

Lipase-Catalyzed Phospha-Michael Addition Reactions under Mild Conditions

 and
and 
Abstract
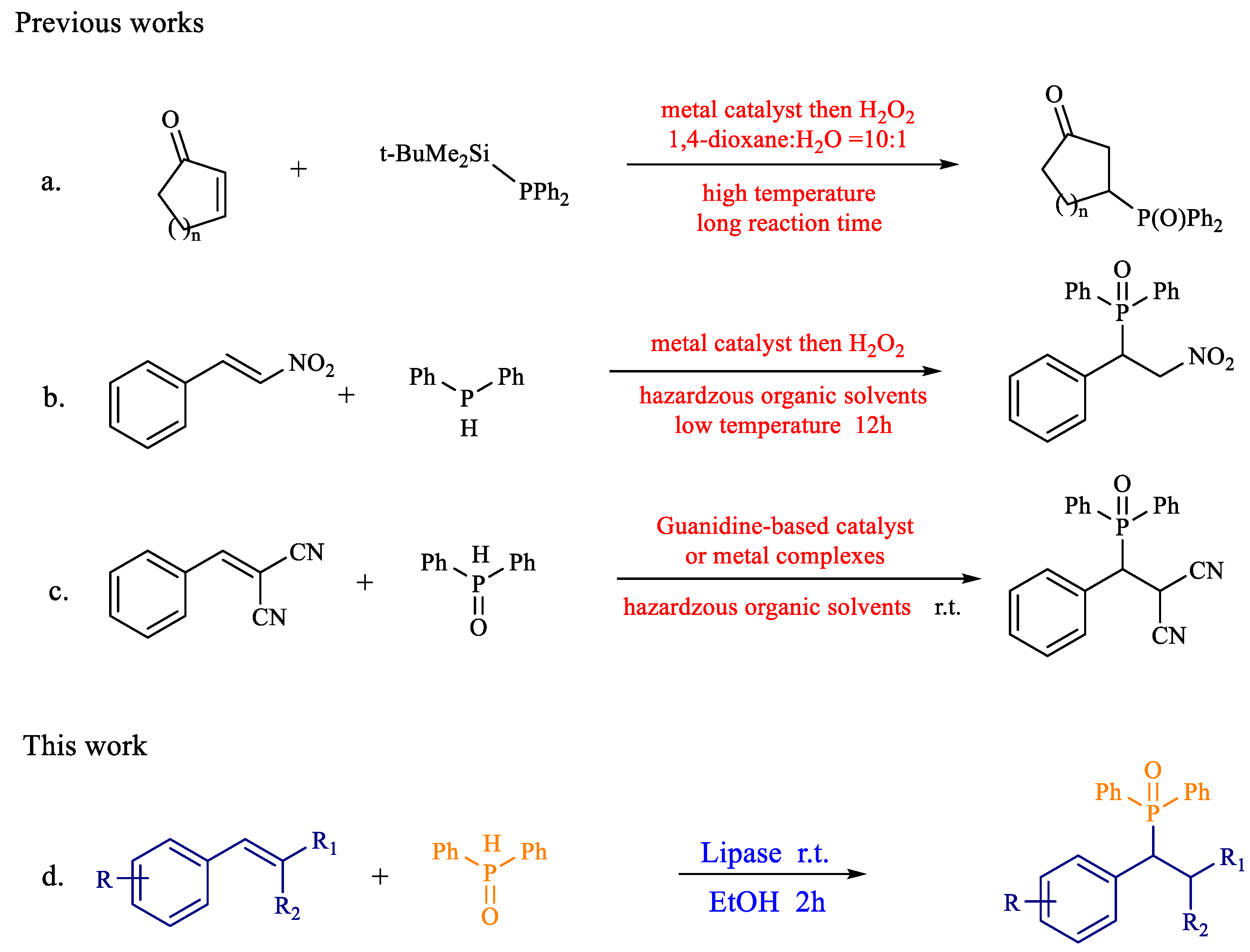
1. Introduction
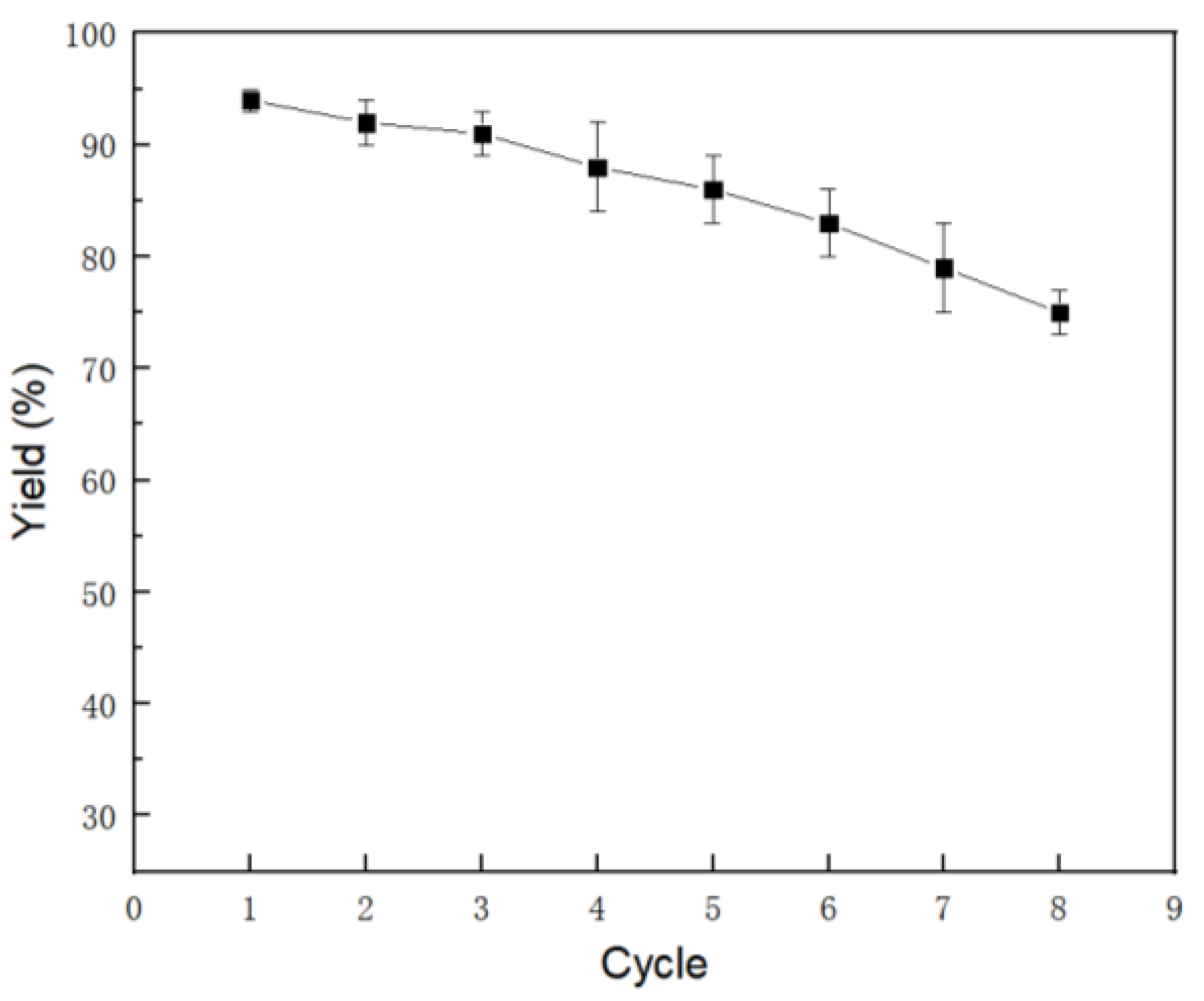
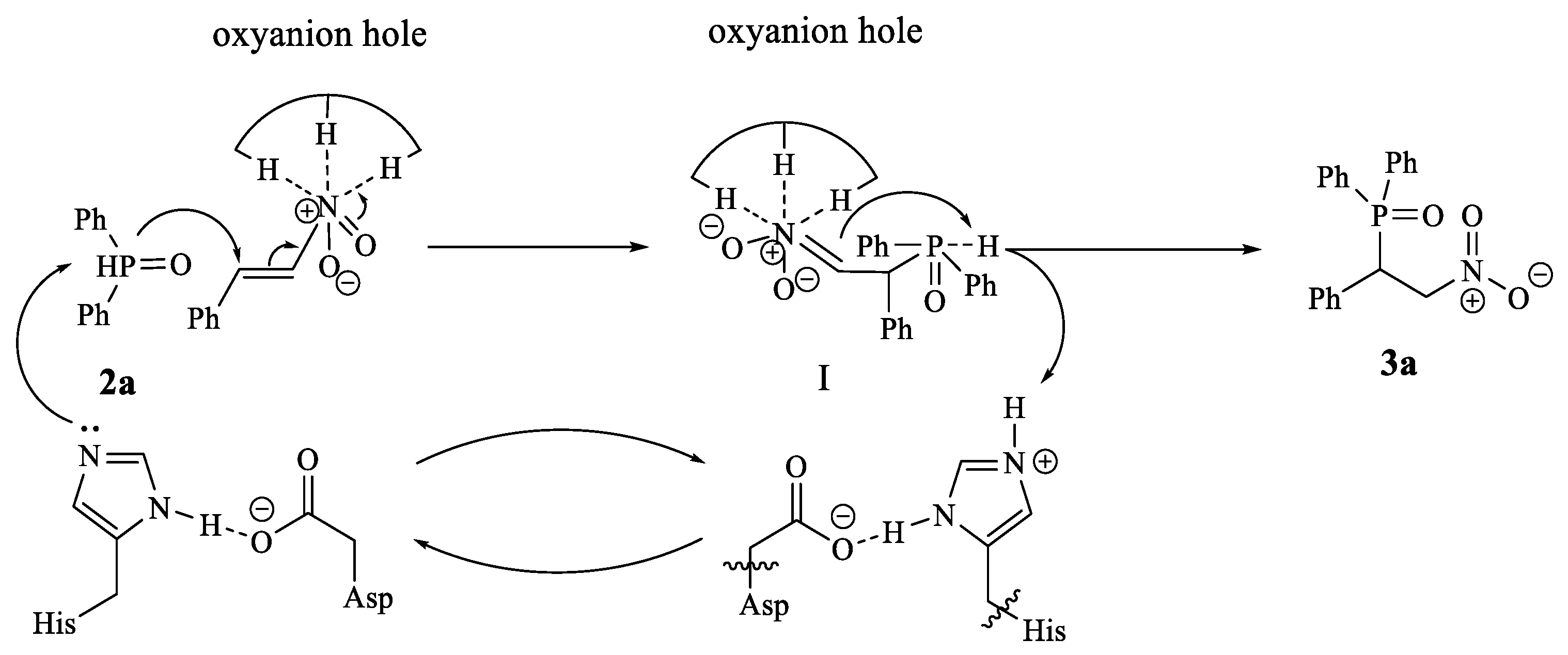
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of 3
3.3. General Procedure for the Synthesis of 5
3.4. Synthetic Procedures of Benzylidene Malono-Nitrile 4
3.5. The Denaturation of Novozym 435 Treated by PMSF
3.5.1. 3a ((2-nitro-1-phenylethyl) diphenylphosphine oxide)
3.5.2. 3b ((2-nitro-1-(p-tolyl) ethyl) diphenylphosphine oxide)
3.5.3. 3c ((1-(4-methoxyphenyl)-2-nitroethyl) diphenylphosphine oxide)
3.5.4. 3d ((1-(4-isopropylphenyl)-2-nitroethyl) diphenylphosphine oxide)
3.5.5. 3e ((1-(4-fluorophenyl)-2-nitroethyl) diphenylphosphine oxide)
3.5.6. 3f ((1-(4-chlorophenyl)-2-nitroethyl) diphenylphosphine oxide)
3.5.7. 3g ((1-(4-bromophenyl)-2-nitroethyl) diphenylphosphine oxide)
3.5.8. 3h ((1-(2-chlorophenyl)-2-nitroethyl) diphenylphosphine oxide)
3.5.9. 3i ((1-(3-chlorophenyl)-2-nitroethyl) diphenylphosphine oxide)
3.5.10. 3j (1-(3,4-dimethoxyphenyl)-2-nitroethyl) diphenylphosphine oxide
3.5.11. 5a (2-((diphenylphosphoryl)(phenyl)methyl) malononitrile)
3.5.12. 5b (2-((2-chlorophenyl) (diphenylphosphoryl)methyl) malononitrile)
3.5.13. 5c (2-((3-chlorophenyl) (diphenylphosphoryl)methyl) malononitrile)
3.5.14. 5d (2-((diphenylphosphoryl)(3-nitrophenyl) methyl) malononitrile)
3.5.15. 5e (2-((diphenylphosphoryl)(m-tolyl) methyl) malononitrile)
3.5.16. 5f (2-((diphenylphosphoryl)(3-methoxyphenyl) methyl) malononitrile)
3.5.17. 5g 2-((diphenylphosphoryl)(4-fluorophenyl) methyl) malononitrile
3.5.18. 5h 2-((4-bromophenyl) (diphenylphosphoryl)methyl) malononitrile
3.5.19. 5i (2-((3,4-dimethoxyphenyl) (diphenylphosphoryl)methyl) malononitrile)
3.5.20. 5j (2-((diphenylphosphoryl)(naphthalen-2-yl) methyl) malononitrile)
3.5.21. 5k (2-((diphenylphosphoryl)(thiophen-2-yl) methyl) malononitrile)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yu, H.; Yang, H.; Shi, E.; Tang, W. Development and Clinical Application of Phosphorus-Containing Drugs. Med. Drug Discov. 2020, 8, 100063. [Google Scholar] [CrossRef] [PubMed]
