
Machine Learning Models to Predict Protein–Protein Interaction Inhibitors


Abstract
1. Introduction
2. Methods
2.1. Data Sets
2.2. Molecular Representations
2.3. Machine Learning Models
2.4. Training Models
2.4.1. Data Proportions
2.4.2. Parameter Settings
- (A)
- RF is an algorithm that generates many decision trees and then assembles their outputs [16]. The parameters explored for this algorithm are: the number of trees in the forest (100, 500, 1000) and gini and entropy as functions to measure the quality of a split. Details of the RF setup are summarized in Table S2 in the Supplementary Materials.
- (B)
- LRG [32] is a linear classification model. In this model, the probabilities describing the possible outcomes of a single trial are modeled using a logistic function. Solver parameters have a major impact on results. Five different solvers included in scikit-learn were used: newton-cg, lbfgs, liblinear, sag, and saga (Table S3 in the Supplementary Materials).
- (C)
- SVM [33] solves classification problems because of its ability to handle high-dimensional data using a kernel function [34]. In SVM, the kernel function is used to map data into high-dimensional space by finding an optimally separating hyperplane. For this study, four different kernels were used: linear, poly, rbf, and sigmoid (Table S4 in the Supplementary Materials).
2.5. The Automated Pipeline
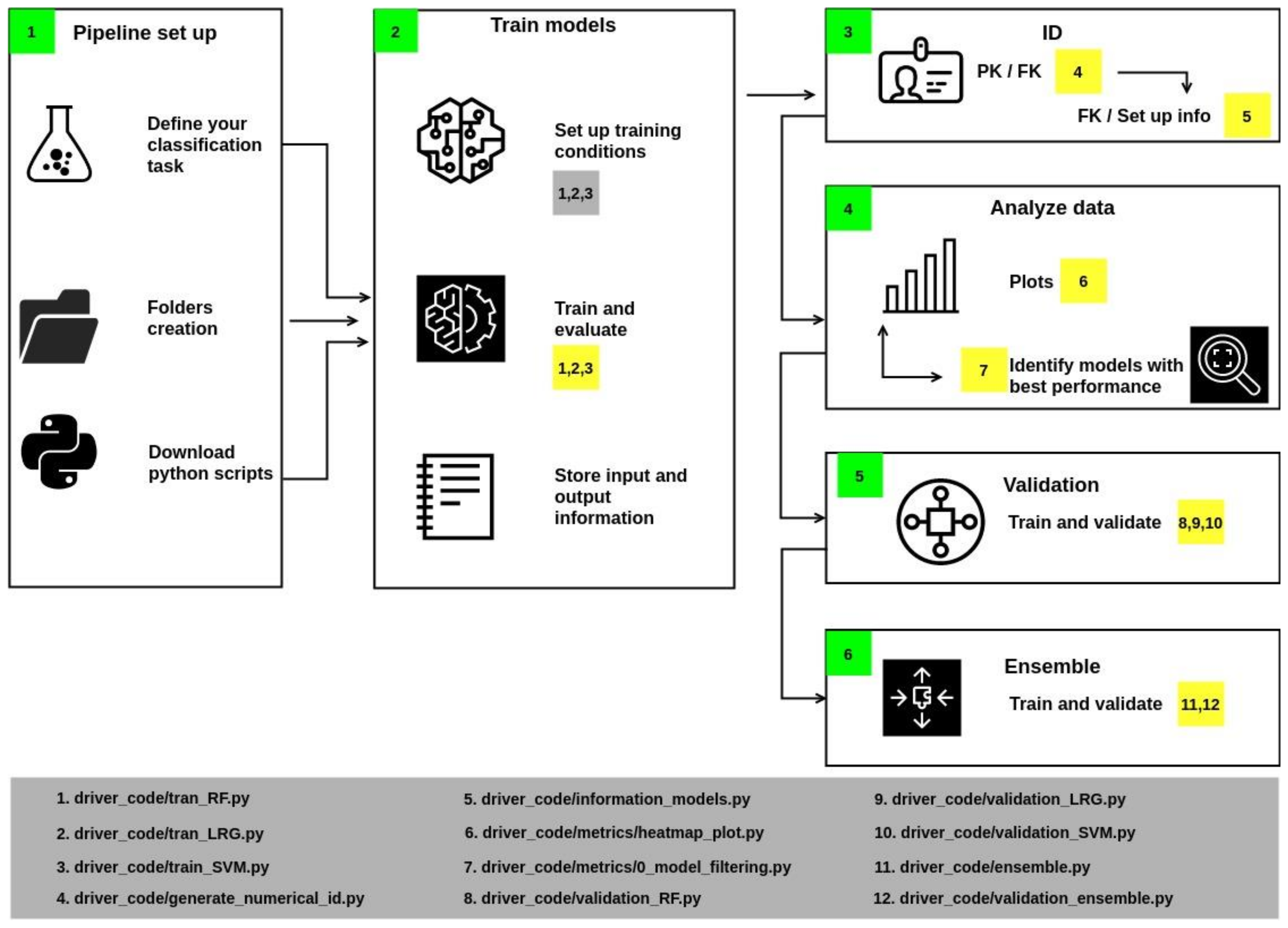
- In Section 1 of the workflow (Figure 2), it is necessary to create the folders and download the python files.
