Abstract
Halogen bonds play an important role in many fields, such as biological systems, drug design and crystal engineering. In this work, the structural characteristics of the halogen bond between heteronuclear halogen XD (ClF, BrCl, IBr, ICl, BrF and IF) and benzene were studied using density functional theory. The structures of the complexes between heteronuclear halogen and benzene have Cs symmetry. The interaction energies of the complexes between heteronuclear halogen XD (ClF, BrCl, IBr, ICl, BrF and IF) and benzene range from −27.80 to −37.18 kJ/mol, increasing with the increases in the polarity between the atoms of X and D, and are proportional to the angles of a between the Z axis and the covalent bond of heteronuclear halogen. The electron density (ρ) and corresponding Laplacian (∇2ρ) values indicate that the interaction of the heteronuclear halogen and benzene is a typical long-range weak interaction similar to a hydrogen bond. Independent gradient model analysis suggests that the van der Waals is the main interaction between the complexes of heteronuclear halogen and benzene. Symmetry-adapted perturbation theory analysis suggests that the electrostatic interaction is the dominant part in the complexes of C6H6⋯ClF, C6H6⋯ICl, C6H6⋯BrF and C6H6⋯IF, and the dispersion interaction is the main part in the complexes of C6H6⋯BrCl, C6H6⋯IBr.
1. Introduction
A halogen bond is a noncovalent interaction [1,2] similar to the typical hydrogen bond [3,4,5,6]. In the case of a halogen bond, a halogen atom is shared both by a donor D and an acceptor A [7]. The halogen bond interaction can be depicted by:
where the X can be chlorine, bromine or iodine and the angle of D-X⋯A is close to 180°.
D-X---A
Halogen bonding plays an important role in many fields, such as biological systems [8,9,10,11,12], drug design [13,14,15], crystal engineering [16,17,18,19] and function materials [20,21,22,23]. Auffinger et al. explored the function of halogen bonds in ligand binding, recognition, conformational equilibria and molecular binding [24]. Johnson et al. found that the 4, 5, 6, 7-tetrabromobenzotriazole could displace charged ATP from its binding site on phospho-CDK2-cyclin by the halogen bond of Br–O interaction [25]. As a result, it is of great practical significance to study the halogen bond deeply.
Due to the influence of conjugation, right above the center of the benzene ring is an electron-rich region [26], which can combine with the halogen molecules to form stable complexes. Similar systems have been studied in recent years. Tsuzuki et al. studied the size and direction of halogen bonds of molecules containing halogen atoms interacting with benzene [27]. Schwabedissen et al. studied the action rules of halogen bonds in crystals [28]. Yu et al. studied the effect of halogen bonds on molecular fluorescence [29]. Otte et al. studied the competing weak interactions in the process of complex formation [30]. In addition, Oliveira, Kraka and Cremer et al. have published extensive work on analyzing halogen bonds based on vibrational spectroscopy, leading to a quantitative measure of the interaction strength in these systems and providing a rigorous and comprehensive discussion of halogen bonds on a quantum chemical level [31,32,33,34,35,36,37,38,39,40].
Although these studies have given the geometric structures and interaction energies of the complexes between halogen and benzene [41], to our knowledge, the details of their interaction have not been reported until now. In this work, we studied the structural characteristics of the halogen bond between heteronuclear halogen XD (ClF, BrCl, IBr, ICl, BrF and IF) and benzene using density functional theory (DFT). The calculation results indicate that heteronuclear halogen and benzene can form stable complexes with Cs symmetry. The interaction energy ranges from −27.80 to −37.18 kJ/mol, which is proportional to the angle of a. Topological properties of the electron density based on the atoms in molecules (AIM) theory show that bond critical points (BCPs) between the heteronuclear halogen and benzene exist in all the six complexes. The electron density (ρ) and corresponding Laplacian (∇2ρ) values indicate that the interaction of the heteronuclear halogen and benzene is a non-covalent intermolecular interaction similar to a hydrogen bond. Independent gradient model (IGM) analysis of the interaction between the heteronuclear halogen and benzene suggests that van der Waals is the main interaction.