- Shevchuk, M.; Wang, Q.; Pajkert, R.; Xu, J.; Mei, H.; Röschenthaler, G.-V.; Han, J. Recent Advances in Synthesis of Difluoromethylene Phosphonates for Biological Applications. Adv. Synth. Catal. 2021, 363, 2912–2968. [Google Scholar] [CrossRef]
- Fields, S.C. Synthesis of natural products containing a C-P bond. Tetrahedron 1999, 55, 12237–12273. [Google Scholar] [CrossRef]
- Walsh, C.T.; Tu, B.P.; Tang, Y. Eight Kinetically Stable but Thermodynamically Activated Molecules that Power Cell Metabolism. Chem. Rev. 2018, 118, 1460–1494. [Google Scholar] [CrossRef]
- Fernández-García, C.; Coggins, A.J.; Powner, M.W. A Chemist’s Perspective on the Role of Phosphorus at the Origins of Life. Life 2017, 7, 31. [Google Scholar] [CrossRef]
- Miller, R.C.; Bradley, J.S.; Hamilton, L.A. Disubstituted Phosphine Oxides. III. Addition to α,β-Unsaturated Nitriles and Carbonyl Compounds1. J. Am. Chem. Soc. 1956, 78, 5299–5303. [Google Scholar] [CrossRef]
- Bodalski, R.; Pietrusiewicz, K. A new route to the phospholane ring-system. Tetrahedron Lett. 1972, 13, 4209–4212. [Google Scholar] [CrossRef]
- Enders, D.; Mirjafary, Z.; Saeidian, H. Efficient diastereo- and enantioselective synthesis of α,β-disubstituted γ-phosphono sulfonates. Tetrahedron Asymmetry 2009, 20, 2429–2431. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W. Recent advances in organocatalytic asymmetric Michael reactions. Catal. Sci. Technol. 2012, 2, 42–53. [Google Scholar] [CrossRef]
- Strappaveccia, G.; Bianchi, L.; Ziarelli, S.; Santoro, S.; Lanari, D.; Pizzo, F.; Vaccaro, L. PS-BEMP as a basic catalyst for the phospha-Michael addition to electron-poor alkenes. Org. Biomol. Chem. 2016, 14, 3521–3525. [Google Scholar] [CrossRef]
- Lenz, J.; Pospiech, D.; Komber, H.; Paven, M.; Albach, R.; Mentizi, S.; Langstein, G.; Voit, B. Synthesis of the H-phosphonate dibenzo[d,f][1,3,2]dioxaphosphepine 6-oxide and the phospha-Michael addition to unsaturated compounds. Tetrahedron 2019, 75, 1306–1310. [Google Scholar] [CrossRef]
- Tessema, E.; Elakkat, V.; Chiu, C.-F.; Zheng, J.-H.; Chan, K.L.; Shen, C.-R.; Zhang, P.; Lu, N. Recoverable Phospha-Michael Additions Catalyzed by a 4-N,N-Dimethylaminopyridinium Saccharinate Salt or a Fluorous Long-Chained Pyridine: Two Types of Reusable Base Catalysts. Molecules 2021, 26, 1159. [Google Scholar] [CrossRef] [PubMed]
