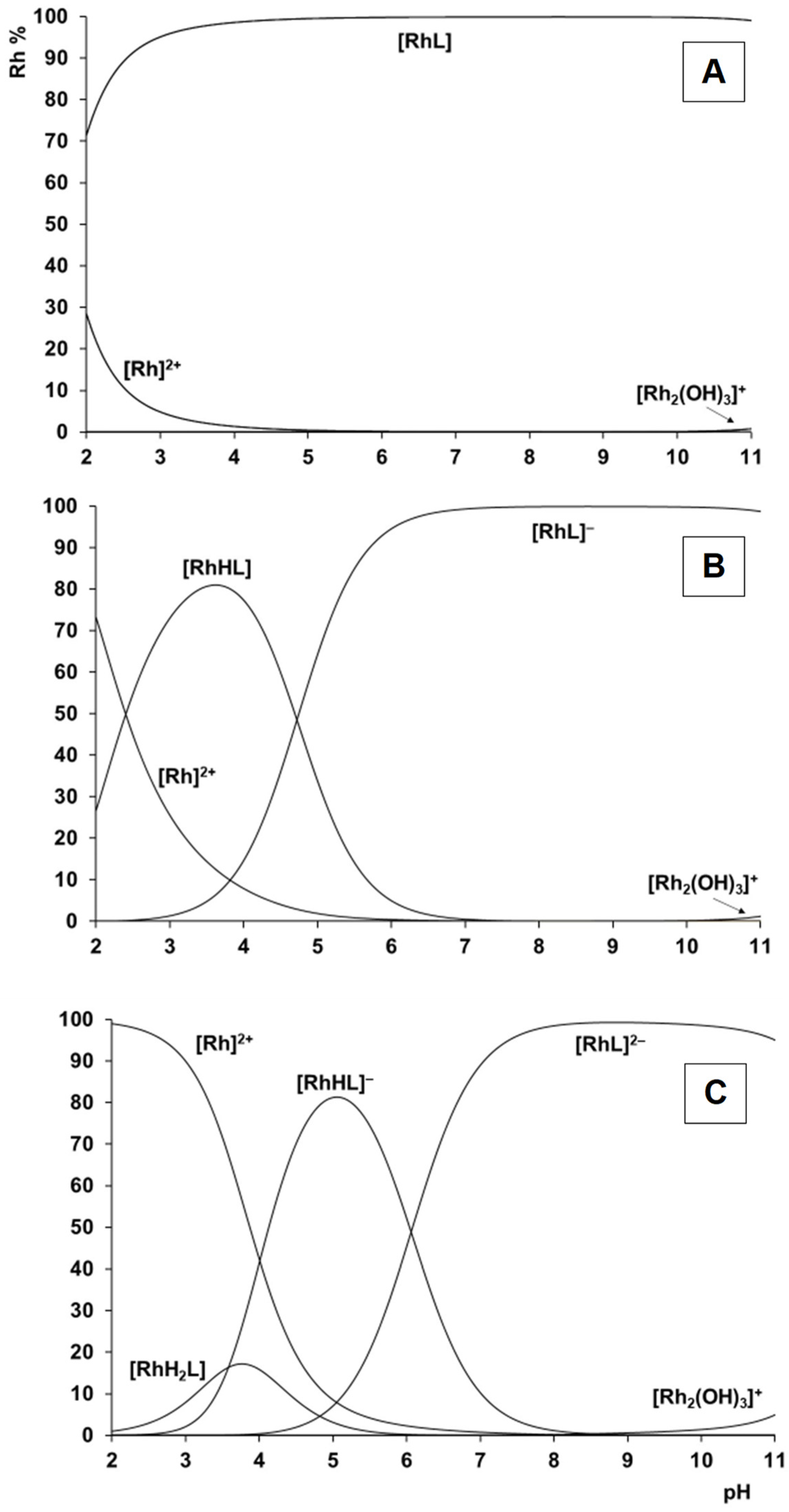
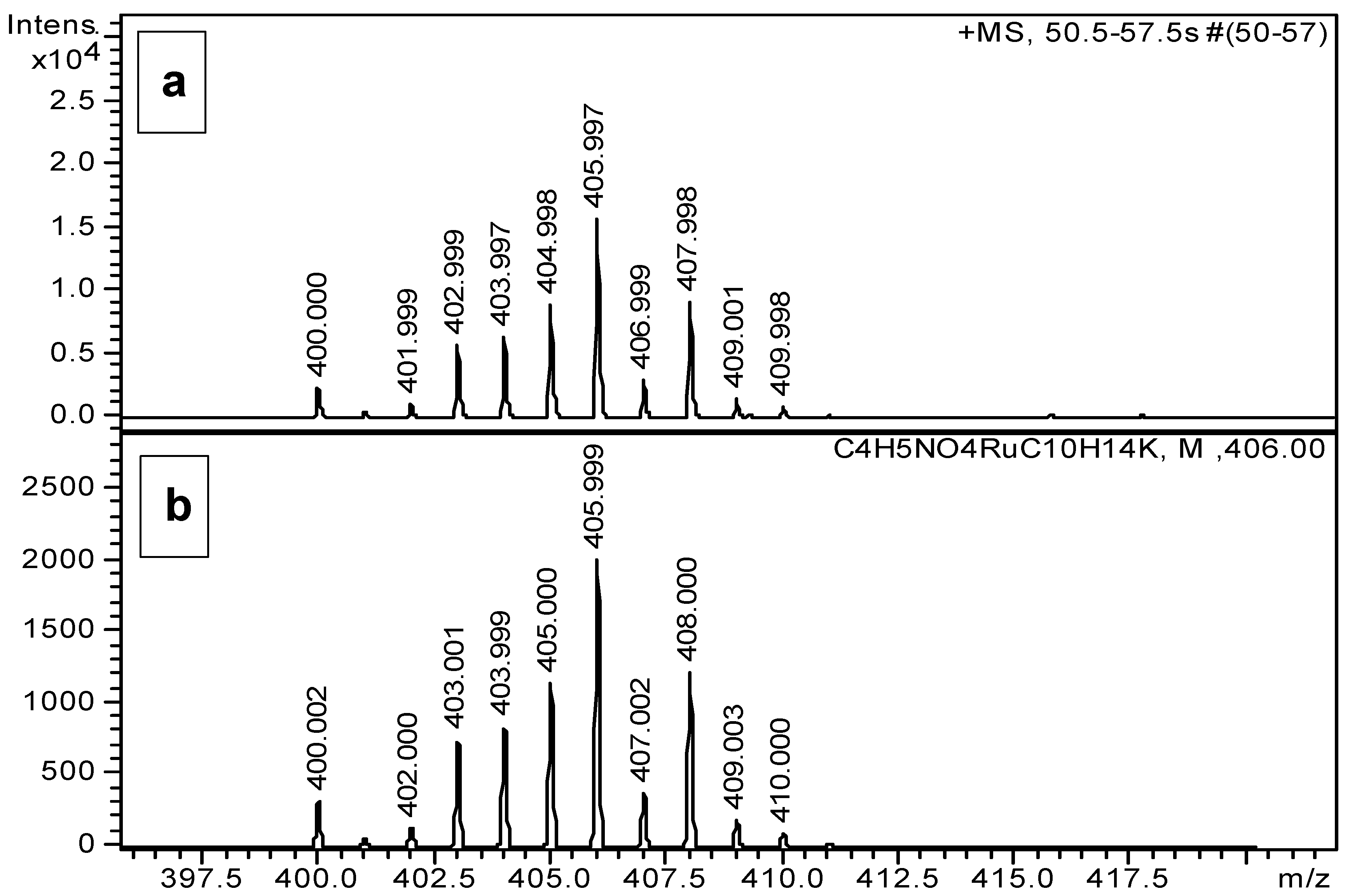
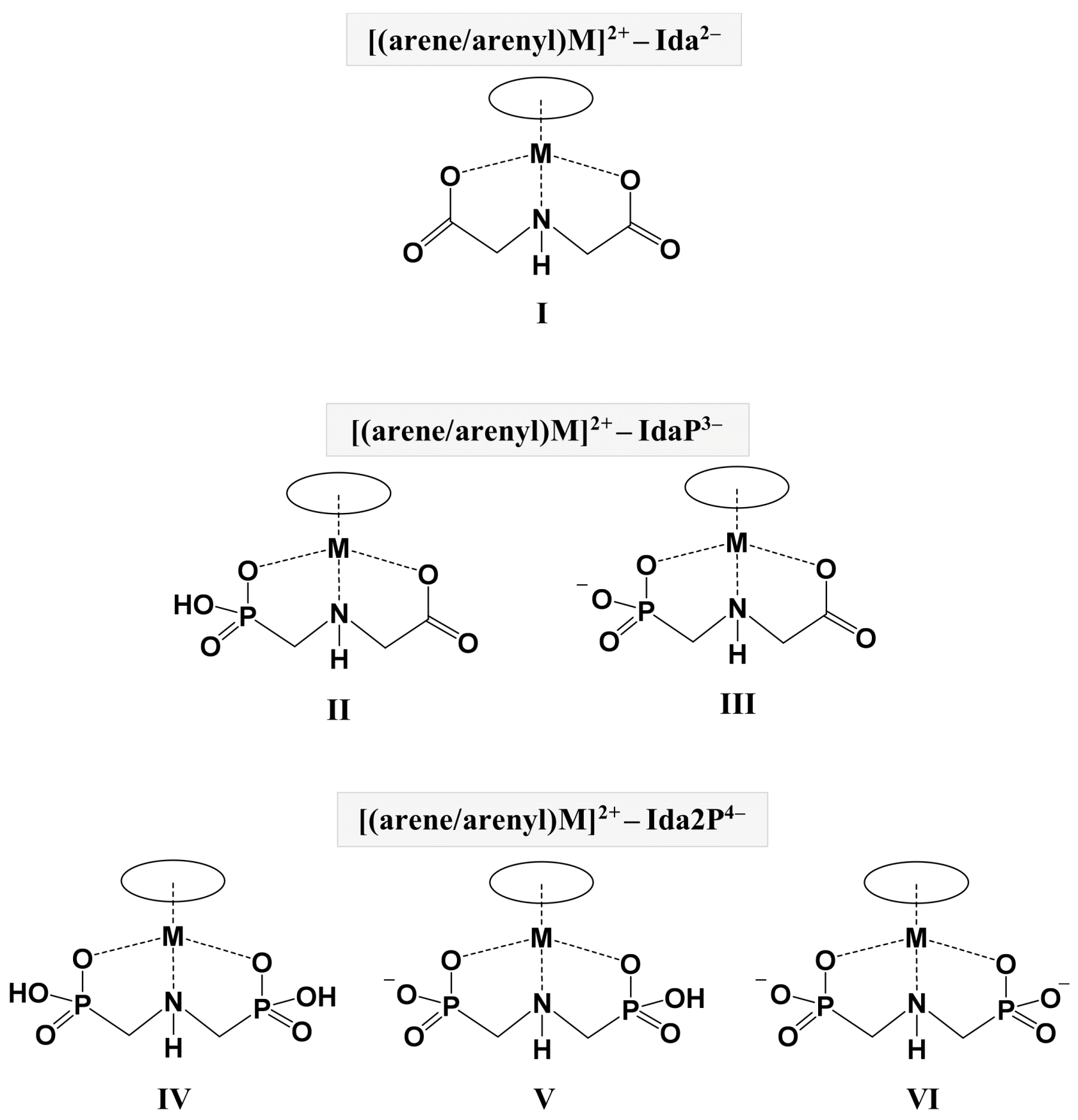
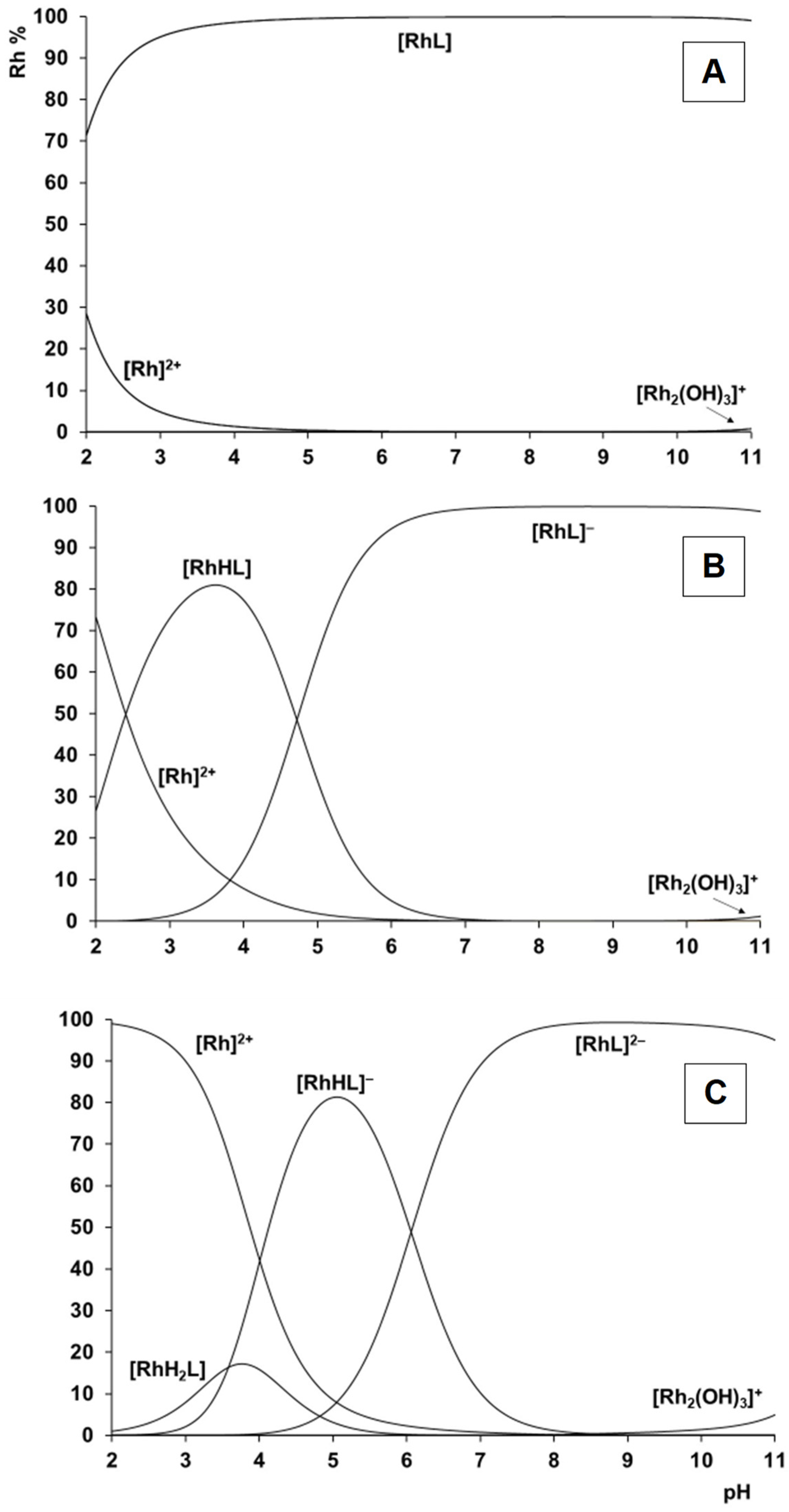
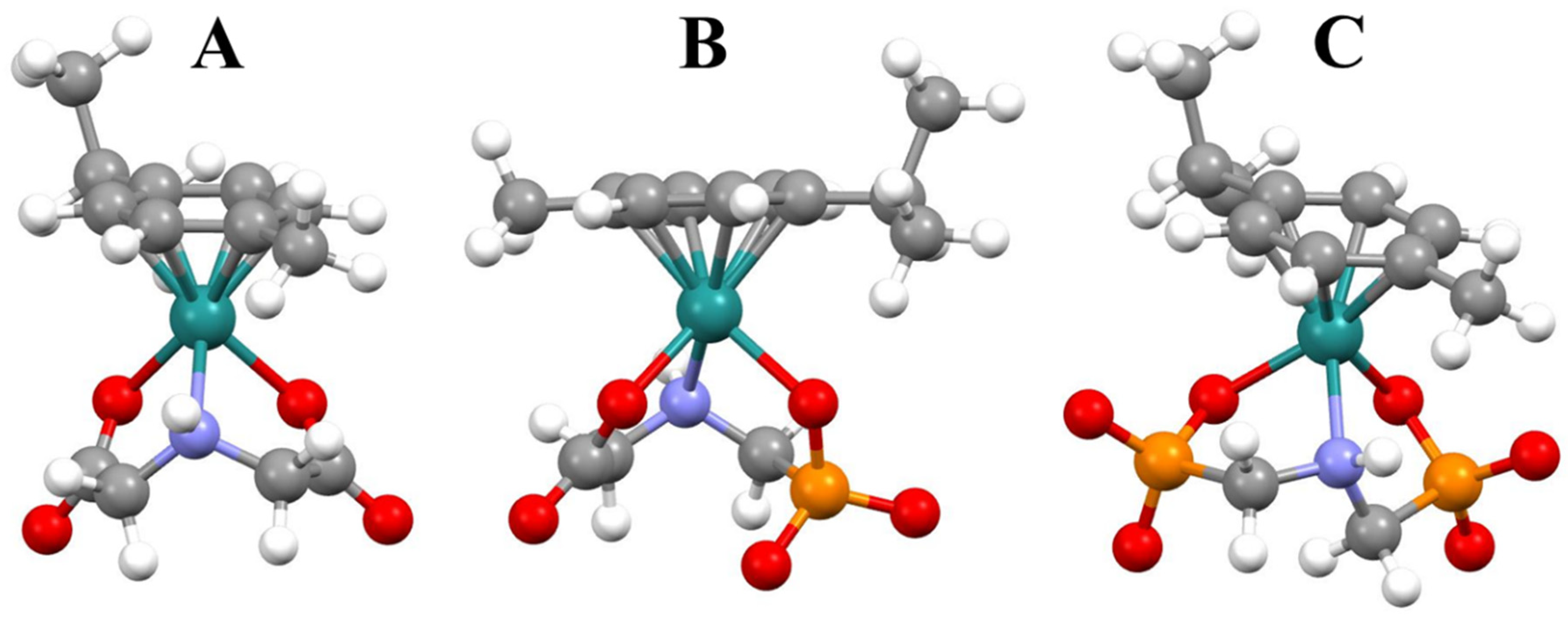
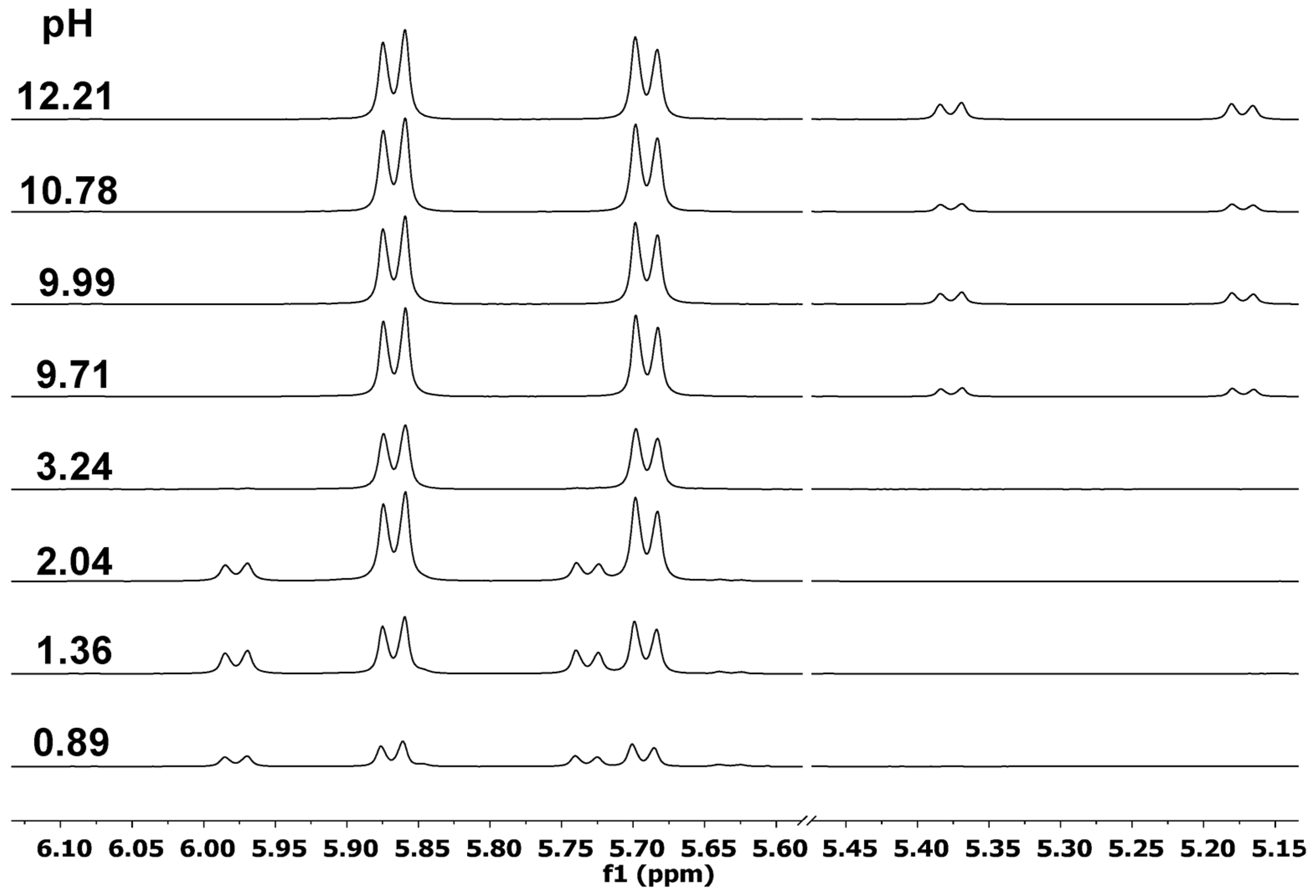
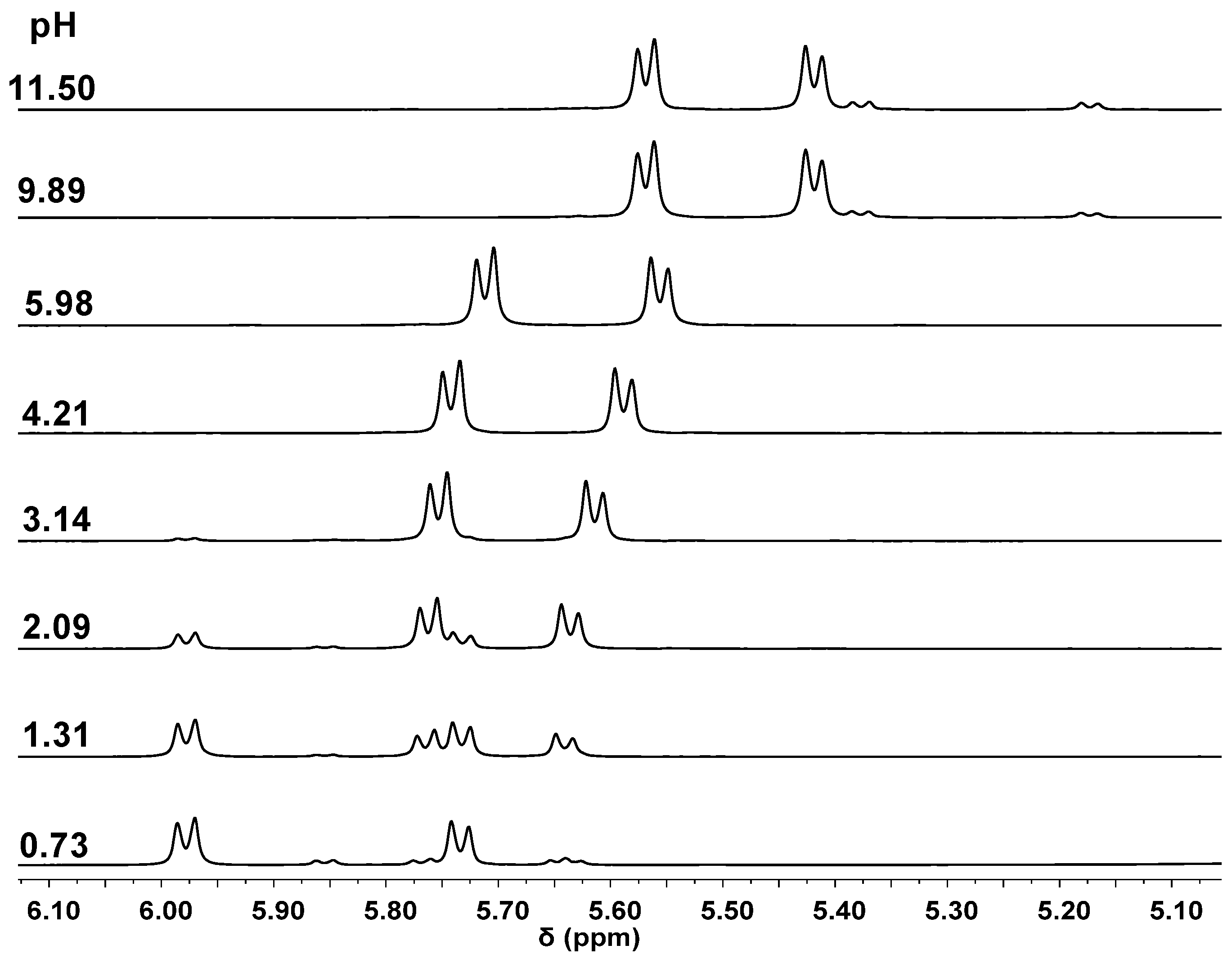
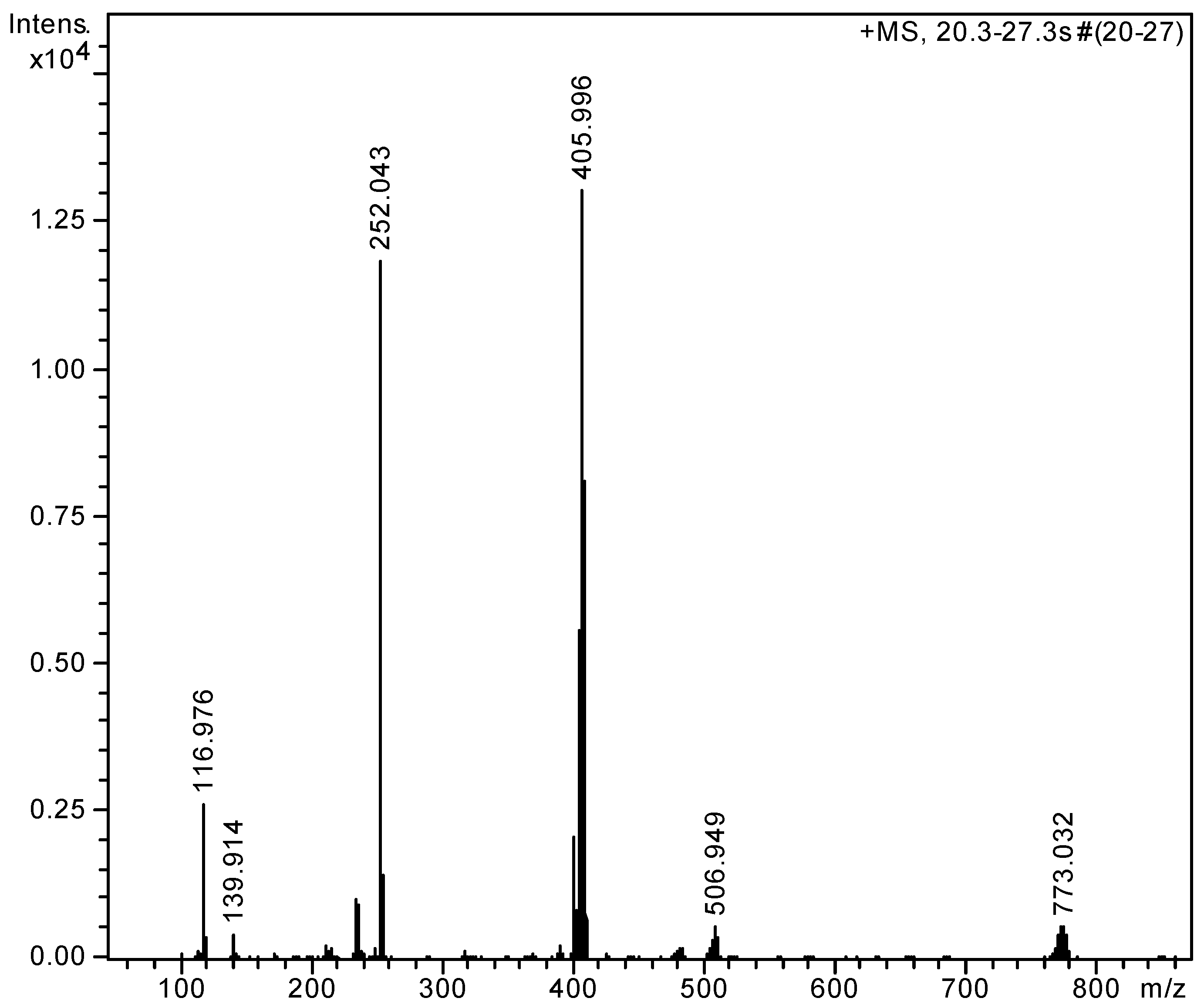
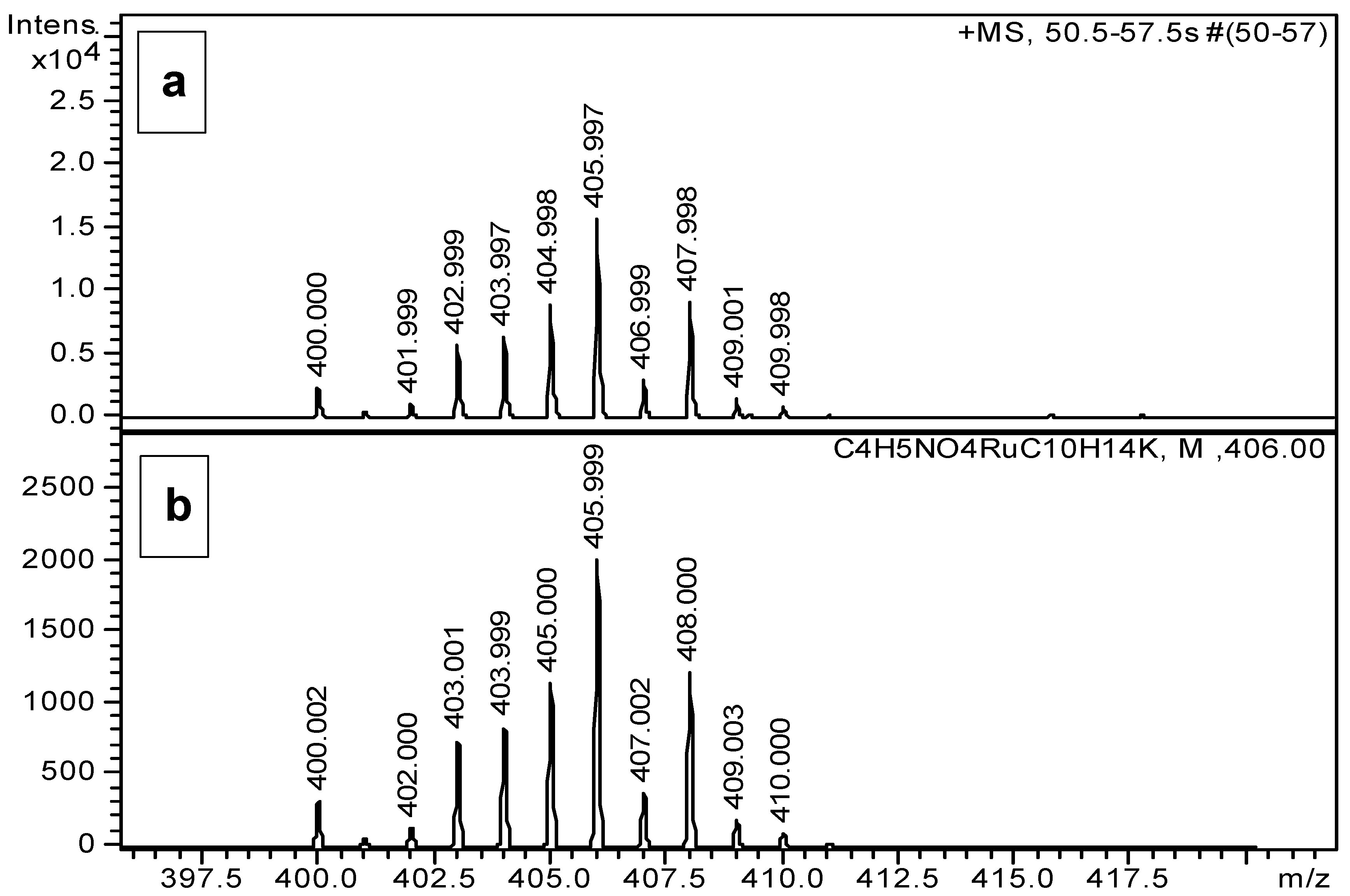
2.2. Complexation Processes with the Organometallic Cations
3. Materials and Methods
3.1. Materials
3.2. Solution Studies
3.3. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Graham, R.; Russell, G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef]
- Galezowska, J. Interactions between Clinically Used Bisphosphonates and Bone Mineral: From Coordination Chemistry to Biomedical Applications and Beyond. ChemMedChem 2018, 13, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Heidel, K.M.; Dowd, C.S. Phosphonate prodrugs: An overview and recent advances. Future Med. Chem. 2019, 11, 1625–1643. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.B.; Alves de Melo, J.; Maheshwari, S.; Queiroz de Medeiros, W.M.T.; Wysllas de Freitas Oliveira, J.; Moreno, C.J.; Amzel, L.M.; Gabelli, S.B.; Silva, M.S. Bisphosphonate-Based Molecules as Potential New Antiparasitic Drugs. Molecules 2020, 25, 2602. [Google Scholar] [CrossRef] [PubMed]
- Ebetino, F.H.; Sun, S.; Cherian, P.; Roshandel, S.; Neighbors, J.D.; Hu, E.; Dunford, J.E.; Sedghizadeh, P.P.; McKenna, C.E.; Srinivasan, V.; et al. Bisphosphonates: The role of chemistry in understanding their biological actions and structure-activity relationships, and new directions for their therapeutic use. Bone 2022, 156, 116289. [Google Scholar] [CrossRef]