2. Results and Discussion
2.1. Geometry and Interaction Energy
Figure 1 shows the structure of the complexes formed by heteronuclear halogen (ClF, BrCl, IBr, ICl, BrF and IF) and benzene. As can be seen from the Figure 1, the heteronuclear halogen in the complexes is located above the benzene ring, but not perpendicular to the plane of the benzene ring. All the six complexes belong to the Cs symmetry, and the more electronegative atom of the heteronuclear halogen in the complexes is far away from the benzene ring.
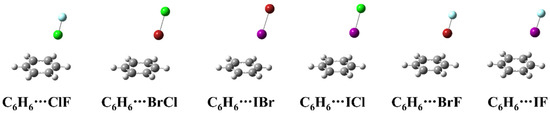
Figure 1.
The geometric structures of the complexes between heteronuclear halogen and benzene.
Figure 2 shows the schematic diagram of the complexes. To better illustrate the relationship between the interaction energy and the structure of the complexes, the direction that passes through the X atom and is perpendicular to the plane of the benzene ring is defined as the z-axis, and the angle between the X−D bond and the z-axis is defined as a.
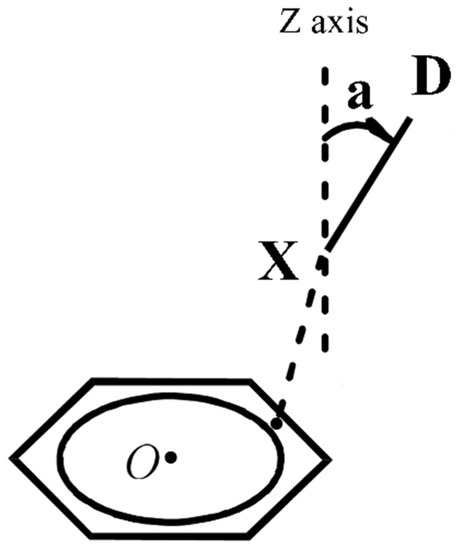
Figure 2.
The structure diagram of the complexes between heteronuclear halogens and benzene.
The values of the interaction energy and the angle of a are shown in Table 1. As can be seen from Table 1, the interaction energy is related to the magnitude of a, and increases with the increase in a. In addition, when the atom of D is fixed, the interaction energy of the complexes increases with the increase in the polarity between the atoms of X and D, which is inconsistent with the calculations from Sugibayashi, et al. [41].

Table 1.
The values of the interaction energy and the angle of a.
As can be seen from Figure 3, the interaction energy and the angle of a have good linearity. The correlation coefficient reaches 99.5%, indicating that the interaction between heteronuclear halogen and benzene tends to be perpendicular to the plane. The interaction energy ranges from −27.80 to −37.18 kJ/mol, which is a typical long-range weak interaction similar to a hydrogen bond.
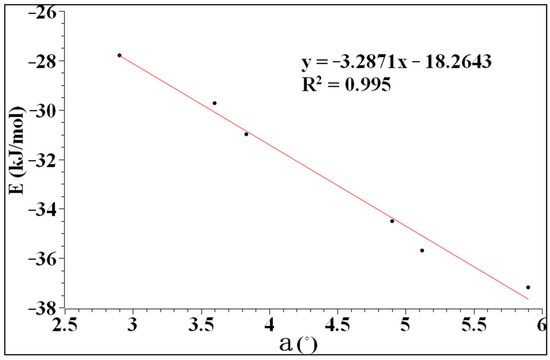
Figure 3.
The relationship between the interaction energy and a.
2.2. Electrostatic Potential
Figure 4 shows the electrostatic potential of each monomer in the complexes. According to the electrostatic potential diagram, the central region of the benzene ring shows a negative electrostatic potential region. However, the heteronuclear halogen (ClF, BrCl, IBr, ICl, BrF and IF) in the complexes does not lie directly above the benzene ring but has a silent deviation from the benzene ring center. The reason for the deviation indicates that the interaction between the two monomers in the complexes is not only electrostatic. In the process of complex formation, a variety of interactions exert influence and achieve a reasonable state, resulting in complexes with Cs symmetry.
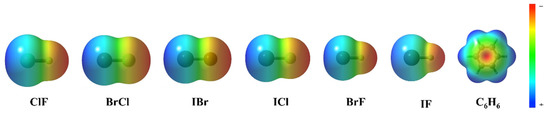
Figure 4.