- Trepohl, V.T.; Mori, S.; Itami, K.; Oestreich, M. Palladium(II)-Catalyzed Conjugate Phosphination of Electron-Deficient Acceptors. Org. Lett. 2009, 11, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Trepohl, V.T.; Fröhlich, R.; Oestreich, M. Conjugate phosphination of cyclic and acyclic acceptors using Rh(I)–phosphine or Rh(I)–carbene complexes. Probing the mechanism with chirality at the silicon atom or the phosphorus atom of the Si–P reagent. Tetrahedron 2009, 65, 6510–6518. [Google Scholar] [CrossRef]
- Yan, J.; Wang, Y.-B.; Hou, S.; Shi, L.; Zhu, X.; Hao, X.-Q.; Song, M.-P. NCC Pincer Ni (II) Complexes Catalyzed Hydrophosphination of Nitroalkenes with Diphenylphosphine. Appl. Organomet. Chem. 2020, 34, e5954. [Google Scholar] [CrossRef]
- Hao, X.-Q.; Huang, J.-J.; Wang, T.; Lv, J.; Gong, J.-F.; Song, M.-P. PCN Pincer Palladium(II) Complex Catalyzed Enantioselective Hydrophosphination of Enones: Synthesis of Pyridine-Functionalized Chiral Phosphine Oxides as NCsp3O Pincer Preligands. J. Org. Chem 2014, 79, 9512–9530. [Google Scholar] [CrossRef]
- Miao, H.; Wang, S.; Zhu, X.; Zhou, S.; Wei, Y.; Yuan, Q.; Mu, X. Synthesis, characterization and catalytic activity of rare-earth metal amides incorporating cyclohexyl bridged bis(β-diketiminato) ligands. RSC Adv. 2017, 7, 42792–42799. [Google Scholar] [CrossRef]
- Ding, B.; Zhang, Z.; Xu, Y.; Liu, Y.; Sugiya, M.; Imamoto, T.; Zhang, W. P-Stereogenic PCP Pincer–Pd Complexes: Synthesis and Application in Asymmetric Addition of Diarylphosphines to Nitroalkenes. Org. Lett. 2013, 15, 5476–5479. [Google Scholar] [CrossRef]
- Feng, J.-J.; Huang, M.; Lin, Z.-Q.; Duan, W.-L. Palladium-Catalyzed Asymmetric 1,4-Addition of Diarylphosphines to Nitroalkenes for the Synthesis of Chiral P,N-Compounds. Adv. Synth. Catal. 2012, 354, 3122–3126. [Google Scholar] [CrossRef]
- Wani, A.A.; Chourasiya, S.S.; Kathuria, D.; Bharatam, P.V. 1,1-Diaminoazines as organocatalysts in phospha-Michael addition reactions. Chem. Commun. 2021, 57, 11717–11720. [Google Scholar] [CrossRef]