- In Section 2, some variables should be associated with specific values, such as allocating the files’ location. Training parameters must be assigned: once configured, run scripts 1, 2, and 3 to train the models. These scripts also calculate accuracy, precision, F1, and recall, which are metrics to describe the quality of the model’s predictions (metrics are computed from the test population). The script also stores the results in an individual report.
- Section 3 generates identifiers if the report’s name corresponds to the Primary Key. Then, a Foreign Keys is generated by the union of the initials of the algorithm and a numerical index. The results are stored in a JSON file.
- Section 4 includes a series of scripts to collect metrics values from reports and generate heatmaps. These plots contain information from those models whose values were greater than or equal to the statistical metric known as Q2 (i.e., the middle of the data set, also termed the 50th percentile).
- Section 5 implements cross-validation of models with values above Q2 with a k equal to 20. As a result, an output file is generated that reports the value of the mean and the deviation of accuracy.
- Section 6 performs the training and validation of a consensus model.
3. Results and Discussion
3.1. RF
3.2. LRG
3.3. SVM
3.4. Consensus Prediction
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Villoutreix, B.O.; Kuenemann, M.A.; Poyet, J.-L.; Bruzzoni-Giovanelli, H.; Labbé, C.; Lagorce, D.; Sperandio, O.; Miteva, M.A. Drug-Like Protein-Protein Interaction Modulators: Challenges and Opportunities for Drug Discovery and Chemical Biology. Mol. Inform. 2014, 33, 414–437. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Protein-protein interaction inhibitors get into the groove. Nat. Rev. Drug Discov. 2012, 11, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Eufracio, B.I.; Naveja, J.J.; Medina-Franco, J.L. Protein-Protein Interaction Modulators for Epigenetic Therapies. Adv. Protein Chem. Struct. Biol. 2018, 110, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Yun, J.S.; Song, H.; Kim, N.H.; Kim, H.S.; Yook, J.I. Exploring the chemical space of protein-protein interaction inhibitors through machine learning. Sci. Rep. 2021, 11, 13369. [Google Scholar] [CrossRef]
- Sperandio, O.; Reynès, C.H.; Camproux, A.-C.; Villoutreix, B.O. Rationalizing the chemical space of protein-protein interaction inhibitors. Drug Discov. Today 2010, 15, 220–229. [Google Scholar] [CrossRef]
- Bosica, F.; Andrei, S.A.; Neves, J.F.; Brandt, P.; Gunnarsson, A.; Landrieu, I.; Ottmann, C.; O’Mahony, G. Design of Drug-Like Protein-Protein Interaction Stabilizers Guided by Chelation-Controlled Bioactive Conformation Stabilization. Chem. Eur. J. 2020, 26, 7131–7139. [Google Scholar] [CrossRef]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef]
- A Study of Idasanutlin with Cytarabine Versus Cytarabine Plus Placebo in Participants with Relapsed or Refractory Acute Myeloid Leukemia (AML)—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02545283?term=idasanutlin&draw=2&rank=4 (accessed on 13 April 2021).
- Venetoclax DrugBank. Available online: https://go.drugbank.com/drugs/DB11581 (accessed on 4 February 2021).