The electrostatic potential of ClF, BrCl, IBr, ICl, BrF, IF and C6H6.
2.3. Topological Properties of the Electron Density
In order to further understand the interaction characteristics, we carried out an AIM theoretical analysis of the six complexes. The results show that there are bond critical points between the heteronuclear halogen and benzene in the six complexes. The values of electron density (ρ), Laplacian of electron density (∇2ρ) and ellipticity (ε) of the saddle point of the bond are listed in Table 2. According to the criteria proposed by Popelier et al. [42,43], the electron density (ρ) at the saddle point of the bond ranges from 0.0025 to 0.035 a.u. and the Laplacian amount of electron density (∇2ρ) is between 0.024 and 0.139. We found that the electron density at the saddle point of the bond and the Laplacian amount of the electron density in the six complexes are in the above range, indicating that the interaction in the complex is a weak interaction similar to that of a hydrogen bond. In addition, the ellipticity value at the saddle point of the key is related to the type of the key. As can be seen from Table 2, the ellipticity (ε) of the six complexes is all greater than zero, showing obvious π-bond characteristics.
Table 2.
The values of electron density (ρ), Laplacian of electron density (▽2ρ) ellipticity (ε) and λ2 of the saddle point.
2.4. IGM Analysis
In order to understand the region, size and species of the weak interaction of the halogen bond between the heteronuclear halogen and benzene in the six complexes, IGM analysis of the six complexes was carried out [44,45]. In IGM theory, the second largest eigenvalue of the electron density Hessian matrix (sign (λ2)) is used to define the type of interaction [46]. When the value of sign (λ2) × ρ is less than zero, it is an attractive interaction. When the value of sign (λ2) × ρ is greater than zero, it is a repulsive interaction. When the value of sign (λ2) × ρ is equal to zero, it is a van der Waals interaction. IGM is calculated by distinguishing the interaction into intramolecular and intermolecular interactions, which are represented by δinter and δintra, respectively. The peak value of δinter is generally less than 0.1 a.u., the van der Waals interaction is generally less than 0.03 a.u., and the hydrogen bond interaction is generally less than 0.1 a.u. The calculated results of the six complexes are shown in Figure 5. As can be seen from Figure 5, all six complexes have different degrees of peak values in the region of sign (λ2) × ρ < 0, indicating that the interactions in the complexes are all in the van der Waals intermolecular interaction region. The peak values of δinter are from 0.0125 to 0.0476 a.u., where C6H6⋯BrF and C6H6⋯ClF exceed 0.003 a.u., indicating that the complexes exhibit a strong van der Waals interaction, and the remaining C6H6⋯BrCl, C6H6⋯ICl, C6H6⋯IBr and C6H6⋯IF shows a general van der Waals effect. From the isosurface color map in Figure 5, the color map of the six complexes shows that the majority of the results are green, which further indicates that the van der Waals is the dominant interaction. At the same time, it can be seen that the central region of the isosurface is blue, indicating that the strongest interaction position is between the X atom and the C-C bond center, which is consistent with the above conclusion of electron density topological analysis.
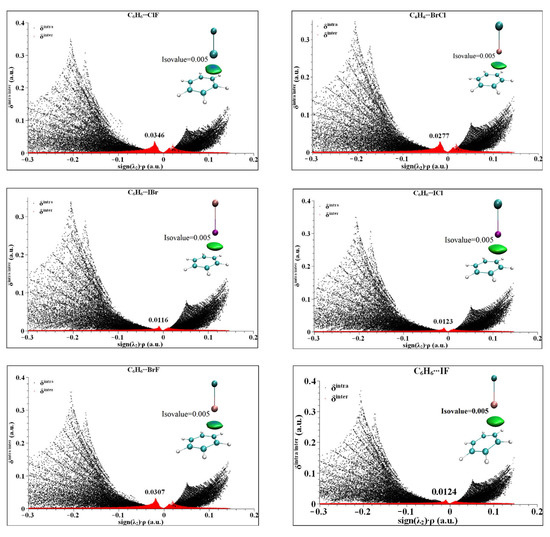
Figure 5.
