


Bridging the Gap in Malaria Parasite Resistance, Current Interventions, and the Way Forward from in Silico Perspective: A Review


Abstract
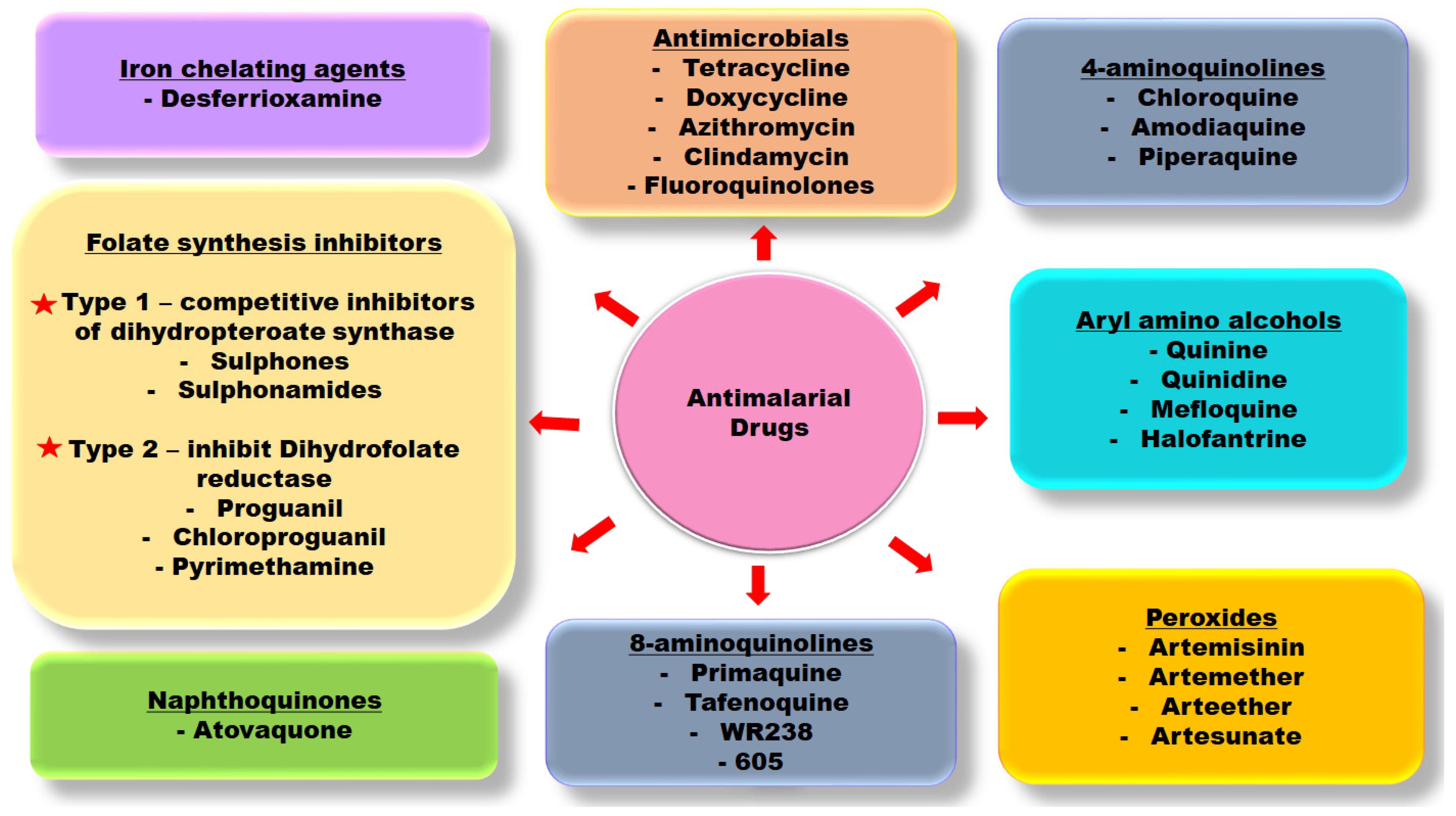
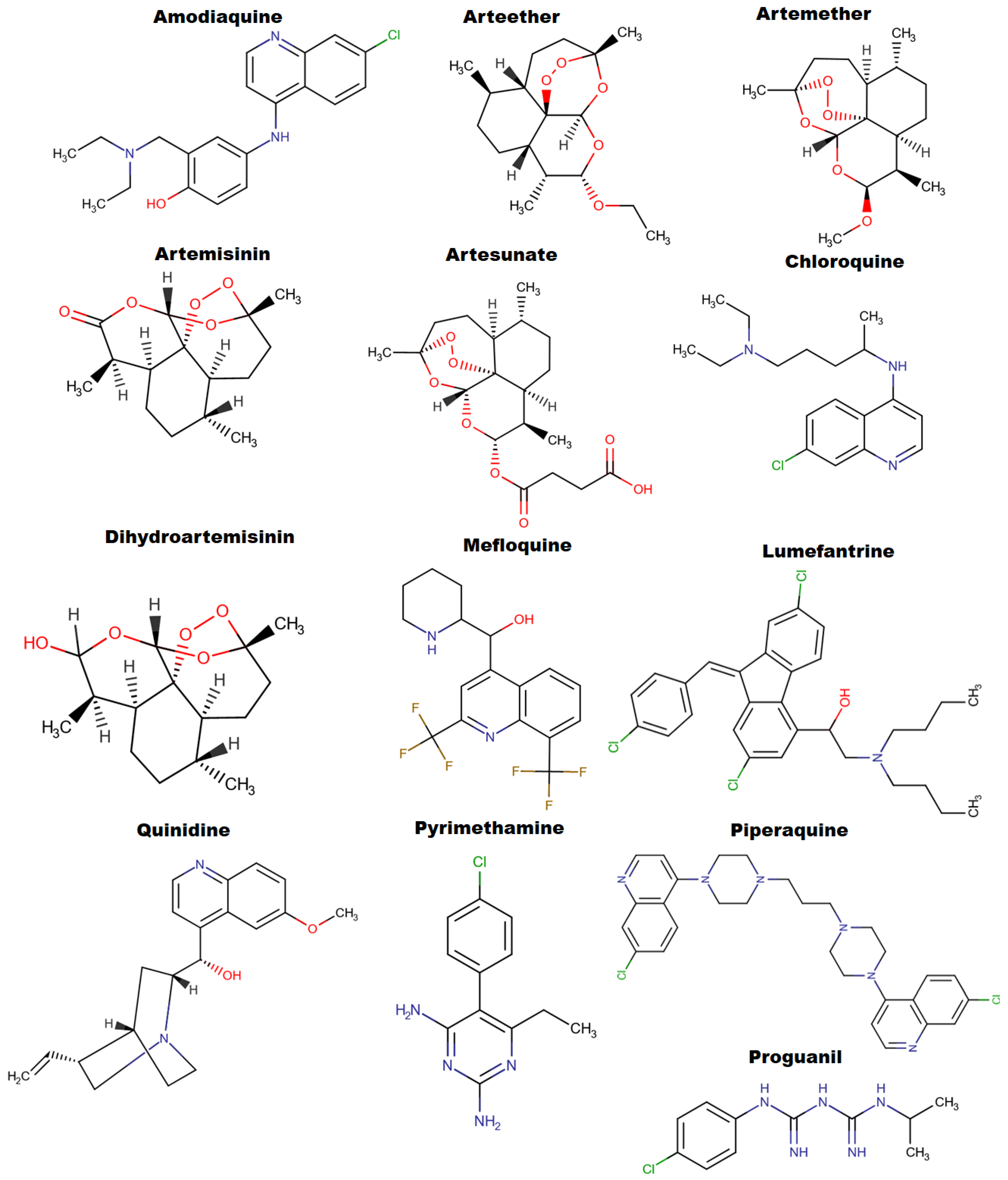
1. Introduction
1.1. Parasite Resistance to Antimalarial Drugs; Persistent Concern
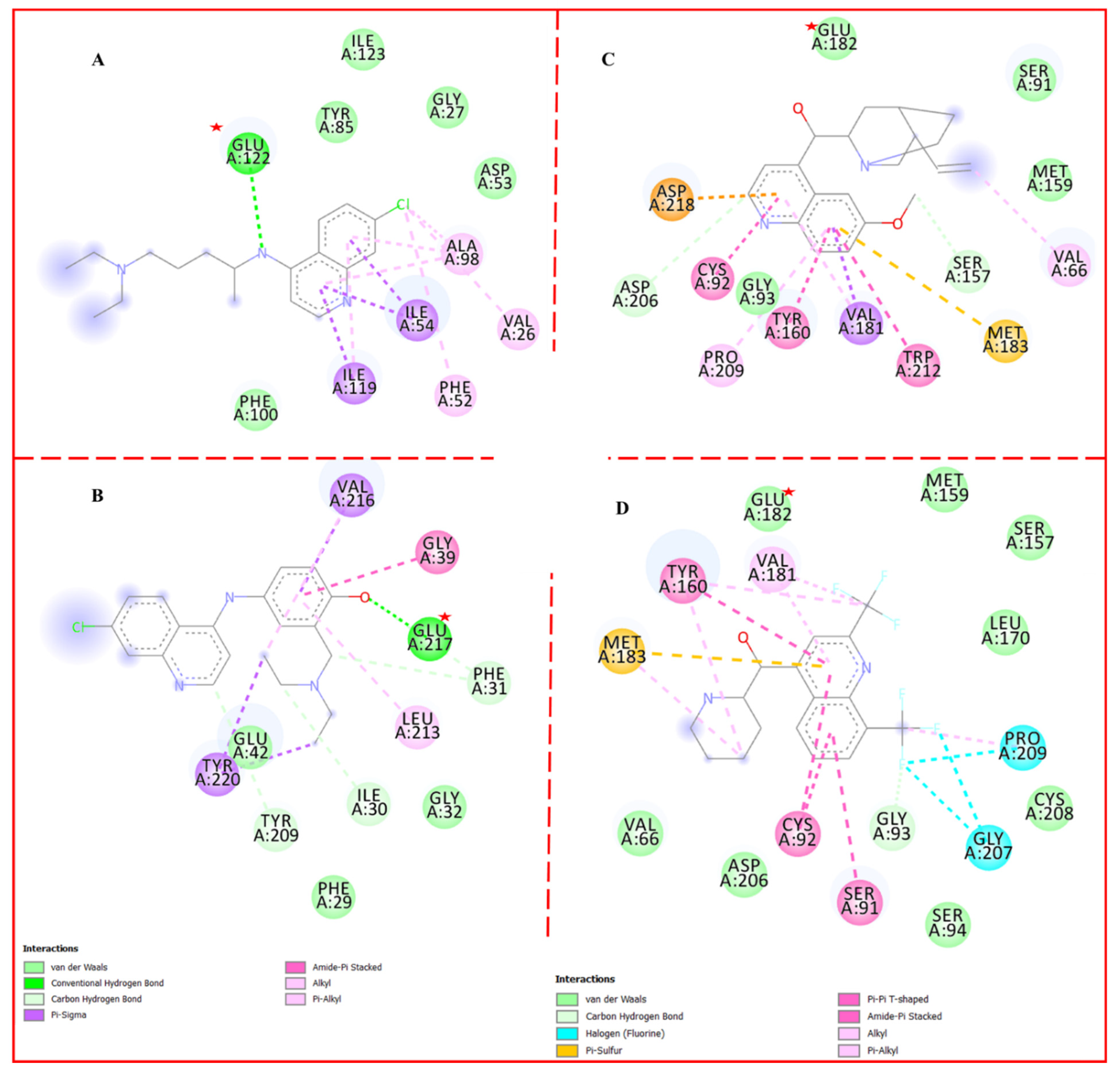
1.2. Binding of Mainstay Antimalarial Drugs Reveals Significant Interaction with Glutamic Acid Residue (Glu) in the Catalytic Domain
2. Computer-Aided Drug Design Approaches to Mitigate the Global Burden of Parasite Resistance
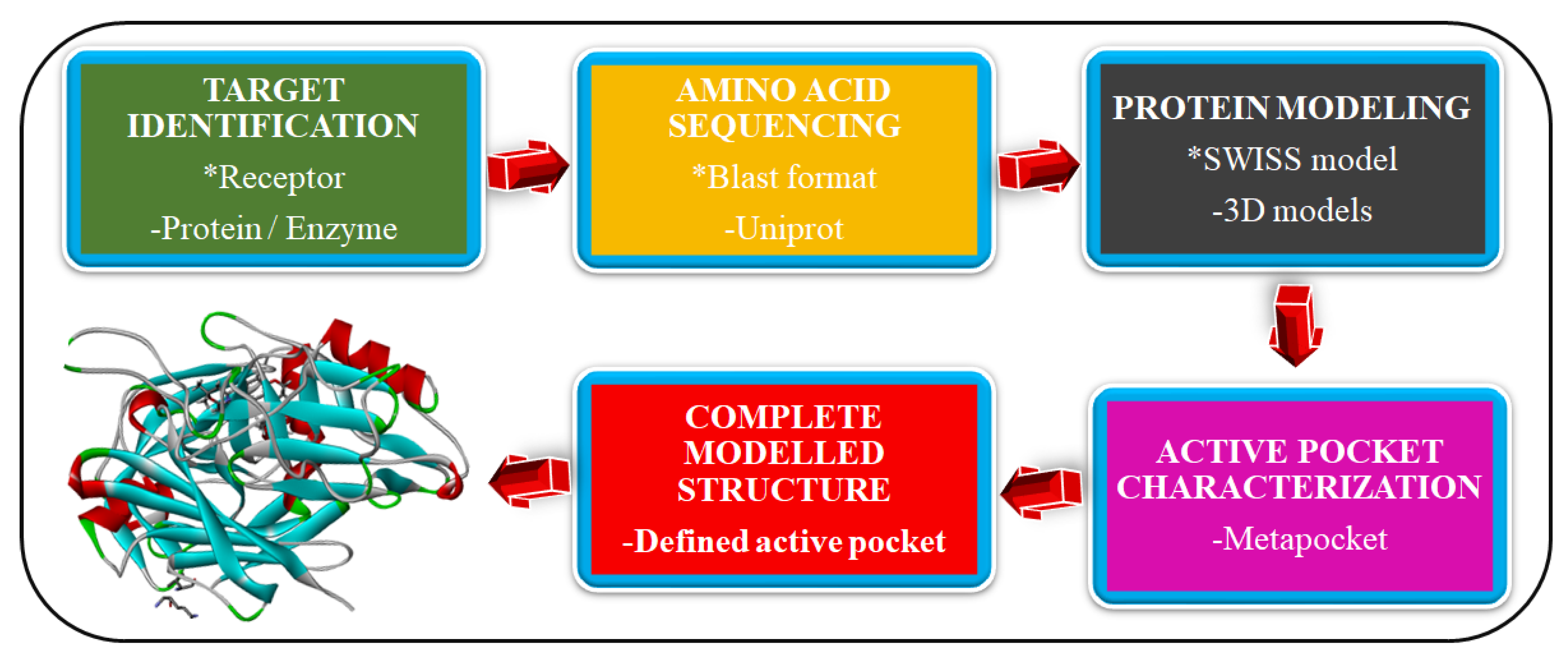
2.1. Homology Modeling of Novel Malaria Drug Targets
2.2. Structure–Activity Relationship (SAR)
2.3. Visual Screening for Antimalarial Compounds
2.4. Structure-Based Virtual Screening (SBVS)
2.5. Ligand-Based Virtual Screening (LBVS)
2.6. Pharmacokinetic Assessment of Antimalarial Compounds
2.7. Molecular Dynamic (MD) Simulation
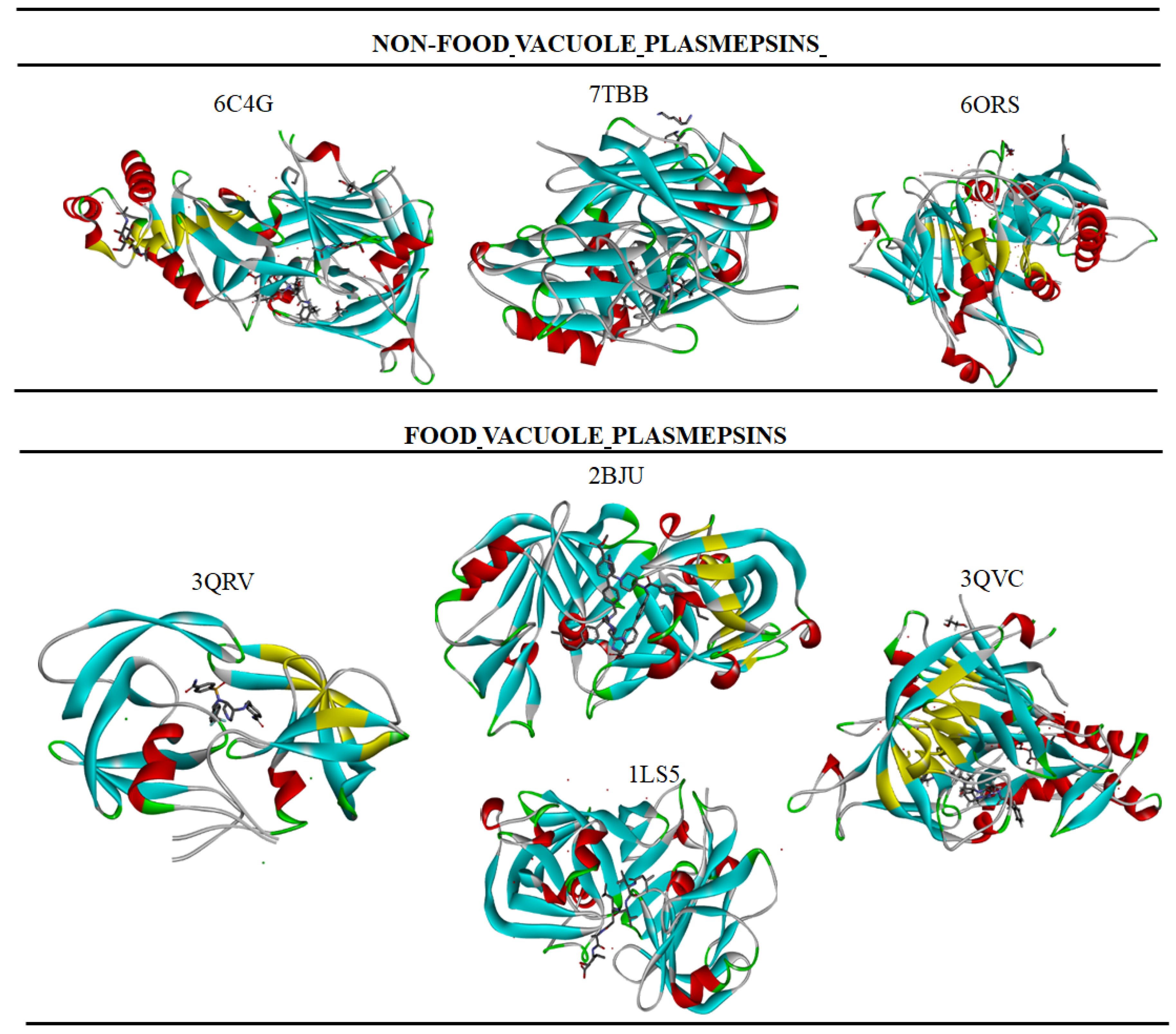
3. Aspartyl Acid Proteases (Plasmepsins) as Novel Drug Targets in Plasmodium falciparum
3.1. Food Vacuole Plasmepsin
3.2. Non-Food Vacuole Plasmepsin
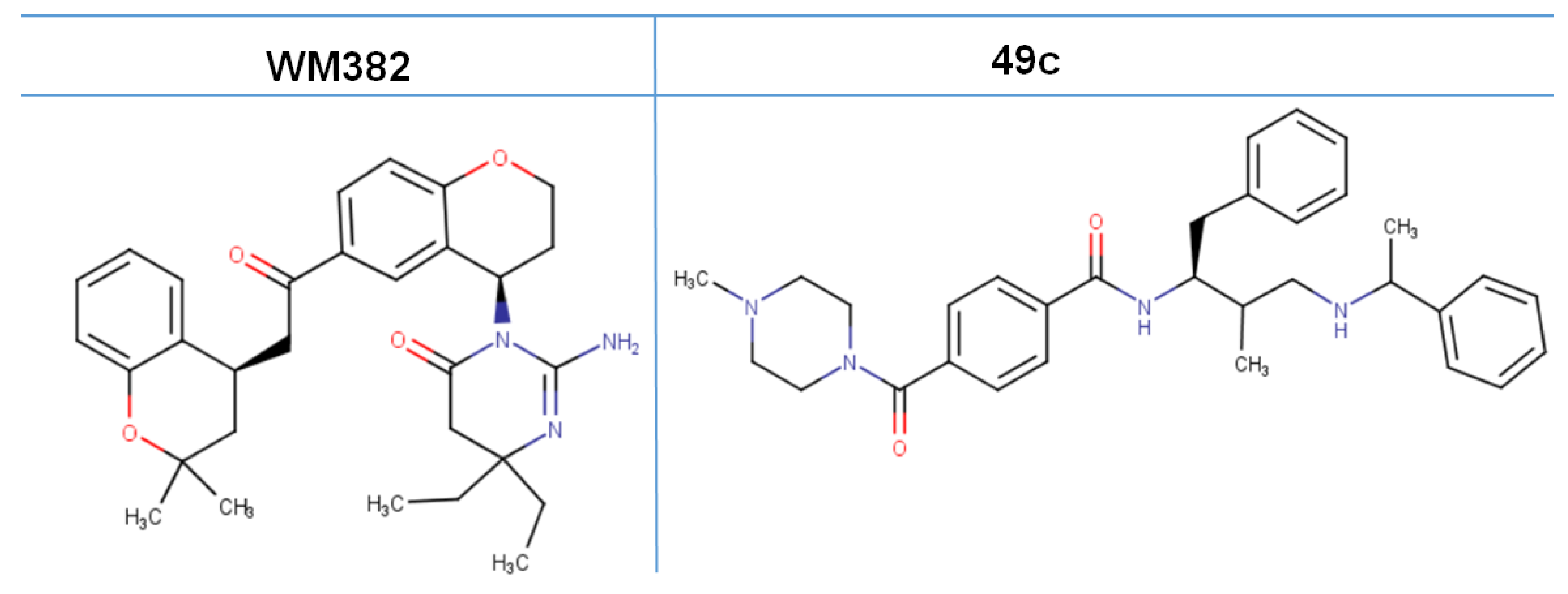
4. Advancement in the Search for Potent Plasmepsin Inhibitors
Limitations of Plasmepsin Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nair, D.N.; Singh, V. Structural Investigation and In-silico Characterization of Plasmepsins from Plasmodium falciparum. J. Proteom. Bioinform. 2016, 9, 181–195. [Google Scholar] [CrossRef]
- Talapko, J.; Škrlec, I.; Alebić, T.; Jukić, M.; Včev, A. Malaria: The Past and the Present. Microorganisms 2019, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, R.S.; Sajid, M.; Ling, I.T.; Tripathi, R.; Pachebat, J.A.; Holder, A.A. Characterization of N-myristoyltransferase from plasmodium falciparum. Biochem. J. 2000, 348, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Laurens, M.B. RTS,S/AS01 vaccine (MosquirixTM): An overview. Hum. Vaccin. Immunother. 2020, 16, 480–489. [Google Scholar] [CrossRef]
