
Progression of Antiviral Agents Targeting Viral Polymerases

Abstract
1. Introduction
2. Common Viruses and Key DNA/RNA Polymerases
2.1. HBV
2.2. HIV
2.3. HCV
2.4. Influenza Virus
2.5. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
3. Viral DNA/RNA Synthases Inhibitors
3.1. Influenza Virus RNA Polymerase Inhibitor
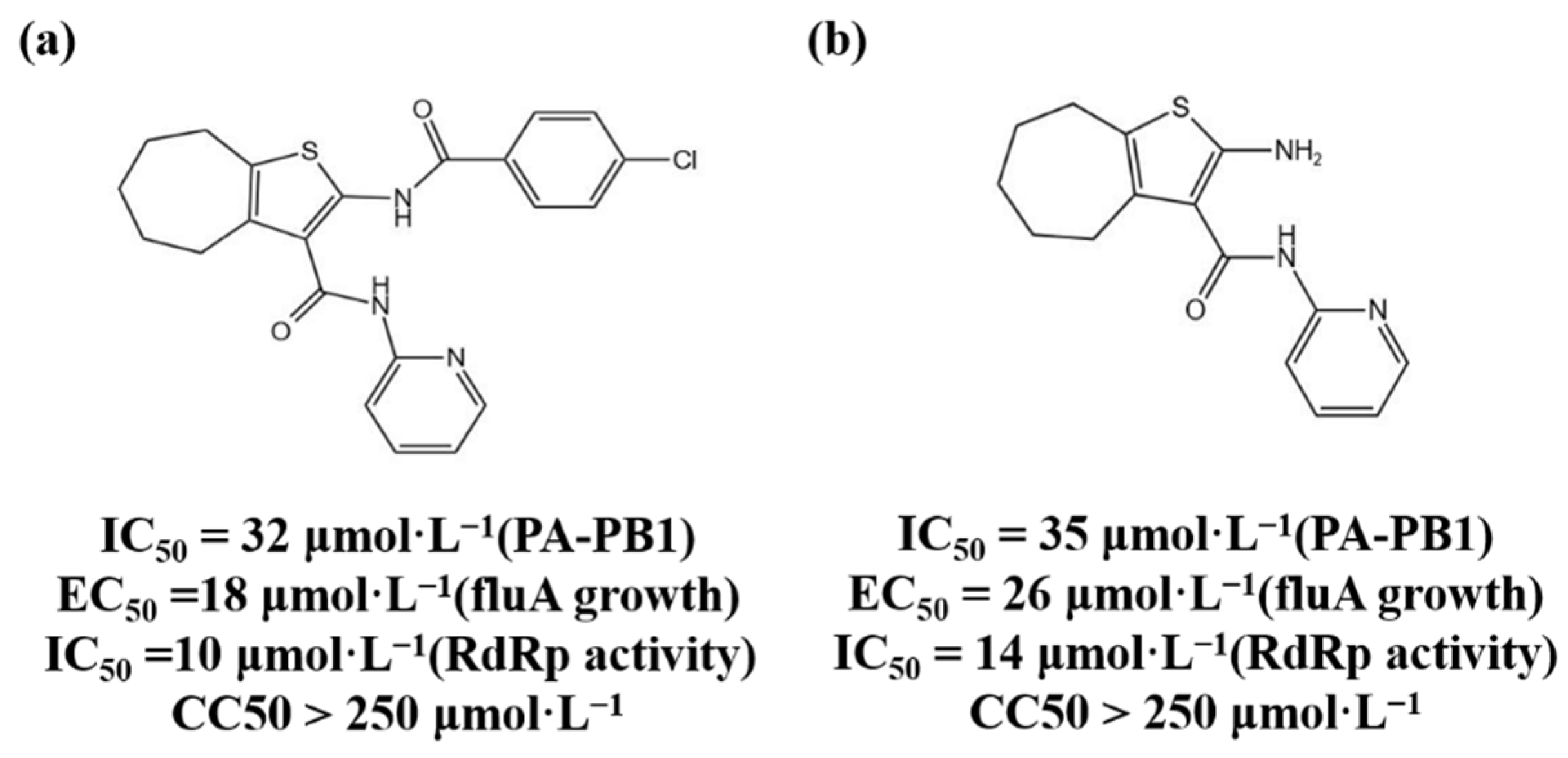
3.1.1. PA Inhibitors
3.1.2. PB1 Inhibitors
3.1.3. PB2 Inhibitors
3.1.4. Protein–Protein Interaction Inhibitors
3.2. HCV-related Inhibitors
3.2.1. NIs
3.2.2. NNIs
3.2.3. NS5A Inhibitors
3.3. HIV-1 Reverse Transcriptase Inhibitors
3.3.1. NRTIs
3.3.2. NNRTIs
3.3.3. HIV-1 Integrase Inhibitors
3.3.4. SARS-CoV-2 Enzyme Inhibitors
4. Research and Development Strategies for Novel Antiviral Drugs
4.1. Nucleic Acid Degradation
4.2. Protein Degradation
4.3. RNA Interference Application Drugs
4.4. Capsid Protein Assembly Regulators
5. Natural Products as Antiviral Drugs
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mateu, M.G. Introduction: The structural basis of virus function. Subcell. Biochem. 2013, 68, 3–51. [Google Scholar] [PubMed]
- Morens, D.M.; Fauci, A.S. Emerging Pandemic Diseases: How We Got to COVID-19. Cell 2020, 182, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Capua, I.; Alexander, D. Human health implications of avian influenza viruses and paramyxoviruses. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D.; Nathanson, N. Antiviral Therapy. In Viral Pathogenesis; Elsevier Ltd.: Amsterdam, The Netherlands, 2016; pp. 271–287. [Google Scholar] [CrossRef]
- Bertino, G.; Ardiri, A.; Proiti, M.; Rigano, G.; Frazzetto, E.; Demma, S.; Ruggeri, M.I.; Scuderi, L.; Malaguarnera, G.; Bertino, N.; et al. Chronic hepatitis C: This and the new era of treatment. World J. Hepatol. 2016, 8, 92–106. [Google Scholar] [CrossRef]
- Vardanyan, R.; Hruby, V. Antiviral Drugs. In Synthesis of Best-Seller Drugs; Academic Press: Cambridge, MA, USA, 2016; pp. 687–736. [Google Scholar] [CrossRef]
- Fenner, F.; Bachmann, P.A.; Gibbs, E.P.J.; Murphy, F.A.; Studdert, M.J.; White, D.O. Viral Replication. In Veterinary Virology; Elsevier: Amsterdam, The Netherlands, 1987; pp. 55–88. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S.J.V. Molecular biology of hepatitis B virus infection. Virology 2015, 479, 672–686. [Google Scholar] [CrossRef]
- Block, T.M.; Guo, H.; Guo, J.-T. Molecular virology of hepatitis B virus for clinicians. Clin. Liver Dis. 2007, 11, 685–706. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.J.P. DNA polymerase κ is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef]
- Jia, H.; Rai, D.; Zhan, P.; Chen, X.; Jiang, X.; Liu, X.J.F. Recent advance of the hepatitis B virus inhibitors: A medicinal chemistry overview. Future Med. Chem. 2015, 7, 587–607. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Hu, J. Hepatitis B virus reverse transcriptase: Diverse functions as classical and emerging targets for antiviral intervention. Emerg. Microbes Infect. 2013, 2, e56. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Seeger, C. Hepadnavirus Genome Replication and Persistence. Cold Spring Harb. Perspect. Med. 2015, 5, a021386. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Protzer, U. Attacking hepatitis B virus cccDNA–The holy grail to hepatitis B cure. J. Hepatol. 2016, 64, S41–S48. [Google Scholar] [CrossRef] [PubMed]
- Merican, I.; Guan, R.; Amarapuka, D.; Alexander, M.; Chutaputti, A.; Chien, R.; Hasnian, S.; Leung, N.; Lesmana, L.; Phiet, P. Chronic hepatitis B virus infection in Asian countries. J. Gastroenterol. Hepatol. 2000, 15, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Campagna, M.R.; Liu, F.; Mao, R.; Mills, C.; Cai, D.; Guo, F.; Zhao, X.; Ye, H.; Cuconati, A.; Guo, H.J. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J. Virol. 2013, 87, 6931–6942. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L.; Álvarez, M.; Pacheco, B. Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: Mechanism of action and resistance. Curr. Opin. Virol. 2014, 8, 1–9. [Google Scholar] [CrossRef]
- De Clercq, E. Antiviral drugs in current clinical use. J. Clin. Virol. 2004, 30, 115–133. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Y.; Wu, X.J.; Li, J.; Hou, F.Q.; Wang, G.Q. Pegylated interferon α-2b up-regulates specific CD8+ T cells in patients with chronic hepatitis B. World J. Gastroenterol. 2010, 16, 6145–6150. [Google Scholar] [CrossRef] [PubMed]
