
Conformational Stability and Denaturation Processes of Proteins Investigated by Electrophoresis under Extreme Conditions


{kind=link}
{kind=link}
{kind=link}
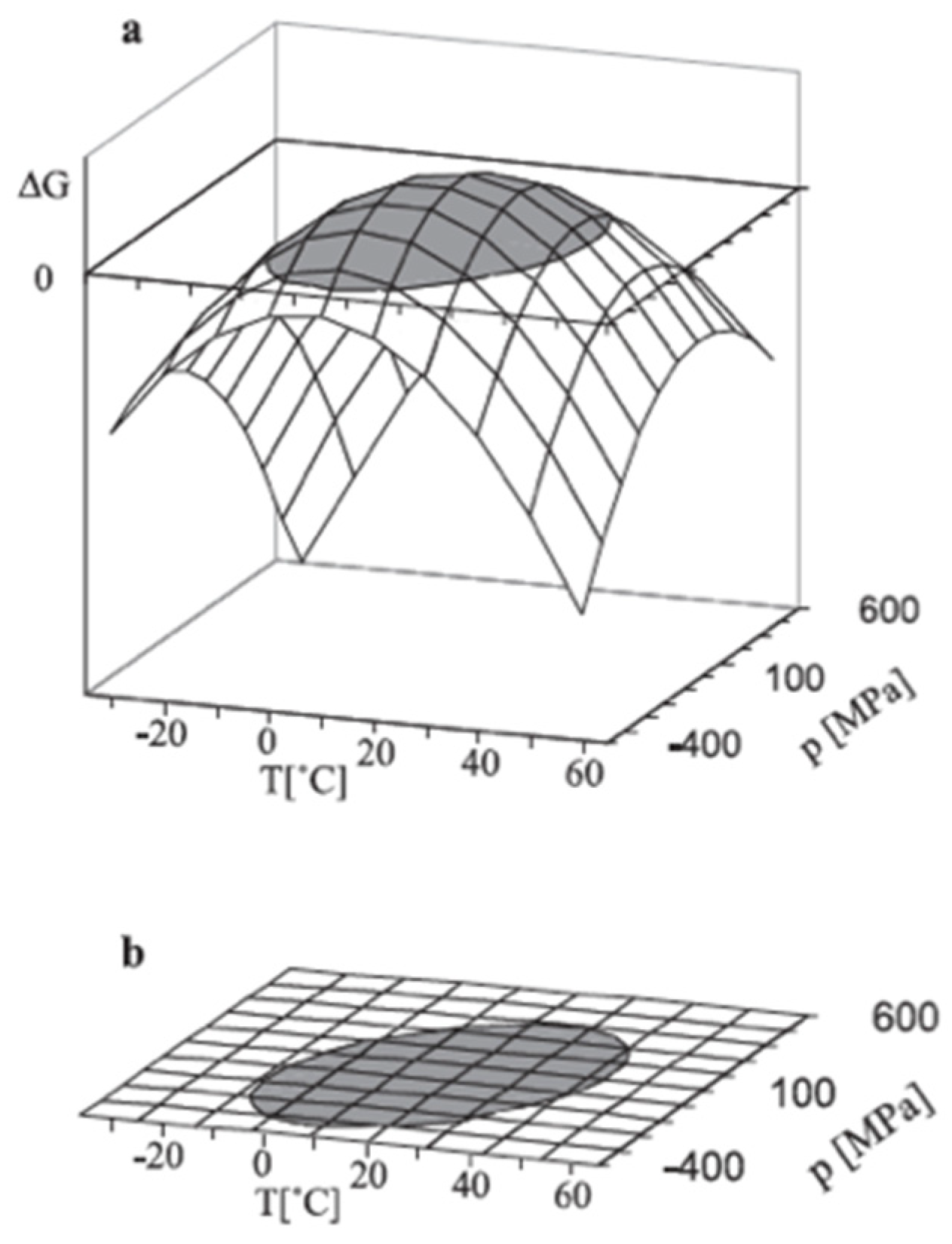{kind=link}
{kind=link}
{kind=link}
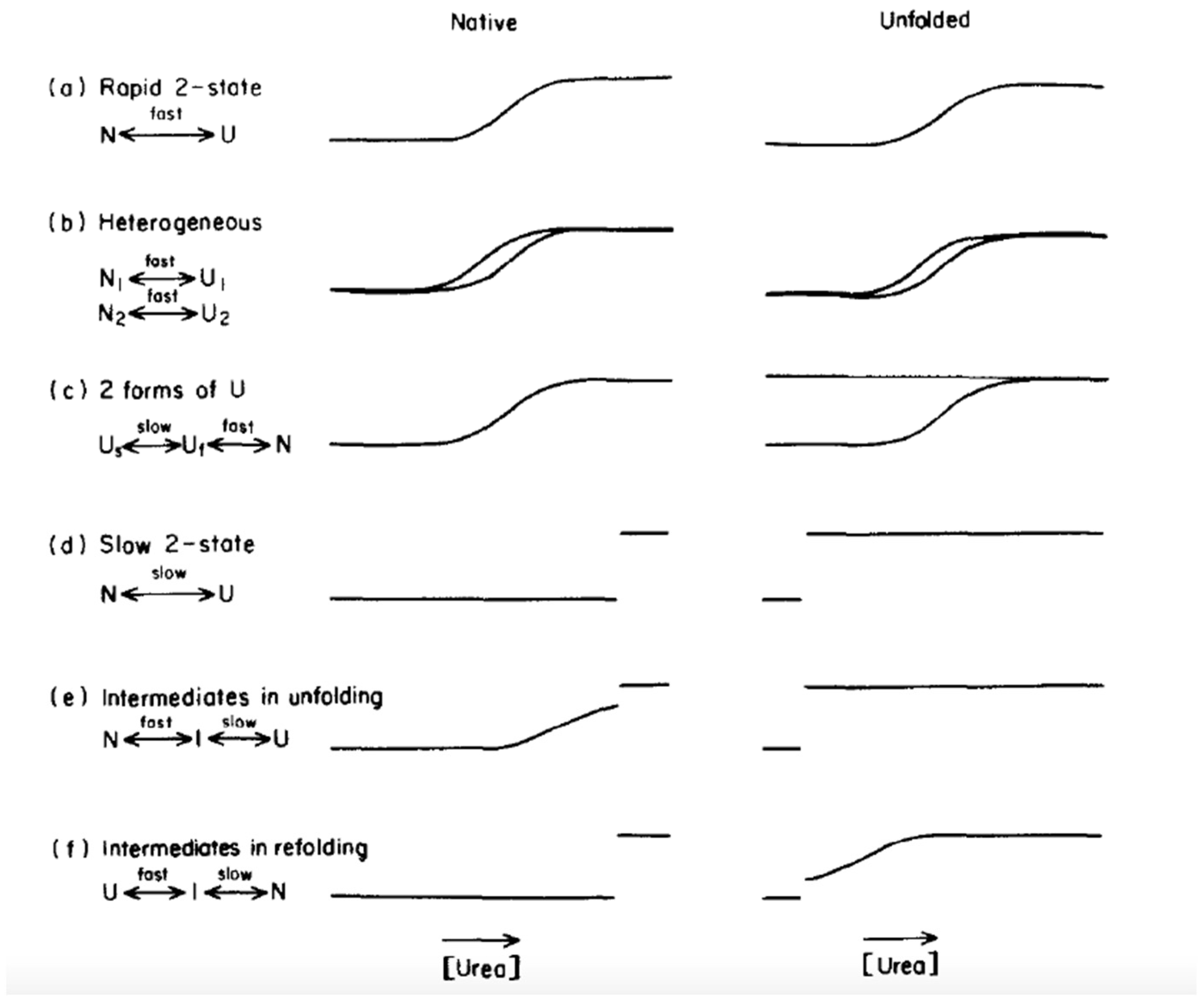{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Structural and Conformational Stability of Proteins
3. Protein Stability, Unfolding, Denaturation, and Deterioration
4. Phenomenology of Protein Denaturation
5. Analysis of Reversible Denaturation Processes
6. Determination of Protein Stability
6.1. Experimental Techniques
6.2. Computational Approaches
7. Electrophoresis of Proteins under Denaturing Conditions
7.1. Electrophoretic Mobility and Denaturation of Proteins
7.2. Electrophoresis under Hydrostatic Pressure
7.3. Electrophoresis in the Presence of Non-Charged Denaturing Agents
7.3.1. Analysis of Denaturation Transitions
7.3.2. Electrophoretic Profiles of Denaturation Transitions
7.3.3. Estimation of the Net Stability of Proteins from TUGGE Transition Curves
7.3.4. Other Denaturing Agents for TdGGE
7.4. Electrophoresis at Different Temperatures
7.4.1. Heat Unfolding Study by Transverse Temperature Gradient Gel Electrophoresis
7.4.2. TTGGE to Study Cold Denaturation of Proteins
7.4.3. Capillary Zone Electrophoresis at Different Temperatures
7.5. Electrophoresis after Exposure to Extreme Physical Conditions
7.5.1. Analysis of Renaturation Processes
7.5.2. Direct Observation of Unfolding Reversibility
7.5.3. Evidence for Irreversible Denaturation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
Units
References
- Pederson, T. Turning a PAGE: The overnight sensation of SDS-polyacrylamide gel electrophoresis. FASEB J. 2008, 22, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Filep, C.; Guttman, A. Capillary sodium dodecyl sulfate gel electrophoresis of proteins: Introducing the three dimensional Ferguson method. Anal. Chim. Acta 2021, 1183, 338958. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, D.; Guttman, A. Capillary Sodium Dodecyl Sulfate Agarose Gel Electrophoresis of Proteins. Gels 2022, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Righetti, P.G.; Verzola, B. Folding/unfolding/refolding of proteins: Present methodologies in comparison with capillary zone electrophoresis. Electrophoresis 2001, 22, 2359–2374. [Google Scholar] [CrossRef]
- Gavina, J.; Britz-McKibbin, P. Protein Unfolding and Conformational Studies by Capillary Electrophoresis. Curr. Anal. Chem. 2007, 3, 17–31. [Google Scholar] [CrossRef]