- Kaboudin, B.; Daliri, P.; Faghih, S.; Esfandiar, H. Hydroxy- and Amino-Phosphonates and -Bisphosphonates: Synthetic Methods and Their Biological Applications. Front. Chem. 2022, 10, 890696. [Google Scholar] [CrossRef] [PubMed]
- Naydenova, E.D.; Todorov, P.T.; Troev, K.D. Recent synthesis of aminophosphonic acids as potential biological importance. Amino Acids 2010, 38, 23–30. [Google Scholar] [CrossRef]
- Kiss, T.; Lázár, I.; Kafarski, P. Chelating Tendencies of Bioactive Aminophosphonates. Metal Based Drugs 1994, 1, 248. [Google Scholar] [CrossRef]
- Buglyó, P.; Kiss, T.; Dyba, M.; Jezowska-Bojczuk, M.; Kozlowski, H.; Bouhsina, S. Complexes of aminophosphonates—Copper(II) complexes of phosphonic derivatives of iminodiacetate and nitrilotriacetate. Polyhedron 1997, 16, 3447–3454. [Google Scholar] [CrossRef]
- Jezowska-Bojczuk, M.; Kiss, T.; Kozcowski, H.; Decock, P.; Barycki, J. Complexes of aminophosphonates—Copper(II) complexes of N-(phosphonomethyl)amino acids and related compounds. J. Chem. Soc. Dalton Trans. 1994, 811–817. [Google Scholar] [CrossRef]
- Madsen, H.E.L.; Christensen, H.H.; Gottlieb-Petersen, C. Stability Constants of Copper(II), Zinc, Manganese(II), Calcium, and Magnesium Complexes of N-(Phosphonomethyl)glycine (Glyphosate). Acta Chem. Scand. A 1978, 32, 79–83. [Google Scholar] [CrossRef]
- Sanna, D.; Bódi, I.; Bouhsina, S.; Micera, G.; Kiss, T. Oxovanadium(IV) complexes of phosphonic derivatives of iminodiacetic and nitrilotriacetic acids. J. Chem. Soc. Dalton Trans. 1999, 3275–3282. [Google Scholar] [CrossRef]
