
Chitosan Encapsulated Meloxicam Nanoparticles for Sustained Drug Delivery Applications: Preparation, Characterization, and Pharmacokinetics in Wistar Rats
 ,
,
Abstract
1. Introduction
2. Results and Discussion
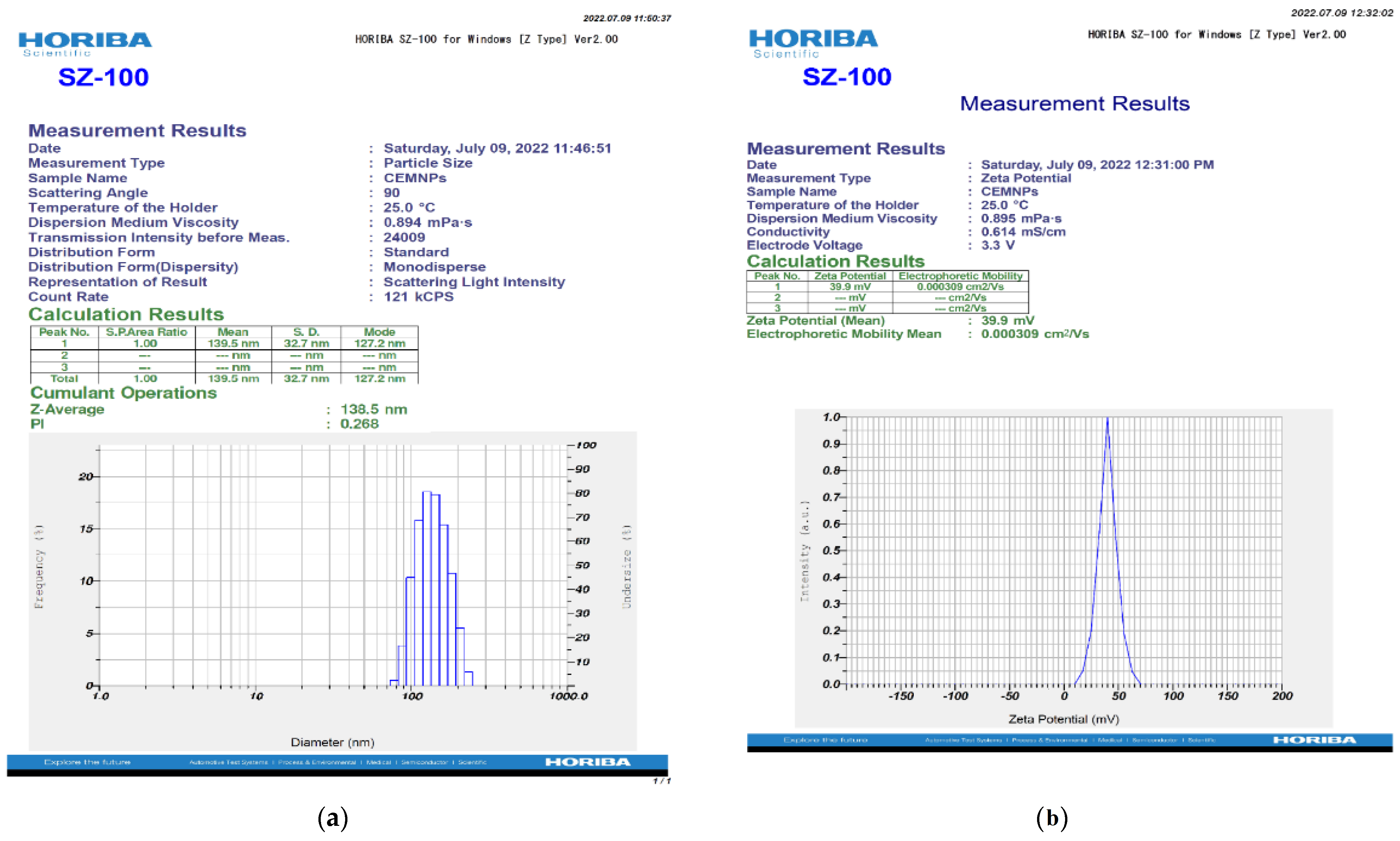
2.1. Preparation and Characterization of CEMNPs
2.2. Encapsulation Efficiency (%)
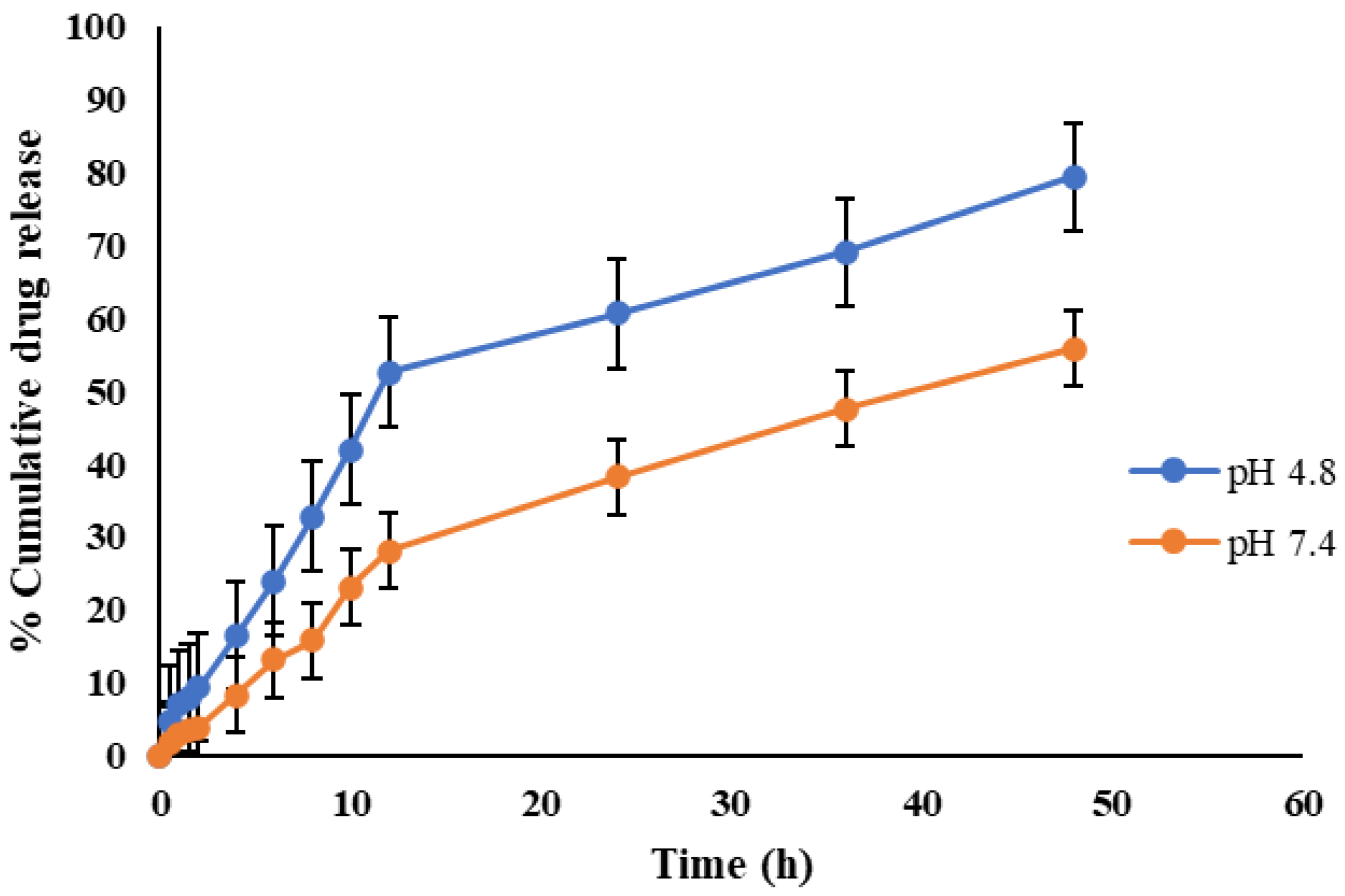
2.3. In Vitro Release Kinetics
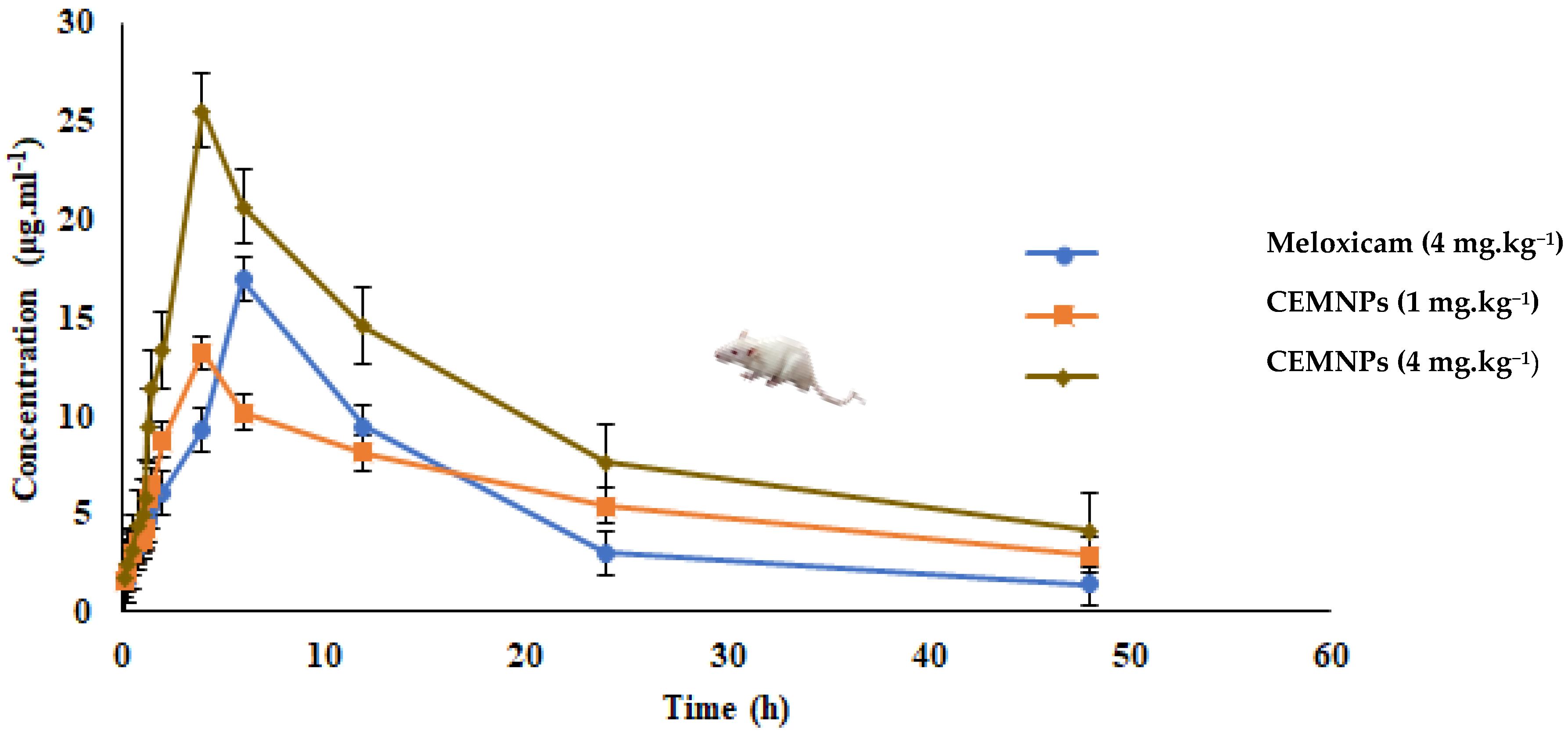
2.4. In Vivo Pharmacokinetics
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Preparation of Chitosan-Encapsulated Meloxicam Nanoparticles (CEMNPs)
3.3. Characterization of CEMNPs
3.3.1. Transmission Electron Microscopic (TEM) and Scanning Electron Microscopic (SEM) with Energy Dispersive Spectroscopy (EDS) Analysis
3.3.2. Zetasizer Analysis
3.3.3. UV-Visible Spectra Analysis
3.3.4. Fourier Transform Infrared (FTIR) Spectroscopy
3.3.5. Encapsulation Efficiency
3.3.6. In Vitro Release Kinetic Studies
3.4. Meloxicam Determination
3.4.1. Instrumentation
3.4.2. Standardization and Validation of Assay
3.5. Pharmacokinetic Studies following Single Oral (p.o) Administration
3.5.1. Experimental Animals
3.5.2. Blood Sampling
3.5.3. Plasma Samples for HPLC Analysis
3.5.4. Pharmacokinetic (PK) Analysis of Meloxicam
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mellor, D.J. Mouth pain in horses: Physiological foundations, behavioural indices, welfare implications, and a suggested solution. Animals 2020, 10, 572. [Google Scholar] [CrossRef] [PubMed]
- Coleman, G.J. Public perceptions of Animal Pain and Animal Welfare. In Scientific Assessment and Management of Animal Pain, 1st ed.; Mellor, D.J., Thornber, P.M., Bayvel, D., Kahn, S., Eds.; OIE (World Organisation for Animal Health): Paris, France, 2008; pp. 26–37. [Google Scholar]
- Lees, P. Analgesic, antiinflammatory, antipyretic drugs. In Veterinary Pharmacology and Therapeutics, 10th ed.; Riviere, J.E., Papich, M.G., Eds.; Iowa State University Press: Ames, IA, USA, 2018; pp. 467–500. [Google Scholar]
- Sinha, M.; Gautam, L.; Shukla, P.K.; Kaur, P.; Sharma, S.; Singh, T.P. Current perspectives in NSAID-induced gastropathy. Mediat. Inflamm. 2013, 2013, 258209. [Google Scholar] [CrossRef] [PubMed]