- Okello, D.; Kang, Y. Exploring Antimalarial Herbal Plants across Communities in Uganda Based on Electronic Data. Evid. -Based Complement. Altern. Med. 2019, 2019, 3057180. [Google Scholar] [CrossRef]
- Randrianarivelojosia, M.; Rasidimanana, V.T.; Rabarison, H.; Cheplogoi, P.K.; Ratsimbason, M.; Mulholland, D.A.; Mauclère, P. Plants traditionally prescribed to treat tazo (malaria) in the eastern region of Madagascar. Malar. J. 2003, 2, 1–9. [Google Scholar] [CrossRef]
- Ridley, R.G.; Hudson, A.T. Chemotherapy of malaria. Curr. Opin. Infect. Dis. 1998, 11, 691–705. [Google Scholar] [CrossRef]
- Noedl, H.; Se, Y.; Sriwichai, S.; Schaecher, K.; Teja-Isavadharm, P.; Smith, B.; Rutvisuttinunt, W.; Bethell, D.; Surasri, S.; Fukuda, M.M.; et al. Artemisinin Resistance in Cambodia: A Clinical Trial Designed to Address an Emerging Problem in Southeast Asia. Clin. Infect. Dis. 2010, 51, e82–e89. [Google Scholar] [CrossRef]
- Cravo, P.; Napolitano, H.; Culleton, R. How genomics is contributing to the fight against artemisinin-resistant malaria parasites. Acta Trop. 2015, 148, 1–7. [Google Scholar] [CrossRef]
- Blasco, B.; Leroy, D.; Fidock, D.A. Antimalarial drug resistance: Linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 2017, 23, 917–928. [Google Scholar] [CrossRef]
- Shibeshi, M.A.; Kifle, Z.D.; Atnafie, S.A. Resistance is the ability of a parasite strain to survive and/or multiply despite the proper administration and absorption of an antimalarial medicine in the dose normally recommended.21 Plasmodium vivax is continued to put a substantial burden on the mal. Infect. Drug Resist. 2020, 13, 4047–4060. [Google Scholar] [CrossRef] [PubMed]