The IGM analysis and color plots of complexes between heteronuclear halogen XD (ClF, BrCl, IBr, ICl, BrF and IF) and benzene.
2.5. SAPT Calculation Analysis
In order to further understand the tendency and characteristics of the van der Waals interaction between heteronuclear halogen and benzene, the intermolecular interaction energy was decomposed and calculated using symmetry-adapted perturbation theory (SAPT). The calculation results are listed in Table 3. In the six complexes, the electrostatic and dispersive energies are both larger than the induced energies, indicating that the electrostatic and dispersive effects are dominant in the six complexes. Further studies reveal that in C6H6⋯ClF, C6H6⋯ICl, C6H6⋯BrF and C6H6⋯IF, electrostatic energy accounts for the largest proportions, which are 36.7%, 37.3%, 37.9% and 38.9%, respectively, indicating that the electrostatic interaction of these four complexes is dominant. In C6H6⋯BrCl and C6H6⋯IBr, the dispersive energy has the largest proportions of the interaction energy, which are 37.9% and 38.6%, respectively, indicating that the dispersive interaction in the two complexes is the main part. The dispersion effect is more obvious in the complexes with large radius halogen atoms, which might be related to the degree of electron dispersion in halogen atoms.
Table 3.
The calculation analysis using symmetry matching perturbation theory (SAPT).
In order to further study the role of the interaction energies, such as induction, dispersion, exchange and electrostatic energy in complex formation, the variation of each energy with the distance of its monomer centroid was calculated. The calculation results are shown in Figure 6. It can be seen in Figure 6 that the variation in each interaction energy with the distance from the centroid of the monomer is not completely the same, but the variation trend is similar. In the range of R < 0.3 nm, the exchange and induction effects are significantly affected by the molecular spacing, and the repulsive and attractive effects are basically equal. At this time, the intermolecular interaction shows electrostatic interaction and dispersion interaction, but the dispersion interaction line is always below the electrostatic interaction line, indicating that the dispersion interaction is obviously affected. At R = 0.4 nm, the six complexes reach equilibrium. Finally, C6H6⋯ClF, C6H6⋯ICl, C6H6⋯BrF and C6H6⋯IF are dominated by electrostatic interaction, while the dispersion interaction in C6H6⋯BrCl, C6H6⋯IBr is dominant. Among the six complexes, the dispersion interaction line is the flattest, indicating that the intermolecular distance is the least affected. In C6H6⋯ICl and C6H6⋯IBr, the electrostatic action line and the dispersion action line are very close, indicating that the electrostatic action and dispersion action are similar.
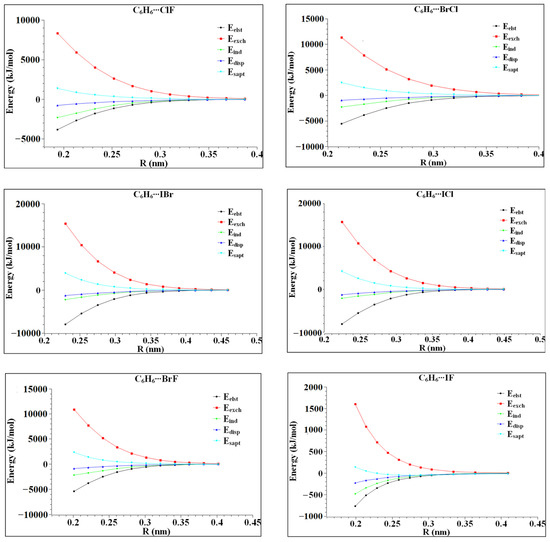
Figure 6.
The interaction energy varies with the distance from the centroid of heteronuclear halogen XD.