- Scarim, C.B.; de Oliveira, V.E.; dos Santos, J.L.; Chin, C.M. NSAIDs and natural products interactions: Mechanism and clinical implications. J. Immunol. Clin. Res. 2017, 4, 1040. [Google Scholar]
- Bertolini, A.; Ottani, A.; Sandrini, M. Selective COX-2 inhibitors and dual acting anti-inflammatory drugs: Critical remarks. Curr. Med. Chem. 2002, 9, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Hawash, M.; Jaradat, N.; Hameedi, S.; Mousa, A. Design, synthesis and biological evaluation of novel benzodioxole derivatives as COX inhibitors and cytotoxic agents. BMC Chem. 2020, 14, 54. [Google Scholar] [CrossRef]
- Engelhardt, G.; Homma, D.; Schlegel, K.; Utzmann, R.; Schnitzler, C. Anti-inflammatory, analgesic, antipyretic and related properties of meloxicam, a new non-steroidal anti-inflammatory agent with favourable gastrointestinal tolerance. Inflamm. Res. 1995, 44, 423–433. [Google Scholar] [CrossRef]
- Davies, N.M.; Skjodt, N.M. Clinical pharmacokinetics of meloxicam. A cyclo-oxygenase-2 preferential nonsteroidal anti-inflammatory drug. Clin. Pharmacokinet. 1999, 36, 115–126. [Google Scholar] [CrossRef]
- Hawkey, C.J. COX-1 and COX-2 inhibitors. Best practice & research. J. Clin. Gastroenterol. 2001, 15, 801–820. [Google Scholar] [CrossRef]
- Mahaprabhu, R.; Bhandarkar, A.G.; Jangir, B.L.; Rahangadale, S.P.; Kurkure, N.V. Ameliorative effect of Ocimum sanctum on meloxicam induced toxicity in Wistar rats. Toxicol. Int. 2011, 18, 130–136. [Google Scholar] [CrossRef][Green Version]
- Fleischmann, R.; Iqbal, I.; Slobodin, G. Meloxicam. Expert Opin. Pharmacother. 2002, 3, 1501–1512. [Google Scholar] [CrossRef]
- Wisher, D. Martindale: The Complete Drug Reference. 37th ed. J. Med. Libr. Assoc. 2012, 100, 75–76. [Google Scholar] [CrossRef]
- Burukoglu, D.; Baycu, C.; Taplamacioglu, F.; Sahin, E.; Bektur, E. Effects of nonsteroidal anti-inflammatory meloxicam on stomach, kidney, and liver of rats. Toxicol. Ind. Health 2016, 32, 980–986. [Google Scholar] [CrossRef]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef] [PubMed]
- Del Tacca, M.; Colucci, R.; Fornai, M.; Blandizzi, C. Efficacy and tolerability of meloxicam, a COX-2 preferential nonsteroidal antiinflamatory drug: A review. Clin. Drug Investig. 2002, 22, 799–818. [Google Scholar] [CrossRef]