- Schopfer, L.M.; Lockridge, O. Mass Spectrometry Identifies Isopeptide Cross-Links Promoted by Diethylphosphorylated Lysine in Proteins Treated with Chlorpyrifos Oxon. Chem. Res. Toxicol. 2019, 32, 762–772. [Google Scholar] [CrossRef]
- Silman, I. The multiple biological roles of the cholinesterases. Prog. Biophys. Mol. Biol. 2021, 162, 41–56. [Google Scholar] [CrossRef]
- Xing, S.; Li, Q.; Xiong, B.; Chen, Y.; Feng, F.; Liu, W.; Sun, H. Structure and therapeutic uses of butyrylcholinesterase: Application in detoxification, Alzheimer’s disease, and fat metabolism. Med. Res. Rev. 2021, 41, 858–901. [Google Scholar] [CrossRef]
- Schopfer, L.M.; Delacour, H.; Masson, P.; Leroy, J.; Krejci, E.; Lockridge, O. The C5 Variant of the Butyrylcholinesterase Tetramer Includes a Noncovalently Bound 60 kDa Lamellipodin Fragment. Molecules 2017, 22, 1083. [Google Scholar] [CrossRef]
- Boyko, K.M.; Baymukhametov, T.N.; Chesnokov, Y.M.; Hons, M.; Lushchekina, S.V.; Konarev, P.V.; Lipkin, A.V.; Vasiliev, A.L.; Masson, P.; Popov, V.O.; et al. 3D structure of the natural tetrameric form of human butyrylcholinesterase as revealed by cryoEM, SAXS and MD. Biochimie 2019, 156, 196–205. [Google Scholar] [CrossRef]
- Leung, M.R.; van Bezouwen, L.S.; Schopfer, L.M.; Sussman, J.L.; Silman, I.; Lockridge, O.; Zeev-Ben-Mordehai, T. Cryo-EM structure of the native butyrylcholinesterase tetramer reveals a dimer of dimers stabilized by a superhelical assembly. Proc. Natl. Acad. Sci. USA 2018, 115, 13270–13275. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Blum, N.T.; Wu, J.; Lin, J.; Huang, P. Weaving Enzymes with Polymeric Shells for Biomedical Applications. Adv. Mater. 2021, 33, e2008438. [Google Scholar] [CrossRef] [PubMed]
- Bawa, R.; Szebeni, J.; Webster, T.J.; Audette, G.F. (Eds.) Immune Aspects of Biopharmaceuticals and Nanomedicines; Pan Stanford Publishing Pte. Ltd.: Singapore, 2019; p. 994. ISBN 978-981-4774-52-9. [Google Scholar] [CrossRef]
- Meunier, S.; de Bourayne, M.; Hamze, M.; Azam, A.; Correia, E.; Menier, C.; Maillere, B. Specificity of the T Cell Response to Protein Biopharmaceuticals. Front. Immunol. 2020, 11, 1550. [Google Scholar] [CrossRef]
- Wu, H. Studies on Denaturation of Proteins XIII. A Theory of Denaturation. In Advances in Protein Chemistry; Academic Press: Cambridge, MA, USA, 1995; Volume 46, pp. 6–26. [Google Scholar] [CrossRef]
- Kauzmann, W. Some Factors in the Interpretation of Protein Denaturation. In Advances in Protein Chemistry; Academic Press: Cambridge, MA, USA, 1959; Volume 14, pp. 1–63. [Google Scholar] [CrossRef]