- Higueruelo, A.P.; Jubb, H.; Blundell, T.L. TIMBAL v2: Update of a database holding small molecules modulating protein-protein interactions. Database 2013, 2013, bat039. [Google Scholar] [CrossRef]
- Labbé, C.M.; Kuenemann, M.A.; Zarzycka, B.; Vriend, G.; Nicolaes, G.A.F.; Lagorce, D.; Miteva, M.A.; Villoutreix, B.O.; Sperandio, O. iPPI-DB: An online database of modulators of protein-protein interactions. Nucleic Acids Res. 2016, 44, D542–D547. [Google Scholar] [CrossRef]
- Bosc, N.; Muller, C.; Hoffer, L.; Lagorce, D.; Bourg, S.; Derviaux, C.; Gourdel, M.-E.; Rain, J.-C.; Miller, T.W.; Villoutreix, B.O.; et al. Fr-PPIChem: An Academic Compound Library Dedicated to Protein-Protein Interactions. ACS Chem. Biol. 2020, 15, 1566–1574. [Google Scholar] [CrossRef]
- Cicaloni, V.; Trezza, A.; Pettini, F.; Spiga, O. Applications of in Silico Methods for Design and Development of Drugs Targeting Protein-Protein Interactions. Curr. Top. Med. Chem. 2019, 19, 534–554. [Google Scholar] [CrossRef]
- Mak, K.-K.; Pichika, M.R. Artificial intelligence in drug development: Present status and future prospects. Drug Discov. Today 2019, 24, 773–780. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing drug discovery via artificial intelligence. Trends Pharmacol. Sci. 2019, 40, 592–604. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Grys, B.T.; Lo, D.S.; Sahin, N.; Kraus, O.Z.; Morris, Q.; Boone, C.; Andrews, B.J. Machine learning and computer vision approaches for phenotypic profiling. J. Cell Biol. 2017, 216, 65–71. [Google Scholar] [CrossRef]
- Lee, J.-G.; Jun, S.; Cho, Y.-W.; Lee, H.; Kim, G.B.; Seo, J.B.; Kim, N. Deep learning in medical imaging: General overview. Korean J. Radiol. 2017, 18, 570–584. [Google Scholar] [CrossRef]
- Prieto-Martínez, F.D.; López-López, E.; Eurídice Juárez-Mercado, K.; Medina-Franco, J.L. Computational drug design methods—Current and future perspectives. In In Silico Drug Design; Elsevier: Amsterdam, The Netherlands, 2019; pp. 19–44. ISBN 9780128161258. [Google Scholar]
- Gastegger, M.; Marquetand, P. Molecular Dynamics with Neural Network Potentials. In Machine Learning Meets Quantum Physics; Schütt, K.T., Chmiela, S., von Lilienfeld, O.A., Tkatchenko, A., Tsuda, K., Müller, K.-R., Eds.; Lecture Notes in Physics; Springer International Publishing: Cham, Switzerland, 2020; Volume 968, pp. 233–252. ISBN 978-3-030-40244-0. [Google Scholar]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Alanis-Lobato, G.; Andrade-Navarro, M.A.; Schaefer, M.H. HIPPIE v2.0: Enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res. 2017, 45, D408–D414. [Google Scholar] [CrossRef]