- Kürti, L.; Gáspár, R.; Márki, Á.; Kápolna, E.; Bocsik, A.; Veszelka, S.; Bartos, C.; Ambrus, R.; Vastag, M.; Deli, M.A.; et al. In vitro and in vivo characterization of meloxicam nanoparticles designed for nasal administration. Eur. J. Pharm. Sci. 2013, 50, 86–92. [Google Scholar] [CrossRef]
- Nagai, N.; Ogata, F.; Otake, H.; Kawasaki, N. Oral administration system based on meloxicam nanocrystals: Decreased dose due to high bioavailability attenuates risk of gastrointestinal side effects. Pharmaceutics 2020, 12, 313. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, D.C. Piroxicam pharmacokinetics: Recent clinical results relating kinetics and plasma levels to age, sex, and adverse effects. Am. J. Med. 1986, 81, 22–28. [Google Scholar]
- Nilsen, O.G. Clinical pharmacokinetics of tenoxicam. Clin. Pharmacokinet. 1994, 26, 16–43. [Google Scholar] [CrossRef]
- Türck, D.; Busch, U.; Heinzel, G.; Narjes, H. Clinical pharmacokinetics of meloxicam. Arzneim. Forsch. 1997, 47, 253–258. [Google Scholar]
- Aguilar-Mariscal, H.; Patiño-Camacho, S.I.; Rodríguez-Silverio, J.; Torres-López, J.E.; Flores-Murrieta, F.J. Oral pharmacokinetics of meloxicam in the rat using a high-performance liquid chromatography method in micro-whole-blood samples. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 587–591. [Google Scholar] [CrossRef]
- Hawash, M.; Jaradat, N.; Eid, A.M.; Abubaker, A.; Mufleh, O.; Al-Hroub, Q.; Sobuh, S. Synthesis of novel isoxazole-carboxamide derivatives as promising agents for melanoma and targeted nano-emulgel conjugate for improved cellular permeability. BMC Chem. 2022, 16, 47. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.; Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.M.; Hawash, M. Biological evaluation of safrole oil and safrole oil nanoemulgel as antioxidant, antidiabetic, antibacterial, antifungal and anticancer. BMC Complement. Altern. Med. 2021, 21, 159. [Google Scholar] [CrossRef] [PubMed]
- Begines, B.; Ortiz, T.; Pérez-Aranda, M.; Martínez, G.; Merinero, M.; Argüelles-Arias, F.; Alcudia, A. Polymeric nanoparticles for drug delivery: Recent developments and future prospects. Nanomaterials 2020, 10, 1403. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. Role of nanobiotechnology in drug delivery. Methods Mol. Biol. 2020, 2059, 55–73. [Google Scholar] [CrossRef]