- Klenner, T.; Wingen, F.; Keppler, B.K.; Krempien, B.; Schmahl, D. Anticancer-agent-linked phosphonates with antiosteolytic and antineoplastic properties: A promising perspective in the treatment of bone-related malignancies? J. Cancer Res. Clin. Oncol. 1990, 116, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Qui, J.-M.; Li, J.-F.; Ye, M.-Y.; Huang, R.-Z.; Xu, Q.; Pan, Y.-M.; Wang, H.-S.; Yao, G.-Y. Synthesis and antitumor activities of novel diacerein α-aminophosphonates conjugates. Indian J. Chem. 2014, 53, 1584–1595. [Google Scholar]
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure–activity relationships for ruthenium and osmium anticancer agents—Towards clinical development. Chem. Soc. Rev. 2018, 47, 909. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs-A New Paradigm in Cancer Drug Treatment Regimens. Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kim, C.Y.; Nam, T.-G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des. Dev. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef]
- Sun, Q.; Li, Y.; Shi, H.; Wang, Y.; Zhang, J.; Zhang, Q. Ruthenium Complexes as Promising Candidates against Lung Cancer. Molecules 2021, 26, 4389. [Google Scholar] [CrossRef]
- Štarha, P. Multinuclear biologically active Ru, Rh, Os and Ir arene complexes. Coord. Chem. Rev. 2021, 431, 213690. [Google Scholar] [CrossRef]
- Máliková, K.; Masaryk, L.; Štarha, P. Anticancer Half-Sandwich Rhodium(III) Complexes. Inorganics 2021, 9, 26. [Google Scholar] [CrossRef]
- Kacsir, I.; Sipos, A.; Ujlaki, G.; Buglyó, P.; Somsák, L.; Bai, P.; Bokor, É. Ruthenium Half-Sandwich Type Complexes with Bidentate Monosaccharide Ligands Show Antineoplastic Activity in Ovarian Cancer Cell Models through Reactive Oxygen Species Production. Int. J. Mol. Sci. 2021, 22, 10454. [Google Scholar] [CrossRef] [PubMed]