- Fu, X.; Jiang, Z.; Tan, C.-H. Bicyclic guanidine-catalyzed enantioselective phospha-Michael reaction: Synthesis of chiral β-aminophosphine oxides and β-aminophosphines. Chem. Commun. 2007, 47, 5058–5060. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhang, Y.; Ye, W.; Tan, C.-H. P–C Bond formation via direct and three-component conjugate addition catalyzed by 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). Tetrahedron Lett. 2007, 48, 51–54. [Google Scholar] [CrossRef]
- Xu, Y.; Li, F.; Zhao, N.; Su, J.; Wang, C.; Wang, C.; Li, Z.; Wang, L. Environment-friendly and efficient synthesis of 2-aminobenzo-xazoles and 2-aminobenzothiazoles catalyzed by Vitreoscilla hemoglobin incorporating a cobalt porphyrin cofactor. Green Chem. 2021, 23, 8047–8052. [Google Scholar] [CrossRef]
- Li, F.; Wang, C.; Xu, Y.; Gao, X.; Xu, Y.; Xie, H.; Chen, P.; Wang, L. Lipase-Catalyzed Synthesis of Anthrone Functionalized Benzylic Amines via a Multicomponent Reaction in Supercritical Carbon Dioxide. ChemistrySelect 2022, 7, e202104517. [Google Scholar] [CrossRef]
- Hanefeld, U.; Hollmann, F.; Paul, C.E. Biocatalysis making waves in organic chemistry. Chem. Soc. Rev. 2022, 51, 594–627. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Gao, C.; Zhang, Z.; Wu, S.; Zhao, J.; Li, A. A Chemoenzymatic Strategy for the Synthesis of Steroid Drugs Enabled by P450 Monooxygenase-Mediated Steroidal Core Modification. ACS Catal. 2022, 12, 2907–2914. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D. Broadening the Scope of Biocatalysis in Sustainable Organic Synthesis. ChemSusChem 2019, 12, 2859–2881. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.H. Innovation by Evolution: Bringing New Chemistry to Life (Nobel Lecture). Angew. Chem. Int. Ed. 2019, 58, 14420–14426. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Kazlauskas, R.J. Catalytic promiscuity in biocatalysis: Using old enzymes to form new bonds and follow new pathways. Angew. Chem. Int. Ed. 2004, 43, 6032–6040. [Google Scholar] [CrossRef]
- Woodley, J.M. Integrating protein engineering into biocatalytic process scale-up. Trends Chem. 2022, 4, 371–373. [Google Scholar] [CrossRef]
- Arana-Peña, S.; Rios, N.S.; Carballares, D.; Goncalves, L.R.; Fernandez-Lafuente, R. Immobilization of lipases via interfacial activation on hydrophobic supports: Production of biocatalysts libraries by altering the immobilization conditions. Catal. Today 2021, 362, 130–140. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D.; Bode, M.L. The Hitchhiker's guide to biocatalysis: Recent advances in the use of enzymes in organic synthesis. Chem. Sci. 2020, 11, 2587–2605. [Google Scholar] [CrossRef]