- Privalov, P.L.; Gill, S.J. Stability of protein structure and hydrophobic interaction. Adv. Protein Chem. 1988, 39, 191–234. [Google Scholar] [CrossRef]
- Tanford, C. Protein denaturation. Adv. Protein Chem. 1968, 23, 121–282. [Google Scholar] [CrossRef]
- Tanford, C. Protein denaturation. C. Theoretical models for the mechanism of denaturation. Adv. Protein Chem. 1970, 24, 1–95. [Google Scholar] [CrossRef]
- Ptitsyn, O.B. Molten globule and protein folding. Adv. Protein Chem. 1995, 47, 83–229. [Google Scholar] [CrossRef]
- Sanchez-Ruiz, J.M. Protein kinetic stability. Biophys. Chem. 2010, 148, 1–15. [Google Scholar] [CrossRef]
- Hlodan, R.; Craig, S.; Pain, R.H. Protein Folding and its Implications for the Production of Recombinant Proteins. Biotechnol. Genet. Eng. Rev. 1991, 9, 47–88. [Google Scholar] [CrossRef]
- Sawada, S.; Akiyoshi, K. Nano-encapsulation of lipase by self-assembled nanogels: Induction of high enzyme activity and thermal stabilization. Macromol. Biosci. 2010, 10, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Protein Misfolding Diseases. Annu. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Privalov, P.L. Stability of Proteins Small Globular Proteins. Adv. Protein Chem. 1979, 33, 167–241. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Balny, C. Conformational plasticity of butyrylcholinesterase as revealed by high pressure experiments. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1990, 1041, 223–231. [Google Scholar] [CrossRef]
- Sanchez-Ruiz, J.M. Theoretical analysis of Lumry-Eyring models in differential scanning calorimetry. Biophys. J. 1992, 61, 921–935. [Google Scholar] [CrossRef]
- Whitaker, J.R.; Fujimaki, M. (Eds.) Chemical Deterioration of Proteins; American Chemical Society: Washington, DC, USA, 1980; Volume 123, p. 268. ISBN 9780841205437. [Google Scholar] [CrossRef]
- Lapanje, S. Physicochemical Aspects of Protein Denaturation; Wiley: New York, NY, USA, 1978; p. 331. ISBN 0471034096. [Google Scholar]
- Creighton, T.E. Experimental studies of protein folding and unfolding. Prog. Biophys. Mol. Biol. 1979, 33, 231–297. [Google Scholar] [CrossRef]
- Pace, C.N.; Scholtz, J.M. Measuring the conformational stability of a protein. In Protein Structure: A Practical Approach; Creighton, T.E., Ed.; IRL Press: New York, NY, USA, 1997; pp. 299–321. [Google Scholar]
- Righetti, P.G.; Gelfi, C.; Verzola, B.; Castelletti, L. The state of the art of dynamic coatings. Electrophoresis 2001, 22, 603–611. [Google Scholar] [CrossRef]