- Kacsir, I.; Sipos, A.; Bényei, A.; Janka, E.; Buglyó, P.; Somsák, L.; Bai, P.; Bokor, E. Reactive Oxygen Species Production Is Responsible for Antineoplastic Activity of Osmium, Ruthenium, Iridium and Rhodium Half-Sandwich Type Complexes with Bidentate Glycosyl Heterocyclic Ligands in Various Cancer Cell Models. Int. J. Mol. Sci. 2022, 23, 813. [Google Scholar] [CrossRef]
- Bíró, L.; Hüse, D.; Bényei, A.C.; Buglyó, P. Interaction of [Ru(η6-p-cym)(H2O)3]2+ with citrate and tricarballate ions in aqueous solution; X-ray crystal structure of novel half-sandwich Ru(II)-citrato complexes. J. Inorg. Biochem. 2012, 116, 116–125. [Google Scholar] [CrossRef]
- Patalenszki, J.; Bíró, L.; Bényei, A.C.; Muchova, T.R.; Kasparkova, J.; Buglyó, P. Half-sandwich complexes of ruthenium, osmium, rhodium and iridium with DL-methionine or S-methyl-L-cysteine: A solid state and solution equilibrium study. RSC Advances 2015, 5, 8094–8107. [Google Scholar] [CrossRef]
- Mészáros, J.P.; Dömötör, O.; Hackl, C.M.; Roller, A.; Keppler, B.K.; Kandioller, W.; Enyedy, E.A. Structural and solution equilibrium studies on half-sandwich organorhodium complexes of (N,N) donor bidentate ligands. New J. Chem. 2018, 42, 11174–11184. [Google Scholar] [CrossRef]
- Parajdi-Losonczi, P.L.; Buglyó, P.; Skakalova, H.; Kasparkova, J.; Lihi, N.; Farkas, E. Half-sandwich type rhodium–aminohydroxamate complexes: The role of the position of the amino group in metal ion binding. New J. Chem. 2018, 42, 7659–7670. [Google Scholar] [CrossRef]
- Pivarcsik, T.; Dömötör, O.; Mészáros, J.P.; May, N.V.; Spengler, G.; Csuvik, O.; Szatmári, I.; Enyedy, E.A. 8-Hydroxyquinoline-amino acid hybrids and their half-sandwich Rh and Ru complexes: Synthesis, anticancer activities, solution chemistry and interaction with biomolecules. Int. J. Mol. Sci. 2021, 22, 11281. [Google Scholar] [CrossRef] [PubMed]