- Dwivedee, B.P.; Soni, S.; Sharma, M.; Bhaumik, J.; Laha, J.K.; Banerjee, U.C. Promiscuity of lipase-catalyzed reactions for organic synthesis: A recent update. ChemistrySelect 2018, 3, 2441–2466. [Google Scholar] [CrossRef]
- Li, F.; Xu, Y.; Wang, C.; Wang, C.; Zhao, R.; Wang, L. Efficient synthesis of cyano-containing multi-substituted indoles catalyzed by lipase. Bioorg. Chem. 2021, 107, 104583. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, L.; Li, F.; Tang, X.; Ma, Y.; Wang, C.; Wang, Z.; Zhao, R.; Wang, L. A Novel Oxidation of Salicyl Alcohols Catalyzed by Lipase. Catalysts 2017, 7, 354. [Google Scholar] [CrossRef]
- Li, C.; Feng, X.-W.; Wang, N.; Zhou, Y.-J.; Yu, X.-Q. Biocatalytic promiscuity: The first lipase-catalysed asymmetric aldol reaction. Green Chem. 2008, 10, 616–618. [Google Scholar] [CrossRef]
- Bavandi, H.; Habibi, Z.; Yousefi, M. Porcine pancreas lipase as a green catalyst for synthesis of bis-4-hydroxy coumarins. Bioorg. Chem. 2020, 103, 104139. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wu, Q.; Chen, X.; Lin, X.; Wu, Q. A Single Lipase-Catalysed One-Pot Protocol Combining Aminolysis Resolution and Aza-Michael Addition: An Easy and Efficient Way to Synthesise β-Amino Acid Esters. Eur. J. Org. Chem. 2015, 2015, 5393–5401. [Google Scholar] [CrossRef]
- González-Martínez, D.; Gotor, V.; Gotor-Fernández, V. Application of Deep Eutectic Solvents in Promiscuous Lipase-Catalysed Aldol Reactions. Eur. J. Org. Chem. 2016, 2016, 1513–1519. [Google Scholar] [CrossRef]
- Mathebula, N.P.; Sheldon, R.A.; Bode, M.L. Lipase-Catalysed Enzymatic Kinetic Resolution of Aromatic Morita-Baylis-Hillman Derivatives by Hydrolysis and Transesterification. ChemBioChem 2022, 23, e202200435. [Google Scholar] [CrossRef]
- Samsonowicz-Górski, J.; Koszelewski, D.; Kowalczyk, P.; Śmigielski, P.; Hrunyk, A.; Kramkowski, K.; Wypych, A.; Szymczak, M.; Lizut, R.; Ostaszewski, R. Promiscuous Lipase-Catalyzed Knoevenagel–Phospha–Michael Reaction for the Synthesis of Antimicrobial β-Phosphono Malonates. Int. J. Mol. Sci. 2022, 23, 8819. [Google Scholar] [CrossRef]