- Gabuzda, D.H.; Hirsch, M.S. Neurologic manifestations of infection with human immunodeficiency virus: Clinical features and pathogenesis. Ann. Intern. Med. 1987, 107, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Ilina, T.; LaBarge, K.; Sarafianos, S.G.; Ishima, R.; Parniak, M.A.J.B. Inhibitors of HIV-1 reverse transcriptase—Associated ribonuclease H activity. Biology 2012, 1, 521–541. [Google Scholar] [CrossRef] [PubMed]
- Götte, M.; Li, X.; Wainberg, M.A. HIV-1 reverse transcription: A brief overview focused on structure–function relationships among molecules involved in initiation of the reaction. Arch. Biochem. Biophys. 1999, 365, 199–210. [Google Scholar] [CrossRef]
- Kaushik, N.; Rege, N.; Yadav, P.N.; Sarafianos, S.G.; Modak, M.J.; Pandey, V.N. Biochemical analysis of catalytically crucial aspartate mutants of human immunodeficiency virus type 1 reverse transcriptase. Biochemistry 1996, 35, 11536–11546. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Corona, A.; Tramontano, E. HIV-1 reverse transcriptase still remains a new drug target: Structure, function, classical inhibitors, and new inhibitors with innovative mechanisms of actions. Mol. Biol. Int. 2012, 2012, 586401. [Google Scholar] [CrossRef] [PubMed]
- Sluis-Cremer, N.; Temiz, N.A.; Bahar, I. Conformational changes in HIV-1 reverse transcriptase induced by nonnucleoside reverse transcriptase inhibitor binding. Curr. HIV Res. 2004, 2, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Bird, L.; Chamberlain, P.; Stewart-Jones, G.; Stuart, D.; Stammers, D. Structure of HIV-2 reverse transcriptase at 2.35-Å resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc. Natl. Acad. Sci. USA 2002, 99, 14410–14415. [Google Scholar] [CrossRef]
- Hang, J.Q.; Li, Y.; Yang, Y.; Cammack, N.; Mirzadegan, T.; Klumpp, K. Substrate-dependent inhibition or stimulation of HIV RNase H activity by non-nucleoside reverse transcriptase inhibitors (NNRTIs). Biochem. Biophys. Res. Commun. 2007, 352, 341–350. [Google Scholar] [CrossRef]
- Delelis, O.; Carayon, K.; Saïb, A.; Deprez, E.; Mouscadet, J.-F. Integrase and integration: Biochemical activities of HIV-1 integrase. Retrovirology 2008, 5, 114. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Overview of Antiviral Drug Discovery and Development: Viral Versus Host Targets. In Antiviral Discovery for Highly Pathogenic Emerging Viruses; Royal Society of Chemistry: London, UK, 2021; pp. 1–27. [Google Scholar]
- Ramadori, G.; Meier, V. Hepatitis C virus infection: 10 years after the discovery of the virus. Eur. J. Gastroenterol. Hepatol. 2001, 13, 465–471. [Google Scholar] [CrossRef]
- De Francesco, R.; Neddermann, P.; Tomei, L.; Steinkühler, C.; Gallinari, P.; Folgori, A. Biochemical and Immunologic Properties of the Nonstructural Proteins of the Hepatitis C Virus: Implications for Development of Antiviral Agents and Vaccines. In Seminars in Liver Disease; Thieme Medical Publishers, Inc.: New York, NY, USA, 2000; pp. 0069–0084. [Google Scholar]
- Lohmann, V.; Körner, F.; Herian, U.; Bartenschlager, R. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 1997, 71, 8416–8428. [Google Scholar] [CrossRef]
- Powdrill, M.H.; Bernatchez, J.A.; Götte, M. Inhibitors of the hepatitis C virus RNA-dependent RNA polymerase NS5B. Viruses 2010, 2, 2169–2195. [Google Scholar] [CrossRef]
- Sofia, M.J.; Chang, W.; Furman, P.A.; Mosley, R.T.; Ross, B.S. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J. Med. Chem. 2012, 55, 2481–2531. [Google Scholar] [CrossRef]
- Stefanik, M.; Valdes, J.J.; Ezebuo, F.C.; Haviernik, J.; Uzochukwu, I.C.; Fojtikova, M.; Salat, J.; Eyer, L.; Ruzek, D. FDA-approved drugs efavirenz, tipranavir, and dasabuvir inhibit replication of multiple flaviviruses in vero cells. Microorganisms 2020, 8, 599. [Google Scholar] [CrossRef] [PubMed]
- Balish, A.L.; Katz, J.M.; Klimov, A.I. Influenza: Propagation, quantification, and storage. Curr. Protoc. Microbiol. 2013, 29, 15G.1.1–15G.1.24. [Google Scholar] [CrossRef] [PubMed]
- Tellier, R.; Li, Y.; Cowling, B.J.; Tang, J.W. Recognition of aerosol transmission of infectious agents: A commentary. BMC Infect. Dis. 2019, 19, 101. [Google Scholar] [CrossRef] [PubMed]