- Protein-Protein Interaction Databases. Available online: https://openwetware.org/wiki/Protein-protein_interaction_databases (accessed on 5 February 2021).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- López-López, E.; Bajorath, J.; Medina-Franco, J.L. Informatics for chemistry, biology, and biomedical sciences. J. Chem. Inf. Model. 2021, 61, 26–35. [Google Scholar] [CrossRef]
- Géron, A. Hands-On Machine Learning with Scikit-Learn and TensorFlow: Concepts, Tools, and Techniques to Build Intelligent Systems (English Edition), 1st ed.; O’REILLY: Sebastopol, CA, USA, 2017; ISBN 9781492032649. [Google Scholar]
- Plisson, F.; Ramírez-Sánchez, O.; Martínez-Hernández, C. Machine learning-guided discovery and design of non-hemolytic peptides. Sci. Rep. 2020, 10, 16581. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pérez, R.; Vogt, M.; Bajorath, J. Support vector machine classification and regression prioritize different structural features for binary compound activity and potency value prediction. ACS Omega 2017, 2, 6371–6379. [Google Scholar] [CrossRef] [PubMed]
- Mughal, H.; Wang, H.; Zimmerman, M.; Paradis, M.D.; Freundlich, J.S. Random forest model prediction of compound oral exposure in the mouse. ACS Pharmacol. Transl. Sci. 2021, 4, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, F.G.; Arutanti, O.; Arif, A.F.; Hirano, T.; Ogi, T.; Okuyama, K. Correlations between Reduction Degree and Catalytic Properties of WO x Nanoparticles. ACS Omega 2018, 3, 8963–8970. [Google Scholar] [CrossRef]
- Vo, A.H.; Van Vleet, T.R.; Gupta, R.R.; Liguori, M.J.; Rao, M.S. An overview of machine learning and big data for drug toxicity evaluation. Chem. Res. Toxicol. 2020, 33, 20–37. [Google Scholar] [CrossRef]
- Hoffman, J.I.E. Logistic Regression. In Basic Biostatistics for Medical and Biomedical Practitioners; Elsevier: Amsterdam, The Netherlands, 2019; pp. 581–589. ISBN 9780128170847. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The Konstanz information miner. SIGKDD Explor. Newsl. 2009, 11, 26. [Google Scholar] [CrossRef]
- Zhang, C.; Ma, Y. Ensemble Machine Learning: Methods and Applications, 2012th ed.; Springer: New York, NY, USA, 2012; p. 340. ISBN 978-1441993250. [Google Scholar]



{kind=link}
{kind=link}
| ID | Accuracy Mean | Accuracy Std |
|---|---|---|
| RF27 | 0.957 | 0.014 |
| LRG22 | 0.941 | 0.017 |
| LRG24 | 0.941 | 0.017 |
| LRG27 | 0.941 | 0.017 |
| SVM22 | 0.958 | 0.015 |
| Ensemble1 | 0.940 | 0.013 |