- Privalov, P.L. Cold denaturation of proteins. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 281–305. [Google Scholar] [CrossRef]
- Franks, F. Protein Destabilization at Low Temperatures. Adv. Protein Chem. 1995, 46, 105–139. [Google Scholar] [CrossRef]
- Ben-Naim, A. Theory of cold denaturation of proteins. Adv. Biol. Chem. 2013, 3, 29–39. [Google Scholar] [CrossRef]
- Sanfelice, D.; Temussi, P.A. Cold denaturation as a tool to measure protein stability. Biophys. Chem. 2016, 208, 4–8. [Google Scholar] [CrossRef]
- Pace, C.N.; Creighton, T.E. The disulphide folding pathway of ribonuclease T1. J. Mol. Biol. 1986, 188, 477–486. [Google Scholar] [CrossRef]
- Ahmad, F.; Bigelow, C.C. Estimation of the stability of globular proteins. Biopolymers 1986, 25, 1623–1633. [Google Scholar] [CrossRef]
- Pace, C.N.; Trevino, S.; Prabhakaran, E.; Scholtz, J.M. Protein structure, stability and solubility in water and other solvents. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 1225–1234, discussion 1234–1225. [Google Scholar] [CrossRef]
- Myers, J.K.; Pace, C.N.; Scholtz, J.M. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995, 4, 2138–2148. [Google Scholar] [CrossRef]
- Scholtz, J.M.; Grimsley, G.R.; Pace, C.N. Solvent Denaturation of Proteins and Interpretations of the m Value. Methods Enzymol. 2009, 466, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Amsdr, A.; Noudeh, N.D.; Liu, L.; Chalikian, T.V. On urea and temperature dependences of m-values. J. Chem. Phys. 2019, 150, 215103. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A. Reversible pressure--temperature denaturation of chymotrypsinogen. Biochemistry 1971, 10, 2436–2442. [Google Scholar] [CrossRef] [PubMed]
- Smeller, L. Pressure–temperature phase diagrams of biomolecules. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 2002, 1595, 11–29. [Google Scholar] [CrossRef]
- Peters, J. High Hydrostatic Pressure–A Key Element to Investigate Molecular Dynamics in Biosystems. Front. Phys. 2022, 9, 771. [Google Scholar] [CrossRef]
- Pfeil, W. The problem of the stability globular proteins. Mol. Cell. Biochem. 1981, 40, 3–28. [Google Scholar] [CrossRef]
- Douzou, P.; Balny, C. Protein Fractionation At Subzero Temperatures. Adv. Protein Chem. 1978, 32, 77–189. [Google Scholar] [CrossRef] [PubMed]
- Heremans, K. High pressure effects on proteins and other biomolecules. Annu. Rev. Biophys. Bioeng. 1982, 11, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kauzmann, W. Thermodynamics of unfolding. Nature 1987, 325, 763–764. [Google Scholar] [CrossRef]