- Nagy, S.; Ozsváth, A.; Bényei, A.C.; Farkas, E.; Buglyó, P. Donor atom preference of organoruthenium and Organorhodium cations on the interaction with novel ambidentate (N,N) and (O,O) chelating ligands in aqueous solution. Molecules 2021, 26, 3586. [Google Scholar] [CrossRef]
- Mészáros, J.P.; Pape, V.F.S.; Szakács, G.; Németi, G.; Dénes, M.; Holczbauer, T.; May, N.V.; Enyedy, E.A. Half-sandwich organometallic Ru and Rh complexes of (N,N) donor compounds: Effect of ligand methylation on solution speciation and anticancer activity. Dalton Trans. 2021, 50, 8218–8231. [Google Scholar] [CrossRef]
- Domestici, C.; Tensi, L.; Boccalon, E.; Zaccaria, F.; Costantino, F.; Zuccaccia, C.; Macchioni, A. Molecular and Heterogenized Cp*Ir Water Oxidation Catalysts Bearing Glyphosate and Glyphosine as Ancillary and Anchoring Ligands. Eur. J. Inorg. Chem. 2021, 2021, 299–307. [Google Scholar] [CrossRef]
- Hkiri, S.; Gourlaouen, C.; Touil, S.; Samarat, A.; Se’meril, D. 1,3,4-Oxadiazole-functionalized α-amino-phosphonates as ligands for the ruthenium-catalyzed reduction of ketones. New J. Chem. 2021, 45, 11327. [Google Scholar] [CrossRef]
- Dömötör, O.; Aicher, S.; Schmidlehner, M.; Novak, M.S.; Roller, A.; Jakupec, M.A.; Kandioller, W.; Hartinger, C.G.; Keppler, B.K.; Enyedy, E.A. Antitumor pentamethylcyclopentadienyl rhodium complexes of maltol and allomaltol: Synthesis, solution speciation and bioactivity. J. Inorg. Biochem. 2014, 134, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Bíró, L.; Farkas, E.; Buglyó, P. Complex formation between [Ru(η6-p-cym)(H2O)3]2+ and (O,O) donor ligands with biological relevance in aqueous solution. Dalton Trans. 2010, 39, 10272–10278. [Google Scholar] [CrossRef] [PubMed]