- Chinappi, M.; Via, A.; Marcatili, P.; Tramontano, A. On the Mechanism of Chloroquine Resistance in Plasmodium falciparum. PLoS ONE 2010, 5, e14064. [Google Scholar] [CrossRef] [PubMed]
- Mita, T.; Tanabe, K. Evolution of Plasmodium falciparum drug resistance: Implications for the development and containment of artemisinin resistance. Jpn. J. Infect. Dis. 2012, 65, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Wongsrichanalai, C.; Nelson, A.; Wernsdorfer, W.; Meshnick, S. Epidemiology of drug-resistant malaria. Lancet Infect. Dis. 2002, 2, 209–218. [Google Scholar] [CrossRef]
- Weiss, H.F. The Democratic Republic of the Congo. Responsib. Prot. 2009, 13, 115–128. [Google Scholar] [CrossRef]
- Brunner, R.; Aissaoui, H.; Boss, C.; Bozdech, Z.; Brun, R.; Corminboeuf, O.; Delahaye, S.; Fischli, C.; Heidmann, B.; Kaiser, M.; et al. Identification of a new chemical class of antimalarials. J. Infect. Dis. 2012, 206, 735–743. [Google Scholar] [CrossRef]
- Kumi, R.O.; Issahaku, A.R.; Soremekun, O.S.; Agoni, C.; Olotu, F.A.; Soliman, M.E.S. From the Explored to the Unexplored: Computer-Tailored Drug Design Attempts in the Discovery of Selective Caspase Inhibitors. Comb. Chem. High Throughput Screen. 2019, 22, 432–444. [Google Scholar] [CrossRef]
- Munsamy, G.; Agoni, C.; Soliman, M.E.S. A dual target of Plasmepsin IX and X: Unveiling the atomistic superiority of a core chemical scaffold in malaria therapy. J. Cell. Biochem. 2019, 120, 7876–7887. [Google Scholar] [CrossRef]
- Munsamy, G.; Soliman, M.E.S. Unveiling a New Era in Malaria Therapeutics: A Tailored Molecular Approach Towards the Design of Plasmepsin IX Inhibitors. Protein J. 2019, 38, 616–627. [Google Scholar] [CrossRef]