- Priego, J.; Ortíz-Nava, C.; Carrillo-Morales, M.; López-Munguía, A.; Escalante, J.; Castillo, E. Solvent engineering: An effective tool to direct chemoselectivity in a lipase-catalyzed Michael addition. Tetrahedron 2009, 65, 536–539. [Google Scholar] [CrossRef]
- Rivera-Ramírez, J.D.; Escalante, J.; López-Munguía, A.; Marty, A.; Castillo, E. Thermodynamically controlled chemoselectivity in lipase-catalyzed aza-Michael additions. J. Mol. Catal. 2015, 112, 76–82. [Google Scholar] [CrossRef]
- Yang, F.; Wang, Z.; Zhang, X.; Jiang, L.; Li, Y.; Wang, L. A Green Chemoenzymatic Process for the Synthesis of Azoxybenzenes. ChemCatChem 2015, 7, 3450–3453. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, C.; Wang, C.; Shang, W.; Xiao, B.; Duan, S.; Li, F.; Wang, L.; Chen, P. Lipase-catalyzed aza-Michael addition of amines to acrylates in supercritical carbon dioxide. J. Chem. Technol. Biotechnol. 2019, 94, 3981–3986. [Google Scholar] [CrossRef]
- Klibanov, A.M. Improving enzymes by using them in organic solvents. Nature 2001, 409, 241–246. [Google Scholar] [CrossRef]
- Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, C.R.; Abou-Shehada, S.; Dunn, P.J. CHEM21 selection guide of classical- and less classical-solvents. Green Chem. 2016, 18, 288–296. [Google Scholar] [CrossRef]
- Spahn, C.; Minteer, D.S. Enzyme Immobilization in Biotechnology. Recent Pat. Eng. 2008, 2, 195–200. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzyme Microb. Technol. 2007, 40, 1451–1463. [Google Scholar] [CrossRef]
- Sampaio, C.S.; Angelotti, J.A.F.; Fernandez-Lafuente, R.; Hirata, D.B. Lipase immobilization via cross-linked enzyme aggregates: Problems and prospects—A review. Int. J. Biol. Macromol. 2022, 215, 434–449. [Google Scholar] [CrossRef]
- Morellon-Sterling, R.; Tavano, O.; Bolivar, J.M.; Berenguer-Murcia, Á.; Vela-Gutiérrez, G.; Sabir, J.S.M.; Tacias-Pascacio, V.G.; Fernandez-Lafuente, R. A review on the immobilization of pepsin: A Lys-poor enzyme that is unstable at alkaline pH values. Int. J. Biol. Macromol. 2022, 210, 682–702. [Google Scholar] [CrossRef]
- de Albuquerque, T.L.; de Sousa, M.; Gomes e Silva, N.C.; Girão Neto, C.A.C.; Gonçalves, L.R.B.; Fernandez-Lafuente, R.; Rocha, M.V.P. β-Galactosidase from Kluyveromyces lactis: Characterization, production, immobilization and applications—A review. Int. J. Biol. Macromol. 2021, 191, 881–898. [Google Scholar] [CrossRef]