- Mohammed, M.A.; Syeda, J.; Wasan, K.M.; Wasan, E.K. An overview of chitosan nanoparticles and its application in non-parenteral drug delivery. Pharmaceutics 2017, 9, 53. [Google Scholar] [CrossRef]
- Mu, Y.; Fu, Y.; Li, J.; Yu, X.; Li, Y.; Wang, Y.; Wu, X.; Zhang, K.; Kong, M.; Feng, C.; et al. Multifunctional quercetin conjugated chitosan nano-micelles with P-gp inhibition and permeation enhancement of anticancer drug. Carbohydr. Polym. 2019, 203, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Pathomthongtaweechai, N.; Muanprasat, C. Potential applications of chitosan-based nanomaterials to surpass the gastrointestinal physiological obstacles and enhance the intestinal drug absorption. Pharmaceutics 2021, 13, 887. [Google Scholar] [CrossRef]
- Mikušová, V.; Mikuš, P. Advances in chitosan-based nanoparticles for drug delivery. Int. J. Mol. Sci. 2021, 22, 9652. [Google Scholar] [CrossRef] [PubMed]
- Oudah, M.H.; Wais, M.H.F.; Al-lam, S.M. Preparation and evaluation of meloxicam nanoparticles as oral thin film. Int. J. Drug Deliv. Technol. 2021, 11, 676–684. [Google Scholar] [CrossRef]
- Mohamed, H.B.; Attia Shafie, M.A.; Mekkawy, A.I. Chitosan nanoparticles for meloxicam ocular delivery: Development, in vitro characterization, and in vivo evaluation in a rabbit eye model. Pharmaceutics 2022, 14, 893. [Google Scholar] [CrossRef] [PubMed]
- Luger, P.; Daneck, K.; Engel, W.; Trummlitz, G.; Wagner, K. Structure and physicochemical properties of meloxicam, a new NSAID. Eur. J. Pharm. Sci. 1996, 4, 175–187. [Google Scholar] [CrossRef]
- Brar, S.K.; Verma, M. Measurement of nanoparticles by light-scattering techniques. TrAC Trends Anal. Chem. 2011, 30, 4–17. [Google Scholar] [CrossRef]
- Matos, B.N.; Reis, T.A.; Gratieri, T.; Gelfuso, G.M. Chitosan nanoparticles for targeting and sustaining minoxidil sulphate delivery to hair follicles. Int. J. Biol. Macromol. 2015, 75, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Chen, D.; Gao, J.; Hu, Y. Preparation of sertraline-loaded chitosan nanoparticles and the pharmacokinetics studies. Afr. J. Pharmacy Pharmacol. 2015, 10, 26–33. [Google Scholar] [CrossRef]