- Kumi, R.O.; Agoni, C.; Ibrahim, M.A.A.; Soliman, M.E.S. Dual enzymatic inhibitory mechanism of WM382 on plasmepsin IX and X: Atomistic perspectives from dynamic analysis. Inform. Med. Unlocked 2022, 29, 100874. [Google Scholar] [CrossRef]
- Kumi, R.O.; Soremekun, O.S.; Issahaku, A.R.; Agoni, C.; Olotu, F.A.; Soliman, M.E.S. Exploring the ring potential of 2,4-diaminopyrimidine derivatives towards the identification of novel caspase-1 inhibitors in Alzheimer’s disease therapy. J. Mol. Model. 2020, 26, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Avery, M.A.; Muraleedharan, K.M.; Desai, P.V.; Bandyopadhyaya, A.K.; Furtado, M.M.; Tekwani, B.L. Structure-activity relationships of the antimalarial agent artemisinin. 8. design, synthesis, and CoMFA studies toward the development of artemisinin-based drugs against leishmaniasis and malaria. J. Med. Chem. 2003, 46, 4244–4258. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, N.; de Julian-Ortiz, J.V.; Ciceron, L.; Galvez, J.; Mazier, D.; Danis, M.; Derouin, F.; García-Domenech, R. Identification of new antimalarial drugs by linear discriminant analysis and topological virtual screenin. J. Antimicrob. Chemother. 2006, 57, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Krajačić, M.B.; Perić, M.; Smith, K.S.; Ivezić Schönfeld, Z.; Žiher, D.; Fajdetić, A.; Kujundžić, N.; Schönfeld, W.; Landek, G.; Padovan, J.; et al. Synthesis, Structure–Activity Relationship, and Antimalarial Activity of Ureas and Thioureas of 15-Membered Azalides. J. Med. Chem. 2011, 54, 3595–3605. [Google Scholar] [CrossRef] [PubMed]
- Waszkowycz, B.; Perkins, T.D.J.; Sykes, R.A.; Li, J. Large-scale virtual screening for discovering leads in the postgenomic era. IBM Syst. J. 2001, 40, 360–376. [Google Scholar] [CrossRef]