- Lister, P.; Reynolds, F.; Parslow, R.; Chan, A.; Cooper, M.; Plunkett, A.; Riphagen, S.; Peters, M. Swine-origin influenza virus H1N1, seasonal influenza virus, and critical illness in children. Lancet 2009, 374, 605–607. [Google Scholar] [CrossRef]
- Malik, Y.A. Properties of coronavirus and SARS-CoV-2. Malays. J. Pathol. 2020, 42, 3–11. [Google Scholar]
- Seah, I.; Agrawal, R. Can the coronavirus disease 2019 (COVID-19) affect the eyes? A review of coronaviruses and ocular implications in humans and animals. Ocul. Immunol. Inflamm. 2020, 28, 391–395. [Google Scholar] [CrossRef]
- Guarner, J. Three Emerging Coronaviruses in Two Decades: The Story of SARS, MERS, and Now COVID-19; Oxford University Press: Oxford, UK, 2020; Volume 153, pp. 420–421. [Google Scholar]
- Zhu, W.; Chen, C.Z.; Gorshkov, K.; Xu, M.; Lo, D.C.; Zheng, W. RNA-dependent RNA polymerase as a target for COVID-19 drug discovery. Adv. Sci. Drug Discov. 2020, 25, 1141–1151. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef]
- Borbone, N.; Piccialli, G.; Roviello, G.N.; Oliviero, G. Nucleoside analogs and nucleoside precursors as drugs in the fight against SARS-CoV-2 and other coronaviruses. Molecules 2021, 26, 986. [Google Scholar] [CrossRef]
- Villanueva, R.A.; Rouillé, Y.; Dubuisson, J. Interactions between virus proteins and host cell membranes during the viral life cycle. Int. Rev. Cytol. 2005, 245, 171–244. [Google Scholar] [PubMed]
- Razonable, R.R. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo Clin. Proc. 2011, 86, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Te Velthuis, A.J.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef]
- Akkina, R.K.; Chambers, T.M.; Londo, D.R.; Nayak, D.P. Intracellular localization of the viral polymerase proteins in cells infected with influenza virus and cells expressing PB1 protein from cloned cDNA. J. Virol. 1987, 61, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Jones, I.M.; Reay, P.A.; Philpott, K.L. Nuclear location of all three influenza polymerase proteins and a nuclear signal in polymerase PB2. EMBO J. 1986, 5, 2371–2376. [Google Scholar] [CrossRef]
- Smith, G.L.; Levin, J.Z.; Palese, P.; Moss, B. Synthesis and cellular location of the ten influenza polypeptides individually expressed by recombinant vaccinia viruses. Virology 1987, 160, 336–345. [Google Scholar] [CrossRef]
- Wang, P.; Palese, P.; O’Neill, R.E. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. J. Virol. 1997, 71, 1850–1856. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, K.; Meng, G.; Zhang, J.; Zhou, J.; Zhao, G.; Luo, M.; Zheng, X.J. Structural and functional characterization of K339Tsubstitution identified in the PB2 subunit cap-binding pocket of influenza A virus. J. Biol. Chem. 2013, 288, 11013–11023. [Google Scholar] [CrossRef]
- Reich, S.; Guilligay, D.; Cusack, S. An in vitro fluorescence based study of initiation of RNA synthesis by influenza B polymerase. Nucleic Acids Res. 2017, 45, 3353–3368. [Google Scholar]
- Kawaguchi, A.; Momose, F.; Nagata, K. Replication-coupled and host factor-mediated encapsidation of the influenza virus genome by viral nucleoprotein. J. Virol. 2011, 85, 6197–6204. [Google Scholar] [CrossRef]
- Hemerka, J.N.; Wang, D.; Weng, Y.; Lu, W.; Kaushik, R.S.; Jin, J.; Harmon, A.F.; Li, F. Detection and characterization of influenza A virus PA-PB2 interaction through a bimolecular fluorescence complementation assay. J. Virol. 2009, 83, 3944–3955. [Google Scholar] [CrossRef]
- Dufrasne, F. Baloxavir Marboxil: An Original New Drug against Influenza. Pharmaceuticals 2021, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Block, S.L.; Matharu, B.; Burleigh Macutkiewicz, L.; Wildum, S.; Dimonaco, S.; Collinson, N.; Clinch, B.; Piedra, P.A. Baloxavir Marboxil Single-dose Treatment in Influenza-infected Children: A Randomized, Double-blind, Active Controlled Phase 3 Safety and Efficacy Trial (miniSTONE-2). Pediatr. Infect. Dis. J. 2020, 39, 700. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Yen, H.L. Targeting the host or the virus: Current and novel concepts for antiviral approaches against influenza virus infection. Antivir. Res. 2012, 96, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.E. Xofluza (Baloxavir Marboxil) for the Treatment of Acute Uncomplicated Influenza. Pharm. Ther. 2019, 44, 9–11. [Google Scholar]