3. Materials and Methods
The geometric structures of heteronuclear halogen, benzene and their complexes were optimized using DFT with the functional of wB97XD and the basis set of aug-cc-pVTZ. The calculations for iodine were carried out with quasi-relativistic small-core effective potentials (ECPs) and the corresponding Peterson AVTZ basis set. The frequency calculation was conducted with the same base set as structure optimization and the results show that heteronuclear halogen, benzene and their complexes have no imaginary frequency. The interaction energy was calculated with the same level of theory as the geometry optimization. The counterpoise procedure of Boys et al. was used for BSSE correction of the base group overlap errors [47]. The program used for calculation was Gaussian 16 [48]. The AIM theory was used for electron density topological analysis and electrostatic potential analysis on the basis of wave function from Gaussian in order to further discuss the characteristics of the interaction between heteronuclear halogens and benzene [49,50]. SAPT was used to study the composition and interaction of molecular interaction energy with the same base set as structure optimization [51,52]. The calculation program was Psi4 [53].
4. Conclusions
Heteronuclear halogen and benzene can form stable complexes with Cs symmetry. The interaction energy is from −27.80 to −37.18 kJ/mol. All six complexes show obvious hydrogen bond-like properties with a weak long-range interaction. The van der Waals is the main interaction between the complexes of heteronuclear halogen and benzene. The electrostatic interaction is the dominant part in the complexes of C6H6⋯ClF, C6H6⋯ICl, C6H6⋯BrF and C6H6⋯IF, and the dispersion interaction is main part in the complexes of C6H6⋯BrCl, C6H6⋯IBr.
Author Contributions
Conceptualization, J.L. and H.D.; methodology, M.C.; software, J.L.; validation, J.L.; formal analysis, C.Z.; investigation, D.W.; resources, J.L.; data curation, J.L.; writing—original draft preparation, J.L.; writing—review and editing, M.C.; visualization, J.L.; supervision, M.C.; project administration, J.L.; funding acquisition, J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of Science and Technology Department of Guizhou Province ([2019]1297).
Data Availability Statement
Not applicable.
Acknowledgments
We acknowledge Min Liu’s suggestions for revisions to the article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Murray, J.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef] [PubMed]
- Voth, A.; Khuu, P.; Oishi, K.; Ho, P. Halogen bonds as orthogonal molecular interactions to hydrogen bonds. Nat. Chem. 2009, 1, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Yee, N.; Dadvand, A.; Hamzehpoor, E.; Titi, H.M.; Perepichka, D.F. Hydrogen Bonding Versus π-Stacking in Charge-Transfer Co-crystals. Cryst. Growth Des. 2021, 21, 2609–2613. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonds, and σ-hole and π-hole bonds—Mechanisms protecting doublet and octet electron structures. Phys. Chem. Chem. Phys. 2017, 19, 29742–29759. [Google Scholar] [CrossRef]
- Chernysheva, M.V.; Bulatova, M.; Ding, X.; Haukka, M. Influence of Substituents in the Aromatic Ring on the Strength of Halogen Bonding in Iodobenzene Derivatives. Cryst. Growth Des. 2020, 20, 7197–7210. [Google Scholar] [CrossRef]
- Xu, L.; Sang, P.; Zou, J.-W.; Xu, M.-B.; Li, X.-M.; Yu, Q.-S. Evaluation of nucleotide C–Br···O–P contacts from ONIOM calculations: Theoretical insight into halogen bonding in nucleic acids. Chem. Phys. Lett. 2011, 509, 175–180. [Google Scholar] [CrossRef]
- Kurczab, R.; Bojarski, A. The Potential Role of Halogen Bonding in Interactions of Ligands with Class A GPCRs—The β2 Adrenergic Receptor Case Study. Poster 2015. [Google Scholar] [CrossRef]