- Rajashekaraiah, R.; Kumar, P.R.; Prakash, N.; Rao, G.S.; Devi, V.R.; Metta, M.; Narayanaswamy, H.D.; Swamy, M.N.; Satyanarayan, K.; Rao, S.; et al. Anticancer efficacy of 6-thioguanine loaded chitosan nanoparticles with or without curcumin. Int. J. Biol. Macromol. 2020, 148, 704–714. [Google Scholar] [CrossRef]
- Bihari, P.; Vippola, M.; Schultes, S.; Praetner, M.; Khandoga, A.G.; Reichel, C.A.; Coester, C.; Tuomi, T.; Rehberg, M.; Krombach, F. Optimized dispersion of nanoparticles for biological in vitro and in vivo studies. Part. Fibre Toxicol. 2008, 5, 14. [Google Scholar] [CrossRef]
- Hunter, R.J. Applications of the zeta potential. In Zeta Potential in Colloid Science: Principles and Applications; Hunter, R.J., Ed.; Academic Press: London, UK, 1988; pp. 219–257. [Google Scholar]
- Li, P.; Wang, Y.; Zeng, F.; Chen, L.; Peng, Z.; Kong, L.X. Synthesis and characterization of folate conjugated chitosan and cellular uptake of its nanoparticles in HT-29 cells. Carbohydr. Res. 2011, 346, 801–806. [Google Scholar] [CrossRef]
- Fan, W.; Yan, W.; Xu, Z.; Ni, H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids Surf. B Biointerfaces 2012, 90, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Joseph-Charles, J.; Bertucat, M. Determination of meloxicam in tablet formulations by ultraviolet spectrophotometry and high-performance liquid chromatography. Anal. Lett. 1999, 32, 2051–2059. [Google Scholar] [CrossRef]
- Hassan, E.M. Spectrophotometric and fluorimetric methods for the determination of meloxicam in dosage forms. J. Pharm. Biomed. Anal. 2002, 27, 771–777. [Google Scholar] [CrossRef]
- Nemutlu, E.; Kir, S. Validated determination of meloxicam in tablets by using UV spectrophotometry. J. Fac. Pharm. 2004, 24, 13–24. [Google Scholar]
- Dhandapani, B.; Murali, S.E.; Susrutha, N.; Swetha, R.; Rani, S.K.; Babu, T.S.; Seetharamanjaneyulu, G.V.; Baboo, R.V. Spectrophotometric estimation of meloxicam in bulk and its pharmaceutical formulations. Int. J. Pharma Sci. Res. 2010, 1, 217–221. [Google Scholar]
- Khalil, Y.N.; Aldosari, F.K. Meloxicam. In Profiles of Drug Substances, Excipients and Related Methodology; Britan, G.H., Ed.; Academic Press: London, UK, 2020; Volume 45, pp. 159–197. [Google Scholar]