- Seifert, E. OriginPro 9.1: Scientific data analysis and graphing software—Software review. J. Chem. Inf. Model 2014, 54, 1552. [Google Scholar] [CrossRef]
- Nasamu, A.S.; Glushakova, S.; Russo, I.; Vaupel, B.; Oksman, A.; Kim, A.S.; Fremont, D.H.; Tolia, N.; Beck, J.R.; Meyers, M.J.; et al. Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science 2017, 27, 518–522. [Google Scholar] [CrossRef]
- Li, F.; Bounkeua, V.; Pettersen, K.; Vinetz, J.M. Plasmodium falciparum ookinete expression of plasmepsin VII and plasmepsin X. Malar. J. 2016, 15, 111. [Google Scholar] [CrossRef]
- Dash, C.; Kulkarni, A.; Dunn, B.; Rao, M. Aspartic peptidase inhibitors: Implications in drug development. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 89–119. [Google Scholar] [CrossRef]
- Favuzza, P.; de Lera Ruiz, M.; Thompson, J.K.; Triglia, T.; Ngo, A.; Steel, R.W.; Vavrek, M.; Christensen, J.; Healer, J.; Boyce, C.; et al. Dual Plasmepsin-Targeting Antimalarial Agents Disrupt Multiple Stages of the Malaria Parasite Life Cycle. Cell Host Microbe 2020, 27, 642–658.e12. [Google Scholar] [CrossRef]
- Banerjee, R.; Liu, J.; Beatty, W.; Pelosof, L.; Klemba, M.; Goldberg, D.E. Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc. Natl. Acad. Sci. USA 2002, 99, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Moura, P.A.; Dame, J.B.; Fidock, D.A. Role of Plasmodium falciparum digestive vacuole plasmepsins in the specificity and antimalarial mode of action of cysteine and aspartic protease inhibitors. Antimicrob. Agents Chemother. 2009, 53, 4968–4978. [Google Scholar] [CrossRef] [PubMed]