- Agrawal, U.; Raju, R.; Udwadia, Z.F. Favipiravir: A new and emerging antiviral option in COVID-19. Med. J. Armed Forces India 2020, 76, 370–376. [Google Scholar] [CrossRef]
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Hamid Khan, A.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211002621. [Google Scholar] [CrossRef]
- Markland, W.; McQuaid, T.J.; Jain, J.; Kwong, A.D. Broad-spectrum antiviral activity of the IMP dehydrogenase inhibitor VX-497: A comparison with ribavirin and demonstration of antiviral additivity with alpha interferon. Antimicrob. Agents Chemother. 2000, 44, 859–866. [Google Scholar] [CrossRef]
- Keppeke, G.D.; Chang, C.C.; Peng, M.; Chen, L.-Y.; Lin, W.-C.; Pai, L.-M.; Andrade, L.E.C.; Sung, L.-Y.; Liu, J.-L. IMP/GTP balance modulates cytoophidium assembly and IMPDH activity. Cell Div. 2018, 13, 5. [Google Scholar] [CrossRef]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Götte, M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.C.; Chesnokov, A.; Jones, J.; Mishin, V.P.; De La Cruz, J.A.; Nguyen, H.T.; Zanders, N.; Wentworth, D.E.; Davis, T.C.; Gubareva, L.V. Susceptibility of widely diverse influenza a viruses to PB2 polymerase inhibitor pimodivir. Antivir. Res. 2021, 188, 105035. [Google Scholar] [CrossRef] [PubMed]
- King, J.C.; Beigel, J.H.; Ison, M.G.; Rothman, R.E.; Uyeki, T.M.; Walker, R.E.; Neaton, J.D.; Tegeris, J.S.; Zhou, J.A.; Armstrong, K.L.; et al. Clinical Development of Therapeutic Agents for Hospitalized Patients with Influenza: Challenges and Innovations. Open Forum Infect. Dis. 2019, 6, ofz137. [Google Scholar] [CrossRef]
- Mishra, A.; Rathore, A.S. RNA dependent RNA polymerase (RdRp) as a drug target for SARS-CoV2. J. Biomol. Struct. Dyn. 2022, 40, 6039–6051. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein-protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef]
- Massari, S.; Nannetti, G.; Goracci, L.; Sancineto, L.; Muratore, G.; Sabatini, S.; Manfroni, G.; Mercorelli, B.; Cecchetti, V.; Facchini, M.; et al. Structural Investigation of Cycloheptathiophene-3-carboxamide Derivatives Targeting Influenza Virus Polymerase Assembly. J. Med. Chem. 2013, 56, 10118–10131. [Google Scholar] [CrossRef] [PubMed]
- Quezada, E.M.; Kane, C.M. The Hepatitis C Virus NS5A Stimulates NS5B During In Vitro RNA Synthesis in a Template Specific Manner. Open Biochem. J. 2009, 3, 39–48. [Google Scholar] [CrossRef]
- Picarazzi, F.; Vicenti, I.; Saladini, F.; Zazzi, M.; Mori, M. Targeting the RdRp of emerging RNA viruses: The structure-based drug design challenge. Molecules 2020, 25, 5695. [Google Scholar] [CrossRef]
- Zhang, C.; Cai, Z.; Kim, Y.C.; Kumar, R.; Yuan, F.; Shi, P.Y.; Kao, C.; Luo, G. Stimulation of hepatitis C virus (HCV) nonstructural protein 3 (NS3) helicase activity by the NS3 protease domain and by HCV RNA-dependent RNA polymerase. J. Virol. 2005, 79, 8687–8697. [Google Scholar] [CrossRef][Green Version]
- Membreno, F.E.; Lawitz, E.J. The HCV NS5B nucleoside and non-nucleoside inhibitors. Clin. Liver Dis. 2011, 15, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Te, H.S.; Randall, G.; Jensen, D.M. Mechanism of action of ribavirin in the treatment of chronic hepatitis C. Gastroenterol. Hepatol. 2007, 3, 218–225. [Google Scholar]
- Jin, Z.; Kinkade, A.; Behera, I.; Chaudhuri, S.; Tucker, K.; Dyatkina, N.; Rajwanshi, V.K.; Wang, G.; Jekle, A.; Smith, D.B.; et al. Structure-activity relationship analysis of mitochondrial toxicity caused by antiviral ribonucleoside analogs. Antivir. Res. 2017, 143, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.C.; Brown, J.A.; Thorpe, I.F. Allosteric inhibitors have distinct effects, but also common modes of action, in the HCV polymerase. Biophys. J. 2015, 108, 1785–1795. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; He, Y.; Lu, H.P. Interrogating the activities of conformational deformed enzyme by single-molecule fluorescence-magnetic tweezers microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, 13904–13909. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Therapeutic implications of hepatitis C virus resistance to antiviral drugs. Therap. Adv. Gastroenterol. 2009, 2, 205–219. [Google Scholar] [CrossRef]
- Polyak, S.J.; Khabar, K.S.; Paschal, D.M.; Ezelle, H.J.; Duverlie, G.; Barber, G.N.; Levy, D.E.; Mukaida, N.; Gretch, D.R. Hepatitis C virus nonstructural 5A protein induces interleukin-8, leading to partial inhibition of the interferon-induced antiviral response. J. Virol. 2001, 75, 6095–6106. [Google Scholar] [CrossRef]