| Ensemble2 | 0.956 | 0.010 |
| ID | Accuracy | Balanced Accuracy | Precision | Recall | F1 | Confusion Matrix |
|---|---|---|---|---|---|---|
| RF27 | 0.96 | 0.96 | 0.98 | 0.94 | 0.96 | [486 10] [32 401] |
| LRG 22 | 0.95 | 0.95 | 0.95 | 0.94 | 0.94 | [470 23] [27 406] |
| LRG24 | 0.95 | 0.95 | 0.95 | 0.94 | 0.94 | [470 23] [27 406] |
| LRG27 | 0.95 | 0.95 | 0.95 | 0.94 | 0.94 | [470 23] [26 407] |
| SVM22 | 0.95 | 0.95 | 0.98 | 0.92 | 0.95 | [484 9] [33 400] |
| Ensemble1 | 0.95 | 0.95 | 0.95 | 0.94 | 0.94 | [471 22] [27 406] |
| Ensemble2 | 0.95 | 0.95 | 0.98 | 0.91 | 0.94 | [486 7] [41 392] |
| Real | RF27 | RF22 | LRG24 | LRG27 | SVM22 | Ensemble1 | Ensemble2 | |
|---|---|---|---|---|---|---|---|---|
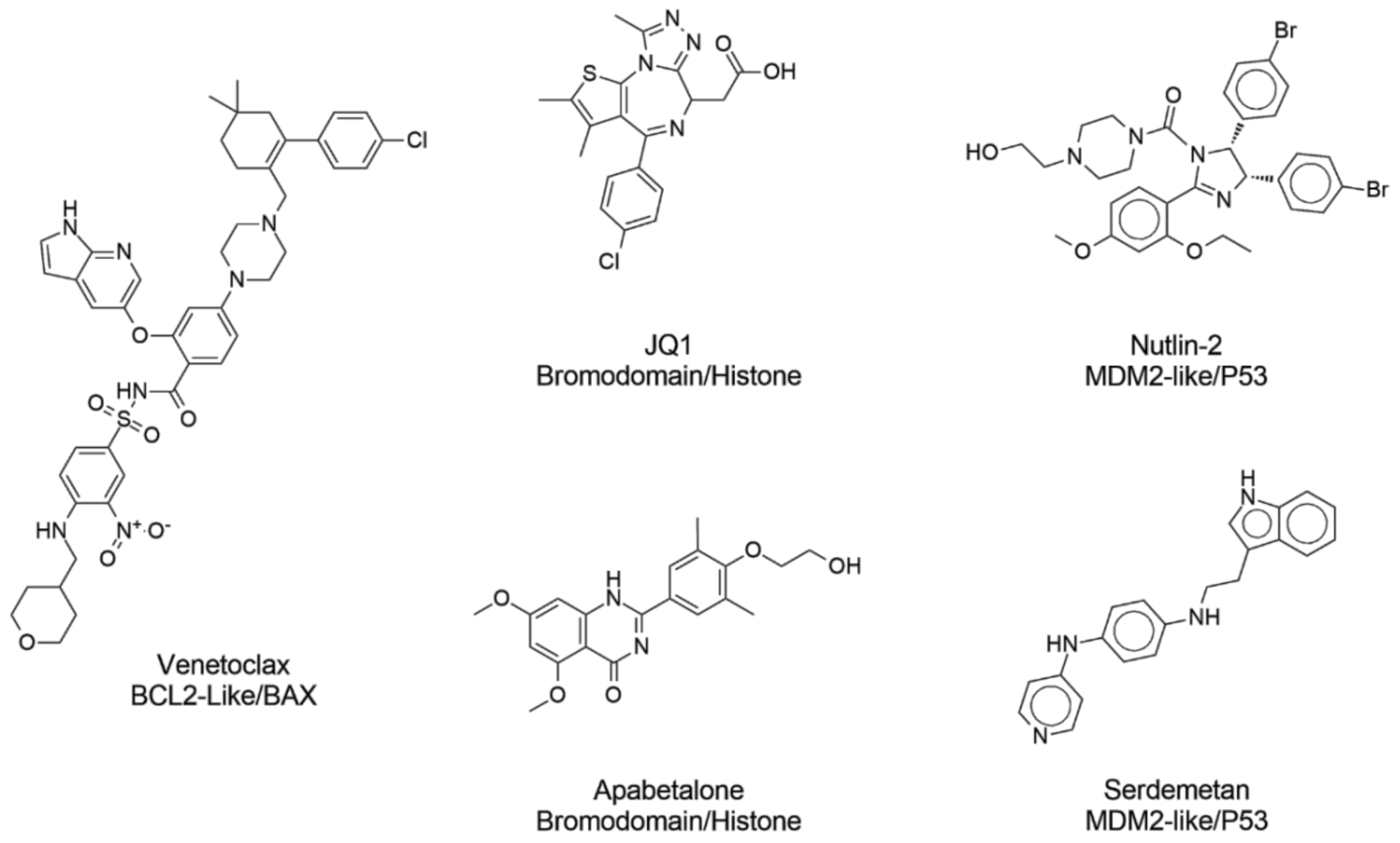
| Venetoclax | Active | Active | Inactive | Inactive | Inactive | Active | Inactive | Active |
| Apabetalone | Active | Inactive | Active | Active | Active | Active | Active | Inactive |
| Idasanutline | Active | Active | Active | Active | Active | Active | Active | Active |
| JQ1 | Active | Active | Active | Active | Active | Active | Active | Active |
| I-BET | Active | Active | Active | Active | Active | Active | Active | Active |
| Nutlin-2 | Active | Active | Active | Active | Active | Active | Active | Active |
| Atorvastatin | Inactive | Inactive | Inactive | Active | Active | Inactive | Active | Inactive |
| Amoxicilin | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| Albuterol | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| Metformin | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| Omeprazol | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
| Losartan | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive | Inactive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Eufracio, B.I.; Medina-Franco, J.L. Machine Learning Models to Predict Protein–Protein Interaction Inhibitors. Molecules 2022, 27, 7986. https://doi.org/10.3390/molecules27227986
Díaz-Eufracio BI, Medina-Franco JL. Machine Learning Models to Predict Protein–Protein Interaction Inhibitors. Molecules. 2022; 27(22):7986. https://doi.org/10.3390/molecules27227986
Chicago/Turabian StyleDíaz-Eufracio, Bárbara I., and José L. Medina-Franco. 2022. "Machine Learning Models to Predict Protein–Protein Interaction Inhibitors" Molecules 27, no. 22: 7986. https://doi.org/10.3390/molecules27227986
APA StyleDíaz-Eufracio, B. I., & Medina-Franco, J. L. (2022). Machine Learning Models to Predict Protein–Protein Interaction Inhibitors. Molecules, 27(22), 7986. https://doi.org/10.3390/molecules27227986