- Asasutjarit, R.; Theerachayanan, T.; Kewsuwan, P.; Veeranodha, S.; Fuongfuchat, A.; Ritthidej, G.C. Development and evaluation of diclofenac sodium loaded-n-trimethyl chitosan nanoparticles for ophthalmic use. AAPS PharmSciTech 2015, 16, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Kasoju, N.; Bora, U. Encapsulation of curcumin in alginate-chitosan-pluronic composite nanoparticles for delivery to cancer cells. Nanomedicine 2010, 6, 153–160. [Google Scholar] [CrossRef]
- Jahromi, M.A.M.; Pirestani, M.; Ramandi, M.F.; Ahmadi, K.; Rajayi, H.; Hassan, Z.M.; Kamali, M.; Mirnejad, R. Curcumin-loaded chitosan tripolyphosphate nanoparticles as a safe, natural and effective antibiotic inhibits the infection of Staphylococcus aureus and Pseudomonas aeruginosa in vivo. Iran. J. Biotech. 2014, 12, 1–8. [Google Scholar] [CrossRef]
- Nair, R.S.; Morris, A.; Billa, N.; Leong, C.O. An evaluation of curcumin-encapsulated chitosan nanoparticles for transdermal delivery. AAPS PharmSciTech 2019, 20, 69. [Google Scholar] [CrossRef] [PubMed]
- Bhoopathy, S.; Inbakandan, D.; Rajendran, T.; Chandrasekaran, K.; Prabha, S.B.; Reddy, B.A.; Kasilingam, R.; Kumar, R.V.; Dharani, G. Dietary supplementation of curcumin-loaded chitosan nanoparticles stimulates immune response in the white leg shrimp Litopenaeus vannamei challenged with Vibrio harveyi. Fish Shellfish Immunol. 2021, 117, 188–191. [Google Scholar] [CrossRef]
- Ahmed, A.B.; Konwar, R.; Sengupta, R. Atorvastatin calcium loaded chitosan nanoparticles: In vitro evaluation and in vivo pharmacokinetic studies in rabbits. Braz. J. Pharm. Sci. 2015, 51, 467–477. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Dahmani, F.Z.; Yin, L.; Zhou, J.; Yao, J. Amphiphilic carboxymethyl chitosan-quercetin conjugate with P-gp inhibitory properties for oral delivery of paclitaxel. Biomaterials 2014, 35, 7654–7665. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Xie, X.; Zhao, Y.N.; Li, Y.; Ruan, J.; Li, H.R.; Jin, T.; Yang, X.L. Chitosan influences the expression of P-gp and metabolism of norfloxacin in grass carp. J. Aquat. Anim. Health 2015, 27, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Tian, F.; Dahmani, F.Z.; Yang, H.; Yue, D.; He, S.; Zhou, J.; Yao, J. Curcumin-carboxymethyl chitosan (CNC) conjugate and CNC/LHR mixed polymeric micelles as new approaches to improve the oral absorption of P-gp substrate drugs. Drug Deliv. 