- Scheel, T.K.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. [Google Scholar] [CrossRef]
- Kohler, J.J.; Nettles, J.H.; Amblard, F.; Hurwitz, S.J.; Bassit, L.; Stanton, R.A.; Ehteshami, M.; Schinazi, R.F. Approaches to hepatitis C treatment and cure using NS5A inhibitors. Infect. Drug Resist. 2014, 7, 41–56. [Google Scholar]
- Sundaram, V.; Kowdley, K.V. Dual daclatasvir and sofosbuvir for treatment of genotype 3 chronic hepatitis C virus infection. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 13–20. [Google Scholar] [CrossRef]
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Rampersad, S.; Tennant, P. Replication and Expression Strategies of Viruses. Viruses 2018, 2018, 55–82. [Google Scholar]
- Schultz, S.J.; Champoux, J.J. RNase H activity: Structure, specificity, and function in reverse transcription. Virus Res. 2008, 134, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; De Clercq, E. HIV genome-wide protein associations: A review of 30 years of research. Microbiol. Mol. Biol. Rev. 2016, 80, 679–731. [Google Scholar] [CrossRef]
- Rai, M.A.; Pannek, S.; Fichtenbaum, C.J. Emerging reverse transcriptase inhibitors for HIV-1 infection. Expert Opin. Emerg. Drugs 2018, 23, 149–157. [Google Scholar] [CrossRef]
- Holec, A.D.; Mandal, S.; Prathipati, P.K.; Destache, C.J. Nucleotide Reverse Transcriptase Inhibitors: A Thorough Review, Present Status and Future Perspective as HIV Therapeutics. Curr. HIV Res. 2017, 15, 411–421. [Google Scholar] [CrossRef]
- Dilmore, C.R.; DeStefano, J.J. HIV Reverse Transcriptase Pre-Steady-State Kinetic Analysis of Chain Terminators and Translocation Inhibitors Reveals Interactions between Magnesium and Nucleotide 3′-OH. ACS Omega 2021, 6, 14621–14628. [Google Scholar] [CrossRef]
- García-Lerma, J.G.; MacInnes, H.; Bennett, D.; Reid, P.; Nidtha, S.; Weinstock, H.; Kaplan, J.E.; Heneine, W. A novel genetic pathway of human immunodeficiency virus type 1 resistance to stavudine mediated by the K65R mutation. J. Virol. 2003, 77, 5685–5693. [Google Scholar] [CrossRef]
- De Clercq, E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antivir. Res. 1998, 38, 153–179. [Google Scholar] [CrossRef]
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: Checkpoint and repair at 30 years. Embo J. 2019, 38, e101801. [Google Scholar] [CrossRef]
- Usach, I.; Melis, V.; Peris, J.E. Non-nucleoside reverse transcriptase inhibitors: A review on pharmacokinetics, pharmacodynamics, safety and tolerability. J Int. AIDS Soc. 2013, 16, 18567. [Google Scholar] [CrossRef] [PubMed]
- Tasara, T.; Maga, G.; Hottiger, M.O.; Hübscher, U. HIV-1 reverse transcriptase and integrase enzymes physically interact and inhibit each other. FEBS Lett. 2001, 507, 39–44. [Google Scholar] [CrossRef]
- Blanco, J.L.; Varghese, V.; Rhee, S.Y.; Gatell, J.M.; Shafer, R.W. HIV-1 integrase inhibitor resistance and its clinical implications. J. Infect. Dis. 2011, 203, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Craigie, R. The molecular biology of HIV integrase. Future Virol. 2012, 7, 679–686. [Google Scholar] [CrossRef]
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Lyumkis, D.; Burke, T.R., Jr.; Hughes, S.H. HIV-1 Integrase Inhibitors That Are Active against Drug-Resistant Integrase Mutants. Antimicrob. Agents Chemother. 2020, 64, e00611–e00620. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P.J.N. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Shannon, A.; Le, N.T.-T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemot, J.-C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 Exonuclease active-sites. Antivir. Res. 2020, 178, 104793. [Google Scholar] [CrossRef]
- Hillen, H.S. Structure and function of SARS-CoV-2 polymerase. Curr. Opin. Virol. 2021, 48, 82–90. [Google Scholar] [CrossRef]
- Tian, L.; Qiang, T.; Liang, C.; Ren, X.; Jia, M.; Zhang, J.; Li, J.; Wan, M.; YuWen, X.; Li, H.; et al. RNA-dependent RNA polymerase (RdRp) inhibitors: The current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021, 213, 113201. [Google Scholar] [CrossRef]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef]
- Maheden, K.; Todd, B.; Gordon, C.J.; Tchesnokov, E.P.; Götte, M. Inhibition of viral RNA-dependent RNA polymerases with clinically relevant nucleotide analogs. Enzymes 2021, 49, 315–354. [Google Scholar] [PubMed]