- Jena, S.; Dutta, J.; Tulsiyan, K.D.; Sahu, A.K.; Choudhury, S.S.; Biswal, H.S. Noncovalent interactions in proteins and nucleic acids: Beyond hydrogen bonding and π-stacking. Chem. Soc. Rev. 2022, 51, 4261–4286. [Google Scholar] [CrossRef]
- Bayse, C.A. Halogen bonding from the bonding perspective with considerations for mechanisms of thyroid hormone activation and inhibition. New J. Chem. 2018, 42, 10623–10632. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Frangville, P.; Meyer, F. Halogen bonding for molecular recognition: New developments in materials and biological sciences. Chem. Commun. 2020, 56, 4970–4981. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Zimmermann, M.O.; Wilcken, R.; Zahn, S.; Boeckler, F.M. Targeting Histidine Side Chains in Molecular Design through Nitrogen–Halogen Bonds. J. Chem. Inf. Model. 2013, 53, 3178–3189. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Zahn, S.; Kirchner, B.; Boeckler, F.M. Addressing Methionine in Molecular Design through Directed Sulfur–Halogen Bonds. J. Chem. Theory Comput. 2011, 7, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shi, T.; Wang, Y.; Yang, H.; Yan, X.; Luo, X.; Jiang, H.; Zhu, W. Halogen Bonding—A Novel Interaction for Rational Drug Design? J. Med. Chem. 2009, 52, 2854–2862. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Ivanov, D.M.; Novikov, A.S.; Ananyev, I.V.; Bokach, N.A.; Kukushkin, V.Y. Metal-involving halogen bond Ar–I···[dz2PtII] in a platinum acetylacetonate complex. Cryst. Eng. Commun. 2020, 22, 554–563. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Ananyev, I.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. π-Hole···dz2[PtII] Interactions with Electron-Deficient Arenes Enhance the Phosphorescence of PtII-Based Luminophores. Inorg. Chem. 2020, 59, 9308–9314. [Google Scholar] [CrossRef]
- Fourmigué, M.; Batail, P. Activation of Hydrogen- and Halogen-Bonding Interactions in Tetrathiafulvalene-Based Crystalline Molecular Conductors. Chem. Rev. 2004, 104, 5379–5418. [Google Scholar] [CrossRef]
- Nemec, V.; Lisac, K.; Bedeković, N.; Fotović, L.; Stilinović, V.; Cinčić, D. Crystal engineering strategies towards halogen-bonded metal–organic multi-component solids: Salts, cocrystals and salt cocrystals. Cryst. Eng. Commun. 2021, 23, 3063–3083. [Google Scholar] [CrossRef]
- Zheng, J.; Suwardi, A.; Wong, C.J.E.; Loh, X.J.; Li, Z. Halogen bonding regulated functional nanomaterials. Nanoscale Adv. 2021, 3, 6342–6357. [Google Scholar] [CrossRef]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef] [PubMed]
- Bunchuay, T.; Docker, A.; Martinez-Martinez, A.J.; Beer, P.D. A Potent Halogen-Bonding Donor Motif for Anion Recognition and Anion Template Mechanical Bond Synthesis. Angew. Chem. Int. Ed. 2019, 58, 13823–13827. [Google Scholar] [CrossRef] [PubMed]
- Serpell, C.J.; Kilah, N.L.; Costa, P.J.; Félix, V.; Beer, P.D. Halogen Bond Anion Templated Assembly of an Imidazolium Pseudorotaxane. Angew. Chem. Int. Ed. 2010, 49, 5322–5326. [Google Scholar] [CrossRef] [PubMed]
- Pascal, A.; Hays, F.; Westhof, E.; Ho, P. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef]
- De Moliner, E.; Brown, N.R.; Johnson, L.N. Alternative binding modes of an inhibitor to two different kinases. Eur. J. Biochem. 2003, 270, 3174–3181. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, S.; Wakisaka, A.; Ono, T.; Sonoda, T. Magnitude and Origin of the Attraction and Directionality of the Halogen Bonds of the Complexes of C6F5X and C6H5X (X = I, Br, Cl and F) with Pyridine. Chem. Eur. J. 2012, 18, 951–960. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Uchimaru, T.; Wakisaka, A.; Ono, T. Magnitude and Directionality of Halogen Bond of Benzene with C6F5X, C6H5X, and CF3X (X = I, Br, Cl, and F). J. Phys. Chem. A 2016, 120, 7020–7029. [Google Scholar] [CrossRef]