2016, 23, 3424–3435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Li, X.; Huang, Y.; Liang, W. A hybrid thermo-sensitive chitosan gel for sustained release of meloxicam. J. Biomater. Sci. Polym. Ed. 2008, 19, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Duse, L.; Baghdan, E.; Pinnapireddy, S.R.; Engelhardt, K.H.; Jedelská, J.; Schaefer, J.; Quendt, P.; Bakowsky, U. Preparation and characterization of curcumin loaded chitosan nanoparticles for photodynamic therapy. Phys. Status Solidi Appl. Mater. Sci. 2017, 215, 1700709. [Google Scholar] [CrossRef]
- Chuah, L.H.; Billa, N.; Roberts, C.J.; Burley, J.C.; Manickam, S. Curcumin-containing chitosan nanoparticles as a potential mucoadhesive delivery system to the colon. Pharm. Dev. Technol. 2013, 18, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.; Hayes, J.; Yarbrough, J.; Veiga-Parga, T.; Greenacre, C. High-performance liquid chromatography determination of meloxicam and piroxicam with ultraviolet detection. Chromatogr. Res. Int. 2014, 2014, 521697. [Google Scholar] [CrossRef]
- Mendyk, A.; Jachowicz, R. Unified methodology of neural analysis in decision support systems built for pharmaceutical technology. Expert Syst. Appl. 2007, 32, 1124–1131. [Google Scholar] [CrossRef]
- Moeremans, I.; Devreese, M.; De Baere, S.; Croubels, S.; Hermans, K. Pharmacokinetics and absolute oral bioavailability of meloxicam in guinea pigs (Cavia porcellus). Vet. Anaesth. Analg. 2019, 46, 548–555. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Meth. Prog. Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | n | R2 | |||||
|---|---|---|---|---|---|---|---|
| Korsmeyer–Peppas Model | Zero Order | First Order | Higuchi | Baker–Lonsdale | |||
| CEMNPs | 4.0 | 0.66 | 0.994 | 0.858 | 0.11 | 0.276 | 0.982 |
| 7.4 | 0.67 | 0.997 | 0.939 | 0.127 | 0.217 | 0.988 | |
| Kinetic Parameter | Unit | Mean ± SE | ||
|---|---|---|---|---|
| Group-1 | Group-2 | Group-3 | ||
| λz | 1/h | 0.0578 ± 0.0008 | 0.0296 ± 0.002 | 0.0404 ± 0.0005 |
| t1/2 | h | 11.980 ± 0.175 a | 23.416 ± 2.471 b | 17.130 ± 0.263 a |
| Tmax(obs.) | h | 6 | 4 | 4 |
| Cmax(obs.) | μg·mL−1 | 16.905 ± 0.267 a | 13.185 ± 1.366 b | 25.52 ± 0.174 c |
| AUC(0–48) | μg/mL·h | 256.569 ± 2.022 a | 289.749 ± 17.993 a | 477.552 ± 7.99 b |
| AUC(0–∞) | μg/mL·h | 280.875 ± 1.60 | 387.299 ± 10.424 | 579.592 ± 16.596 |