- Hassanipour, S.; Arab-Zozani, M.; Amani, B.; Heidarzad, F.; Fathalipour, M.; Martinez-de-Hoyo, R. The efficacy and safety of Favipiravir in treatment of COVID-19: A systematic review and meta-analysis of clinical trials. Sci. Rep. 2021, 11, 11022. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, D.; Sanders, K.C.; Sabot, O.; Hachem, A.; Llanos-Cuentas, A.; Olotu, A.; Gosling, R.; Cutrell, J.B.; Hsiang, M.S. COVID-19 Therapeutics for Low- and Middle-Income Countries: A Review of Candidate Agents with Potential for Near-Term Use and Impact. Am. J. Trop. Med. Hyg. 2021, 105, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Benvindo, C.; Thaler, M.; Tas, A.; Ogando, N.S.; Bredenbeek, P.J.; Ninaber, D.K.; Wang, Y.; Hiemstra, P.S.; Snijder, E.J.; van Hemert, M.J. Suramin Inhibits SARS-CoV-2 Infection in Cell Culture by Interfering with Early Steps of the Replication Cycle. Antimicrob. Agents Chemother. 2020, 64, e00900–e00920. [Google Scholar] [CrossRef]
- Yin, W.; Luan, X.; Li, Z.; Zhou, Z.; Wang, Q.; Gao, M.; Wang, X.; Zhou, F.; Shi, J.; You, E.; et al. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat. Struct. Mol. Biol. 2021, 28, 319–325. [Google Scholar] [CrossRef]
- Venkataraman, S.; Prasad, B.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef]
- Li, H.; Dong, J.; Cai, M.; Xu, Z.; Cheng, X.-D.; Qin, J.-J. Protein degradation technology: A strategic paradigm shift in drug discovery. J. Hematol. Oncol. 2021, 14, 138. [Google Scholar] [CrossRef]
- Costales, M.G.; Childs-Disney, J.L.; Haniff, H.S.; Disney, M.D.J. How we think about targeting RNA with small molecules. J. Med. Chem. 2020, 63, 8880–8900. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef]
- Yang, Z.; Sun, Y.; Ni, Z.; Yang, C.; Tong, Y.; Liu, Y.; Li, H.; Rao, Y. Merging PROTAC and molecular glue for degrading BTK and GSPT1 proteins concurrently. Cell Res. 2021, 31, 1315–1318. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Nganvongpanit, K.; Müller, H.; Rings, F. Targeted suppression of E-cadherin gene expression in bovine preimplantation embryo by RNA interference technology using double-stranded RNA. Mol Reprod Dev. 2006, 73, 153–163. [Google Scholar]
- Schluep, T.; Lickliter, J.; Hamilton, J.; Lewis, D.L.; Lai, C.L.; Lau, J.Y.; Locarnini, S.A.; Gish, R.G.; Given, B.D. Safety, Tolerability, and Pharmacokinetics of ARC-520 Injection, an RNA Interference-Based Therapeutic for the Treatment of Chronic Hepatitis B Virus Infection, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2017, 6, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Schiefke, I.; Yoon, J.H.; Ahn, S.H.; Heo, J.; Kim, J.H.; Lik Yuen Chan, H.; Yoon, K.T.; Klinker, H.; Manns, M.J.H. RNA Interference Therapy With ARC-520 Results in Prolonged Hepatitis B Surface Antigen Response in Patients With Chronic Hepatitis B Infection. Hepatology 2020, 72, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.-F.; Wong, D.K.-H.; Schluep, T.; Lai, C.-L.; Ferrari, C.; Locarnini, S.; Lo, R.C.-L.; Gish, R.G.; Hamilton, J.; Wooddell, C.I. Long-term serological, virological and histological responses to RNA inhibition by ARC-520 in Chinese chronic hepatitis B patients on entecavir treatment. Gut 2022, 71, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, K.; Gane, E.; Cheng, W.; Sievert, W.; Roberts, S.; Ahn, S.H.; Kim, Y.J.; Streinu-Cercel, A.; Denning, J.; Symonds, W.J.H. HBcrAg, HBV-RNA declines in a phase 2a study evaluating the multi-dose activity of ARB-1467 in HBeAg-positive and negative virally suppressed subjects with hepatitis B. J. Hepatol. 2017, 66, S688–S689. [Google Scholar]
- Martinez, M.G.; Villeret, F.; Testoni, B.; Zoulim, F. Can we cure hepatitis B virus with novel directrect Symonds. Liver Int. 2020, 40, 27–34. [Google Scholar] [CrossRef]
- Vaillant, A. HBsAg, subviral particles, and their clearance in establishing a functional cure of chronic hepatitis B virus infection. ACS Infect. Dis. 2020, 7, 1351–1368. [Google Scholar] [CrossRef]
- Diab, A.; Foca, A.; Zoulim, F.; Durantel, D.; Andrisani, O. The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: Implications for the development of HBc-targeting antivirals. Antivir. Res. 2018, 149, 211–220. [Google Scholar] [CrossRef]