- Altinkaynak, C.; Tavlasoglu, S.; ÿzdemir, N.; Ocsoy, I. A new generation approach in enzyme immobilization: Organic-inorganic hybrid nanoflowers with enhanced catalytic activity and stability. Enzyme Microb. Technol. 2016, 93–94, 105–112. [Google Scholar] [CrossRef]
- Palomo, J.M.; Muñoz, G.; Fernández-Lorente, G.; Mateo, C.; Fernández-Lafuente, R.; Guisán, J.M. Interfacial adsorption of lipases on very hydrophobic support (octadecyl–Sepabeads): Immobilization, hyperactivation and stabilization of the open form of lipases. J. Mol. Catal. B Enzym. 2002, 19–20, 279–286. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Woodley, J.M.; Fernandez-Lafuente, R. Is enzyme immobilization a mature discipline? Some critical considerations to capitalize on the benefits of immobilization. Chem. Soc. Rev. 2022, 51, 6251–6290. [Google Scholar] [CrossRef] [PubMed]
- Verdasco-Martín, C.M.; Garcia-Verdugo, E.; Porcar, R.; Fernandez-Lafuente, R.; Otero, C. Selective synthesis of partial glycerides of conjugated linoleic acids via modulation of the catalytic properties of lipases by immobilization on different supports. Food Chem. 2018, 245, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Esteve, F.; Escrig, A.; Porcar, R.; Luis, S.V.; Altava, B.; García-Verdugo, E. Immobilized Supramolecular Systems as Efficient Synzymes for CO2 Activation and Conversion. Adv. Sustain. Syst. 2022, 6, 2100408. [Google Scholar] [CrossRef]
- Cipolatti, E.P.; Rios, N.S.; Sousa, J.S.; Robert, J.d.M.; da Silva, A.A.T.; Pinto, M.C.C.; Simas, A.B.C.; Vilarrasa-García, E.; Fernandez-Lafuente, R.; Gonçalves, L.R.B.; et al. Synthesis of lipase/silica biocatalysts through the immobilization of CALB on porous SBA-15 and their application on the resolution of pharmaceutical derivatives and on nutraceutical enrichment of natural oil. Mol. Catal. 2021, 505, 111529. [Google Scholar] [CrossRef]
- Arana-Peña, S.; Carballares, D.; Morellon-Sterling, R.; Rocha-Martin, J.; Fernandez-Lafuente, R. The combination of covalent and ionic exchange immobilizations enables the coimmobilization on vinyl sulfone activated supports and the reuse of the most stable immobilized enzyme. Int. J. Biol. Macromol. 2022, 199, 51–60. [Google Scholar] [CrossRef]