- Buglyó, P.; Parajdi-Losonczi, P.L.; Bényei, A.C.; Lihi, N.; Bíró, L.; Farkas, E. Versatility of Coordination Modes in Complexes of Monohydroxamic Acids with Half-Sandwich Type Ruthenium, Rhodium, Osmium and Iridium Cations. ChemistrySelect 2017, 2, 8127–8136. [Google Scholar] [CrossRef]
- Bruijnincx, P.C.A.; Sadler, P.J. Controlling platinum, ruthenium, and osmium reactivity for anticancer drug design. Adv. Inorg. Chem. 2009, 61, 1–62. [Google Scholar] [CrossRef] [PubMed]
- Bíró, L.; Godó, A.J.; Bihari, Z.; Garribba, E.; Buglyó, P. Tuning the Hydrolytic Properties of Half-Sandwich-Type Organometallic Cations in Aqueous Solution. Eur. J. Inorg. Chem. 2013, 2013, 3090–3100. [Google Scholar] [CrossRef]
- Bennett, M.A.; Smith, A.K. Arene ruthenium(II) complexes formed by dehydrogenation of cyclohexadienes with ruthenium(III) trichloride. J. Chem. Soc. Dalton Trans. 1974, 233–241. [Google Scholar] [CrossRef]
- Gran, G. Determination of the equivalent point in potentiometric titrations. Acta Chem. Scand. 1950, 4, 559–577. [Google Scholar] [CrossRef]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A study of some problems in determining the stoichiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. SUPERQUAD: An improved general program for computation of formation constants from potentiometric data. J. Chem. Soc. Dalton Trans. 1985, 1195–1200. [Google Scholar] [CrossRef]
- Zékány, L.; Nagypál, I. Computational Methods for the Determination of Formation Constants; Leggett, D.J., Ed.; Springer: Boston, MA, USA, 1985; pp. 291–353. [Google Scholar] [CrossRef]