- Nassal, M. Hepatitis B viruses: Reverse transcription a different way. Virus Res. 2008, 134, 235–249. [Google Scholar] [CrossRef]
- Tan, Z.; Pionek, K.; Unchwaniwala, N.; Maguire, M.L.; Loeb, D.D.; Zlotnick, A.J. The interface between hepatitis B virus capsid proteins affects self-assembly, pregenomic RNA packaging, and reverse transcription. J. Virol. 2015, 89, 3275–3284. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, B.; Katen, S.P.; Francis, S.; Chirapu, S.; Finn, M.; Zlotnick, A.J. Hepatitis B virus capsids have diverse structural responses to small-molecule ligands bound to the heteroaryldihydropyrimidine pocket. J. Virol. 2016, 90, 3994–4004. [Google Scholar] [CrossRef] [PubMed]
- Mak, L.-Y.; Wong, D.K.-H.; Seto, W.-K.; Lai, C.-L.; Yuen, M.F. Hepatitis B core protein as a therapeutic target. Expert Opin. Ther. Targets 2017, 21, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Schlicksup, C. Hepatitis B Virus Capsid Protein as an Antiviral Target; Indiana University: Bloomington, IN, USA, 2020. [Google Scholar]
- Stray, S.J.; Zlotnick, A. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J. Mol. Recognit. JMR 2006, 19, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liu, F.; Zhao, Q.; Ma, X.; Lu, D.; Li, H.; Zeng, Y.; Tong, X.; Zeng, L.; Liu, J. Discovery of phthalazinone derivatives as novel hepatitis B virus capsid inhibitors. J. Med. Chem. 2020, 63, 8134–8145. [Google Scholar] [CrossRef] [PubMed]
- Asres, K.; Seyoum, A.; Veeresham, C.; Bucar, F.; Gibbons, S. Naturally derived anti-HIV agents. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2005, 19, 557–581. [Google Scholar] [CrossRef] [PubMed]
- Kostova, I. Coumarins as inhibitors of HIV reverse transcriptase. Curr. HIV Res. 2006, 4, 347–363. [Google Scholar] [CrossRef]
- Martini, R.; Esposito, F.; Corona, A.; Ferrarese, R.; Ceresola, E.R.; Visconti, L.; Tintori, C.; Barbieri, A.; Calcaterra, A.; Iovine, V. Natural Product Kuwanon-L Inhibits HIV-1 Replication through Multiple Target Binding. ChemBioChem 2017, 18, 374–377. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Rumschlag-Booms, E.; Guan, Y.-F.; Wang, D.-Y.; Liu, K.-L.; Li, W.-F.; Nguyen, V.H.; Cuong, N.M.; Soejarto, D.D.; Fong, H.H. Potent inhibitor of drug-resistant HIV-1 strains identified from the medicinal plant Justicia gendarussa. J. Nat. Prod. 2017, 80, 1798–1807. [Google Scholar] [CrossRef]
- Kashman, Y.; Gustafson, K.R.; Fuller, R.W.; Cardellina, J.H.; McMahon, J.B.; Currens, M.J.; Buckheit, R.W., Jr.; Hughes, S.H.; Cragg, G.M.; Boyd, M.R. HIV inhibitory natural products. Part 7. The calanolides, a novel HIV-inhibitory class of coumarin derivatives from the tropical rainforest tree, Calophyllum lanigerum. J. Med. Chem. 1992, 35, 2735–2743. [Google Scholar] [CrossRef]
- Manvar, D.; Mishra, M.; Kumar, S.; Pandey, V.N. Identification and evaluation of anti hepatitis C virus phytochemicals from Eclipta alba. J. Ethnopharmacol. 2012, 144, 545–554. [Google Scholar] [CrossRef]
- Moghadamtousi, S.Z.; Goh, B.H.; Chan, C.K.; Shabab, T.; Kadir, H.A. Biological activities and phytochemicals of Swietenia macrophylla King. Molecules 2013, 18, 10465–10483. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Chen, Z.; Tao, Y.; Zhang, B.; Wu, X.; Yang, L.; Wang, Q.; Wang, Z. An integrated method for optimized identification of effective natural inhibitors against SARS-CoV-2 3CLpro. Sci. Rep. 2021, 11, 22796. [Google Scholar] [CrossRef] [PubMed]
- Chayama, K.; Hayes, C.N. HCV drug resistance challenges in Japan: The role of pre-existing variants and emerging resistant strains in direct acting antiviral therapy. Viruses 2015, 7, 5328–5342. [Google Scholar] [CrossRef] [PubMed]
- Kurt Yilmaz, N.; Schiffer, C.A. Introduction: Drug Resistance. Chem. Rev. 2021, 121, 3235–3237. [Google Scholar] [CrossRef] [PubMed]
- Pardo, J.; Shukla, A.M.; Chamarthi, G.; Gupte, A. The journey of remdesivir: From Ebola to COVID-19. Drugs Context 2020, 9. [Google Scholar] [CrossRef]
- Shiraki, K.; Daikoku, T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol. Ther. 2020, 209, 107512. [Google Scholar] [CrossRef]
- Tsai, C.-H.; Lee, P.-Y.; Stollar, V.; Li, M.-L. Antiviral therapy targeting viral polymerase. Curr. Pharm. Des. 2006, 12, 1339–1355. [Google Scholar] [CrossRef]
- Hecker, S.J.; Erion, M.D.J. Prodrugs of phosphates and phosphonates. J. Med. Chem. 2008, 51, 2328–2345. [Google Scholar] [CrossRef]