- Nasamu, A.S.; Polino, A.J.; Istvan, E.S.; Goldberg, D.E. Malaria parasite plasmepsins: More than just plain old degradative pepsins. J. Biol. Chem. 2020, 295, 8425–8441. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, M.J.; Moon, R.P.; Klinder, A.; Fowler, S.D.; Rupp, K.; Bur, D.; Ridley, R.G.; Berry, C. The aspartic proteinase from the rodent parasite Plasmodium berghei as a potential model for plasmepsins from the human malaria parasite, Plasmodium falciparum. FEBS Lett. 1999, 463, 43–48. [Google Scholar] [CrossRef]
- Bhaumik, P.; Horimoto, Y.; Xiao, H.; Miura, T.; Hidaka, K.; Kiso, Y.; Wlodawer, A.; Yada, R.Y.; Gustchina, A. Crystal structures of the free and inhibited forms of plasmepsin I (PMI) from Plasmodium falciparum. J. Struct. Biol. 2011, 175, 73–84. [Google Scholar] [CrossRef]
- Cheuka, P.M.; Dziwornu, G.; Okombo, J.; Chibale, K. Plasmepsin Inhibitors in Antimalarial Drug Discovery: Medicinal Chemistry and Target Validation (2000 to Present). J. Med. Chem. 2020, 63, 4445–4467. [Google Scholar] [CrossRef]
- Grädler, U.; Czodrowski, P.; Tsaklakidis, C.; Klein, M.; Werkmann, D.; Lindemann, S.; Maskos, K.; Leuthner, B. Struc-ture-based optimization of non-peptidic Cathepsin D inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4141–4150. [Google Scholar] [CrossRef]
- Meissner, K.A.; Kronenberger, T.; Maltarollo, V.G.; Trossini, G.H.G.; Wrenger, C. Targeting the Plasmodium falciparum plasmepsin V by ligand-based virtual screening. Chem. Biol. Drug Des. 2019, 93, 300–312. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimalarial | Classification | Mode of Action |
|---|---|---|
| Artemisinin and derivatives; Dihydroartemisinin, Arteether, Artesunate, Artemether | Gametocytocidal | Involves the heme-mediated decomposition of the peroxide bridge to produce carbon-centered free radicals |
| Chloroquine | Blood schizonticidal | Chloroquine accumulates in the acidic food vacuole of intraerythrocytic trophozoites and thereby prevents hemoglobin degradation |