- Bíró, L.; Farkas, E.; Buglyó, P. Hydrolytic behaviour and chloride ion binding capability of [Ru(η6-p-cym)(H2O)3]2+: A solution equilibrium study. Dalton Trans. 2012, 41, 285. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Bal, W. A formula for correlating pKa values determined in D2O and H2O. J. Inorg. Biochem. 2004, 98, 161–166. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Rev. B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Ab initio Pseudopotential Study of the First Row Transition Metal Monoxides and Iron Monohydride. J. Chem. Phys. 1987, 86, 2123–2131. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Minenkov, Y.; Singstad, Å.; Occhipinti, G.; Jensen, V.R. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ida2− | IdaP3− | Ida2P4− | |
|---|---|---|---|
| logK1 | 9.25 (1) | 10.01 (1) | 10.81 (1) |
| logK2 | 2.57 (2) | 5.40 (1) | 5.98 (1) |
| logK3 | 1.67 (5) | 2.21 (1) | 4.92 (1) |
| logK4 | – | <1 | <1 |
| logK5 | – | – | <1 |
| [(η6-p-cym)Ru]2+ | [(η5-Cp*)Rh]2+ | [(η6-p-cym)Os]2+ | [(η5-Cp*)Ir]2+ | ||
|---|---|---|---|---|---|
| Ida2− | logβ [MHL]+ | 12.81 (5) | — | — | — |
| logβ [ML] | 10.26 (2) | 11.53 (4) | 10.1 (1) | 11.9 (1) | |
| Fitting parameter # | 0.0117 | 0.00420 | 0.01505 | 0.00887 | |
| Number of data points fitted | 141 | 146 | 184 | 123 | |
| pK[MHL] | 2.55 | — | — | — | |
| IdaP3− | logβ [MHL] | 17.44 (1) | 16.05 (1) | 18.5 (1) | 17.9 (1) |
| logβ [ML]− | 13.00 (1) | 11.33 (2) | 14.9 (1) | 12.6 (1) | |
| Fitting parameter # | 0.00388 | 0.00787 | 0.01645 | 0.00868 | |
| Number of data points fitted | 154 | 207 | 156 | 225 | |
| pK[MHL] | 4.44 | 4.72 | 3.6 | 5.3 | |
| Ida2P4− | logβ [MH2L] | 21.92 (1) | 20.23 (1) | 21.7 (1) | 23.0 (1) |
| logβ [MHL]− | 19.13 (1) | 16.65 (4) | 19.4 (1) | 18.8 (1) | |
| logβ [ML]2− | 13.11 (1) | 10.59 (7) | 13.8 (1) | 13.0 (1) | |
| Fitting parameter # | 0.00526 | 0.00306 | 0.00797 | 0.00853 | |
| Number of data points fitted | 220 | 164 | 284 | 249 | |
| pK[MH2L] | 2.79 | 3.58 | 2.3 | 4.2 | |