- Sugawara, M.; Huang, W.; Fei, Y.J.; Leibach, F.H.; Ganapathy, V.; Ganapathy, M.E.J. Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J. Pharm. Sci. 2000, 89, 781–789. [Google Scholar] [CrossRef]
- Li, F.; Maag, H.; Alfredson, T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J. Pharm. Sci. 2008, 97, 1109–1134. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 2002, 1, 13–25. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef]
- Adamson, C.S.; Chibale, K.; Goss, R.J.; Jaspars, M.; Newman, D.J.; Dorrington, R.A. Antiviral drug discovery: Preparing for the next pandemic. Chem. Soc. Rev. 2021, 50, 3647–3655. [Google Scholar] [CrossRef] [PubMed]
- Christy, M.P.; Uekusa, Y.; Gerwick, L.; Gerwick, W.H. Natural products with potential to treat RNA virus pathogens including SARS-CoV-2. J. Nat. Prod. 2020, 84, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Musarra-Pizzo, M.; Pennisi, R.; Ben-Amor, I.; Mandalari, G.; Sciortino, M.T. Antiviral activity exerted by natural products against human viruses. Viruses 2021, 13, 828. [Google Scholar] [CrossRef]
- Shaneyfelt, M.E.; Burke, A.D.; Graff, J.W.; Jutila, M.A.; Hardy, M.E. Natural products that reduce rotavirus infectivity identified by a cell-based moderate-throughput screening assay. Virol. J. 2006, 3, 68. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Fang, Z.; Li, Z.; Huang, B.; Zhang, H.; Lu, X.; Xu, H.; Zhou, Z.; Ding, X.; Daelemans, D. Design, synthesis, and evaluation of thiophene [3, 2-d] pyrimidine derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors with significantly improved drug resistance profiles. J. Med. Chem. 2016, 59, 7991–8007. [Google Scholar] [CrossRef]
- Zhang, X. Direct anti-HCV agents. Acta Pharm. Sin. B 2016, 6, 26–31. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Viruses | Targets | Approved Drugs | Novel Inhibitors |
|---|---|---|---|
| Human immunodeficiency virus (HIV) | HIV-1 reverse transcriptase (NRTI) | Zidovudine | MK-8583 |
| Didanosine | Rovafovir etalafenamide | ||
| Stavudine | Islatravir (mk-8591) | ||
| Lamivudine | Tenofovir Alafenamide | ||
| Emtricitabine | Tenofovir amibufenamide | ||
| Abacavir Sulfate | Tenofovir Alafenamide Fumarate | ||
| Apricitabine | Abacavir hydroxyacetate | ||
| Tenofovir Disoproxil Fumarate | |||
| Tenofovir Succinate | |||
| Tenofovir | |||
| Azvudine | |||
| HIV-1 reverse transcriptase (NNRTI) | Nevirapine | VM-1500A | |
| Delavirdine mesylate | Dapivirine | ||
| Efavirenz | IQP-0528 | ||
| Etravirine | IQP-0528 | ||
| Elsulfavirine | |||
| Ainuovirine | |||
| Doravirine | |||
| Rilpivirine Hydrochloride | |||
| HIV-1 integrase | Raltegravir Potassium | Bictegravir | |
| Cabotegravir sodium | Fipravirimat | ||
| Dolutegravir Sodium | STP-0404 | ||
| GSK3732394 | |||
| Hepatitis C virus (HCV) | NS5B polymerase (NI) | Sofosbuvir | Adafosbuvir |
| Bemnifosbuvir | |||
| NS5B polymerase (NNI) | Dasabuvir Sodium Hydrate | Radalbuvir | |
| GSK-2878175 | |||
| TMC-647055 | |||
| CC-31244 | |||
| NS5A inhibitors | Emitasvir Phosphate | Ledipasvir | |
| Coblopasvir hydrochloride | |||
| Elbasvir | |||
| Daclatasvir Dihydrochloride | |||
| Cyclophilin A (Cyp) | SCY-635 | ||
| Alisporivir | |||
| EDP-494 | |||
| Human influenza virus | RNA polymerase (PA) | baloxavir marboxil | |
| RNA polymerase (PB2) | Onradivir | ||
| RNA polymerase (PB1) | Favipiravir | ||
| Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) | RdRP polymerase (Nucleoside) | Remdesivir | Remindevir |
| Favipiravir | Galidesivir | ||
| GS-441524 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, S.; Wang, H.; Wang, Z.; Wang, Q. Progression of Antiviral Agents Targeting Viral Polymerases. Molecules 2022, 27, 7370. https://doi.org/10.3390/molecules27217370
Peng S, Wang H, Wang Z, Wang Q. Progression of Antiviral Agents Targeting Viral Polymerases. Molecules. 2022; 27(21):7370. https://doi.org/10.3390/molecules27217370
Chicago/Turabian StylePeng, Siqi, Huizhen Wang, Zhengtao Wang, and Qingzhong Wang. 2022. "Progression of Antiviral Agents Targeting Viral Polymerases" Molecules 27, no. 21: 7370. https://doi.org/10.3390/molecules27217370
APA StylePeng, S., Wang, H., Wang, Z., & Wang, Q. (2022). Progression of Antiviral Agents Targeting Viral Polymerases. Molecules, 27(21), 7370. https://doi.org/10.3390/molecules27217370