- Schwabedissen, J.; Trapp, P.C.; Stammler, H.-G.; Neumann, B.; Lamm, J.-H.; Vishnevskiy, Y.V.; Körte, L.A.; Mitzel, N.W. Halogen Bonds of Halotetrafluoropyridines in Crystals and Co-crystals with Benzene and Pyridine. Chem. Eur. J. 2019, 25, 7339–7350. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, X.; Zhao, H.; Jiang, Z.; Wang, T.; Wang, N.; Huang, X.; Zhou, L.; Hao, H. Enhanced luminescence of single-benzene fluorescent molecules through halogen bond cocrystals. Cryst. Eng. Commun. 2022, 24, 3537–3545. [Google Scholar] [CrossRef]
- Otte, F.; Kleinheider, J.; Hiller, W.; Wang, R.; Englert, U.; Strohmann, C. Weak yet Decisive: Molecular Halogen Bond and Competing Weak Interactions of Iodobenzene and Quinuclidine. J. Am. Chem. Soc. 2021, 143, 4133–4137. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E. Systematic Coupled Cluster Study of Noncovalent Interactions Involving Halogens, Chalcogens, and Pnicogens. J. Phys. Chem. A 2017, 121, 9544–9556. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Kraka, E.; Cremer, D. Quantitative Assessment of Halogen Bonding Utilizing Vibrational Spectroscopy. Inorg. Chem. 2017, 56, 488–502. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. The intrinsic strength of the halogen bond: Electrostatic and covalent contributions described by coupled cluster theory. Phys. Chem. Chem. Phys. 2016, 18, 33031–33046. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Cremer, D. Transition from metal-ligand bonding to halogen bonding involving a metal as halogen acceptor a study of Cu, Ag, Au, Pt, and Hg complexes. Chem. Phys. Lett. 2017, 681, 56–63. [Google Scholar] [CrossRef]
- Yannacone, S.; Oliveira, V.; Verma, N.; Kraka, E. A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit? Inorganics 2019, 7, 47. [Google Scholar] [CrossRef]
- Yannacone, S.; Freindorf, M.; Tao, Y.; Zou, W.; Kraka, E. Local Vibrational Mode Analysis of π–Hole Interactions between Aryl Donors and Small Molecule Acceptors. Crystals 2020, 10, 556. [Google Scholar] [CrossRef]
- Freindorf, M.; Yannacone, S.; Oliveira, V.; Verma, N.; Kraka, E. Halogen Bonding Involving I2 and d8 Transition-Metal Pincer Complexes. Crystals 2021, 11, 373. [Google Scholar] [CrossRef]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative Assessment of Tetrel Bonding Utilizing Vibrational Spectroscopy. Molecules 2018, 23, 2763. [Google Scholar] [CrossRef]
- Oliveira, V.P.; Marcial, B.L.; Machado, F.B.C.; Kraka, E. Metal–Halogen Bonding Seen through the Eyes of Vibrational Spectroscopy. Materials 2020, 13, 55. [Google Scholar] [CrossRef]
- Tao, Y.; Zou, W.; Sethio, D.; Verma, N.; Qiu, Y.; Tian, C.; Cremer, D.; Kraka, E. In Situ Measure of Intrinsic Bond Strength in Crystalline Structures: Local Vibrational Mode Theory for Periodic Systems. J. Chem. Theory Comput. 2019, 15, 1761–1776. [Google Scholar] [CrossRef]
- Sugibayashi, Y.; Hayashi, S.; Nakanishi, W. Behavior of Halogen Bonds of the Y-X…π Type (X, Y = F, Cl, Br, I) in the Benzene pi System, Elucidated by Using a Quantum Theory of Atoms in Molecules Dual-Functional Analysis. Chemphyschem 2016, 17, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Popelier, P.L.A. Characterization of a Dihydrogen Bond on the Basis of the Electron Density. J. Phys. Chem. A 1998, 102, 1873–1878. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A.; Stone, A.J. Conformational dependence of atomic multipole moments. Chem. Phys. Lett. 1995, 238, 253–260. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef]
- Ponce-Vargas, M.; Lefebvre, C.; Boisson, J.-C.; Hénon, E. Atomic Decomposition Scheme of Noncovalent Interactions Applied to Host–Guest Assemblies. J. Chem. Inf. Model. 2020, 60, 268–278. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [PubMed]
- Turney, J.M.; Simmonett, A.C.; Parrish, R.M.; Hohenstein, E.G.; Evangelista, F.A.; Fermann, J.T.; Mintz, B.J.; Burns, L.A.; Wilke, J.J.; Abrams, M.L.; et al. Psi4: An open-source ab initio electronic structure program. WIREs Comput. Mol. Sci. 2012, 2, 556–565. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).