| Piperaquine | Blood schizonticidal | Accumulate in the parasite digestive vacuole and interfere with the detoxification of heme into hemozoin |
| Lumefantrine | Blood schizonticidal | Lumefantrine is believed to inhibit nucleic acid and the formation of beta-hematin by forming a complex with hemin |
| Curcumin | Blood schizonticidal | Curcumin inhibits the activity of enzyme and lipid peroxides |
| Protein | Function | Location | Principal Drugs Affected |
|---|---|---|---|
| chloroquine resistance transporter (CRT) | Transporter | Membrane of food vacuole | Chloroquine Mefloquine, halofantrine, lumefantrine, artemisinin, quinine |
| Pgh1 (P-glycoprotein homologue 1) or MDR1 (multidrug resistance 1) | Transporter | Membrane of food vacuole | Mefloquine, halofantrine, lumefantrine, quinine |
| Minor determinant | |||
| Dihydrofolate synthase (DHPS) | Folate pathway enzyme | Cytoplasm (principally) | Sulfadoxine, dapsone |
| Dihydrofolate reductase (DHFR) | Folate pathway enzyme | Cytoplasm (principally) | Pyrimethamine, proguanil, chlorproguanil |
| Cytochrome b | Subunit of complex III (cytochrome bc1 complex) electron transport chain | Mitochondrion | Atovaquone |
| ATP6 (sarco/endoplasmic reticulum calcium-dependent ATPase [SERCA] orthologue) | Membrane-bound Ca2+ -transporting ATPase | Membranous structures within cytoplasm | Artemisinin |
| Kelch13 | Endocytosis pathway protein | Cytoplasm | Artemisinin |
| Class of Plasmepsin | Function |
|---|---|
| Food vacuole plasmepsin | PMI Hemoglobin digestion and degradation |
| PMII Hemoglobin digestion and degradation. Cytoskeletal protein processing and host cell remodeling | |