| AUMC0-inf(obs.) | μg/mL·h2 | 5230.04 ± 67.82 a | 13230.251 ± 961.288 b | 15395.995 ± 921.371 b |
| MRT0-inf(obs.) | h | 18.62 ± 0.339 a | 34.160 ± 3.000 b | 26.563 ± 0.832 c |
| Vz/F(obs.) | (mg/kg)/(μg/mL) | 0.246 ± 0.004 | 0.0872 ± 0.011 | 0.170 ± 0.002 |
| Cl/F(obs.) | (mg/kg)/(μg/mL)/h | 0.0142 ± 0.00008 | 0.00258198 ± 0.000069 | 0.00690 ± 0.0002 |
| Vdss | L·kg−1 | 0.265 ± 0.006 a | 0.088 ± 0.016 b | 0.183 ± 0.0005 c |
| Concentration in Plasma (µg·mL−1) | Intraday Assay CV (%) | Interday Assay CV (%) |
|---|---|---|
| 1 | 7.64 | 5.94 |
| 10 | 5.12 | 7.15 |
| 25 | 3.15 | 6.49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yegireddy, M.; Nadoor, P.; Rao, S.; Hanumanthu, P.B.; Rajashekaraiah, R.; Ramachandrappa, S.C.; Halemani, G.M.; Mannem, S.; Prasad, T.N.V.K.V.; Ubaradka, S. Chitosan Encapsulated Meloxicam Nanoparticles for Sustained Drug Delivery Applications: Preparation, Characterization, and Pharmacokinetics in Wistar Rats. Molecules 2022, 27, 7312. https://doi.org/10.3390/molecules27217312
Yegireddy M, Nadoor P, Rao S, Hanumanthu PB, Rajashekaraiah R, Ramachandrappa SC, Halemani GM, Mannem S, Prasad TNVKV, Ubaradka S. Chitosan Encapsulated Meloxicam Nanoparticles for Sustained Drug Delivery Applications: Preparation, Characterization, and Pharmacokinetics in Wistar Rats. Molecules. 2022; 27(21):7312. https://doi.org/10.3390/molecules27217312
Chicago/Turabian StyleYegireddy, Muralidhar, Prakash Nadoor, Suguna Rao, Pavithra Balekatte Hanumanthu, Rashmi Rajashekaraiah, Santhosh Chickankandahalli Ramachandrappa, Girish Mallikarjun Halemani, Sravanthi Mannem, Tollamadugu Naga Venkata Krishna Vara Prasad, and Sunilchandra Ubaradka. 2022. "Chitosan Encapsulated Meloxicam Nanoparticles for Sustained Drug Delivery Applications: Preparation, Characterization, and Pharmacokinetics in Wistar Rats" Molecules 27, no. 21: 7312. https://doi.org/10.3390/molecules27217312
APA StyleYegireddy, M., Nadoor, P., Rao, S., Hanumanthu, P. B., Rajashekaraiah, R., Ramachandrappa, S. C., Halemani, G. M., Mannem, S., Prasad, T. N. V. K. V., & Ubaradka, S. (2022). Chitosan Encapsulated Meloxicam Nanoparticles for Sustained Drug Delivery Applications: Preparation, Characterization, and Pharmacokinetics in Wistar Rats. Molecules, 27(21), 7312. https://doi.org/10.3390/molecules27217312