| pK[MHL] | 6.02 | 6.06 | 5.6 | 5.8 |
| Bond Lengths (Å)/Angles (°) | Ida | IdaP | Ida2P |
|---|---|---|---|
| Ru–Ct a | 1.623 | 1.730 | 1.662 |
| Ru–OOC (1) | 2.127 | 2.137 | |
| Ru–OOC (2) | 2.149 | ||
| Ru–OPOO (1) | 2.159 | 2.155 | |
| Ru–OPOO (2) | 2.149 | ||
| Ru–NH | 2.156 | 2.174 | 2.184 |
| (1) COO–Ru–NH | 79.0 | 78.1 | |
| HN–Ru–OOC (2) | 77.2 | ||
| (1) OOPO–Ru–NH | 82.8 | 77.8 | |
| HN–Ru–OPOO (2) | 82.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bíró, L.; Tóth, B.; Lihi, N.; Farkas, E.; Buglyó, P. Interaction between [(η6-p-cym)M(H2O)3]2+ (MII = Ru, Os) or [(η5-Cp*)M(H2O)3]2+ (MIII = Rh, Ir) and Phosphonate Derivatives of Iminodiacetic Acid: A Solution Equilibrium and DFT Study. Molecules 2023, 28, 1477. https://doi.org/10.3390/molecules28031477
Bíró L, Tóth B, Lihi N, Farkas E, Buglyó P. Interaction between [(η6-p-cym)M(H2O)3]2+ (MII = Ru, Os) or [(η5-Cp*)M(H2O)3]2+ (MIII = Rh, Ir) and Phosphonate Derivatives of Iminodiacetic Acid: A Solution Equilibrium and DFT Study. Molecules. 2023; 28(3):1477. https://doi.org/10.3390/molecules28031477
Chicago/Turabian StyleBíró, Linda, Botond Tóth, Norbert Lihi, Etelka Farkas, and Péter Buglyó. 2023. "Interaction between [(η6-p-cym)M(H2O)3]2+ (MII = Ru, Os) or [(η5-Cp*)M(H2O)3]2+ (MIII = Rh, Ir) and Phosphonate Derivatives of Iminodiacetic Acid: A Solution Equilibrium and DFT Study" Molecules 28, no. 3: 1477. https://doi.org/10.3390/molecules28031477
APA StyleBíró, L., Tóth, B., Lihi, N., Farkas, E., & Buglyó, P. (2023). Interaction between [(η6-p-cym)M(H2O)3]2+ (MII = Ru, Os) or [(η5-Cp*)M(H2O)3]2+ (MIII = Rh, Ir) and Phosphonate Derivatives of Iminodiacetic Acid: A Solution Equilibrium and DFT Study. Molecules, 28(3), 1477. https://doi.org/10.3390/molecules28031477