| Histo-aspartic protease (HAP) An active protease with a novel catalytic mechanism in hemoglobin degradation | |
| PMIV Hemoglobin digestion and degradation. Cytoskeletal protein processing and host cell remodeling | |
| Non-food vacuole plasmepsin | PMV Responsible for the export of effector proteins mediated to the host cell. |
| PMVI-VIII Expressed in the vector during the parasite’s intra-erythrocytic stages of motility, formation of sporozoites, and midgut transversal and hence are not directly involved in the transmission of the malarial parasite within the human host | |
| PMIX and PMIX Egress, invasion, and spread of malaria parasite |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumi, R.O.; Oti, B.; Abo-Dya, N.E.; Alahmdi, M.I.; Soliman, M.E.S. Bridging the Gap in Malaria Parasite Resistance, Current Interventions, and the Way Forward from in Silico Perspective: A Review. Molecules 2022, 27, 7915. https://doi.org/10.3390/molecules27227915
Kumi RO, Oti B, Abo-Dya NE, Alahmdi MI, Soliman MES. Bridging the Gap in Malaria Parasite Resistance, Current Interventions, and the Way Forward from in Silico Perspective: A Review. Molecules. 2022; 27(22):7915. https://doi.org/10.3390/molecules27227915
Chicago/Turabian StyleKumi, Ransford Oduro, Belinda Oti, Nader E. Abo-Dya, Mohamed Issa Alahmdi, and Mahmoud E. S. Soliman. 2022. "Bridging the Gap in Malaria Parasite Resistance, Current Interventions, and the Way Forward from in Silico Perspective: A Review" Molecules 27, no. 22: 7915. https://doi.org/10.3390/molecules27227915
APA StyleKumi, R. O., Oti, B., Abo-Dya, N. E., Alahmdi, M. I., & Soliman, M. E. S. (2022). Bridging the Gap in Malaria Parasite Resistance, Current Interventions, and the Way Forward from in Silico Perspective: A Review. Molecules, 27(22), 7915. https://doi.org/10.3390/molecules27227915