- Arana-Peña, S.; Rios, N.S.; Mendez-Sanchez, C.; Lokha, Y.; Gonçalves, L.R.B.; Fernández-Lafuente, R. Use of polyethylenimine to produce immobilized lipase multilayers biocatalysts with very high volumetric activity using octyl-agarose beads: Avoiding enzyme release during multilayer production. Enzyme Microb. Technol. 2020, 137, 109535. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
 | ||||
| Entry | Solvent | Catalyst 2 | Time | Yield (%) 1 |
| 1 | EtOH | Novozym 435 | 2 h | 94 |
| 2 | EtOH | PPL | 4 h | 89 |
| 3 | EtOH | Cal-B | 4 h | 78 |
| 4 | EtOH | CSL | 4 h | 64 |
| 5 | EtOH | TEA (20 mol%) | 12 h | 30 |
| 6 | EtOH | No catalyst | 12 h | N. D. 3 |
| 7 | EtOH | Novozym 435 4 | 12 h | N. D. |
| 8 | EtOH | Novozym 435 5 | 12 h | N. D. |
| 9 | MeOH | Novozym 435 | 2 h | 78 |
| 10 | MeCN | Novozym 435 | 2 h | 72 |
| 11 | CH2Cl2 | Novozym 435 | 2 h | 42 |
| 12 | Dioxane | Novozym 435 | 2 h | 12 |
| 13 | THF | Novozym 435 | 2 h | 28 |
| 14 | Toluene | Novozym 435 | 2 h | 32 |
| 15 | hexane | Novozym 435 | 2 h | 25 |
| 16 | Water | Novozym 435 | 2 h | 40 |
| 17 | EtOH | Novozym 435 (150 U) | 2 h | 81 |
| 18 | EtOH | Novozym 435 (600 U) | 2 h | 95 |
 |
 |
 |
 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Li, F.; Ma, J.; Li, J.; Xie, H.; Wang, C.; Chen, P.; Wang, L. Lipase-Catalyzed Phospha-Michael Addition Reactions under Mild Conditions. Molecules 2022, 27, 7798. https://doi.org/10.3390/molecules27227798
Xu Y, Li F, Ma J, Li J, Xie H, Wang C, Chen P, Wang L. Lipase-Catalyzed Phospha-Michael Addition Reactions under Mild Conditions. Molecules. 2022; 27(22):7798. https://doi.org/10.3390/molecules27227798
Chicago/Turabian StyleXu, Yuelin, Fengxi Li, Jinglin Ma, Jiapeng Li, Hanqing Xie, Chunyu Wang, Peng Chen, and Lei Wang. 2022. "Lipase-Catalyzed Phospha-Michael Addition Reactions under Mild Conditions" Molecules 27, no. 22: 7798. https://doi.org/10.3390/molecules27227798
APA StyleXu, Y., Li, F., Ma, J., Li, J., Xie, H., Wang, C., Chen, P., & Wang, L. (2022). Lipase-Catalyzed Phospha-Michael Addition Reactions under Mild Conditions. Molecules, 27(22), 7798. https://doi.org/10.3390/molecules27227798