- Chalikian, T.V. Volumetric properties of proteins. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 207–235. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Petsko, G.A. Molecular dynamics simulations in biology. Nature 1990, 347, 631–639. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F. Molecular dynamics studies of proteins. Curr. Opin. Struct. Biol. 1993, 3, 277–281. [Google Scholar] [CrossRef]
- Pikkemaat, M.G.; Linssen, A.B.; Berendsen, H.J.; Janssen, D.B. Molecular dynamics simulations as a tool for improving protein stability. Protein Eng. 2002, 15, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Childers, M.C.; Daggett, V. Insights from molecular dynamics simulations for computational protein design. Mol. Syst. Des. Eng. 2017, 2, 9–33. [Google Scholar] [CrossRef]
- Krieger, E.; Nabuurs, S.B.; Vriend, G. Homology Modeling. In Structural Bioinformatics; Bourne, P.E., Weissig, H., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; Volume 44, pp. 509–523. ISBN 9780471721208. [Google Scholar] [CrossRef]
- Franca, T.C. Homology modeling: An important tool for the drug discovery. J. Biomol. Struct. Dyn. 2015, 33, 1780–1793. [Google Scholar] [CrossRef]
- Hameduh, T.; Haddad, Y.; Adam, V.; Heger, Z. Homology modeling in the time of collective and artificial intelligence. Comput. Struct. Biotechnol. J. 2020, 18, 3494–3506. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Delacour, H.; Lushchekina, S.; Mabboux, I.; Bousquet, A.; Ceppa, F.; Schopfer, L.M.; Lockridge, O.; Masson, P. Characterization of a novel BCHE “silent” allele: Point mutation (p.Val204Asp) causes loss of activity and prolonged apnea with suxamethonium. PLoS ONE 2014, 9, e101552. [Google Scholar] [CrossRef] [PubMed]
- Delacour, H.; Lushchekina, S.; Mabboux, I.; Ceppa, F.; Masson, P.; Schopfer, L.M.; Lockridge, O. Characterization of a novel butyrylcholinesterase point mutation (p.Ala34Val), “silen” with mivacurium. Biochem. Pharm. 2014, 92, 476–483. [Google Scholar] [CrossRef]
- Dafferner, A.J.; Lushchekina, S.; Masson, P.; Xiao, G.; Schopfer, L.M.; Lockridge, O. Characterization of butyrylcholinesterase in bovine serum. Chem. Biol. Interact. 2017, 266, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Brazzolotto, X.; Courcelle, S.; Sauvanet, C.; Guillon, V.; Igert, A.; Kononchik, J.; Nachon, F.; Ceppa, F.; Delacour, H. Characterization of four BCHE mutations associated with prolonged effect of suxamethonium. Pharm. J. 2021, 21, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.A.M.; Loccisano, A.E.; Firestine, S.M.; Evanseck, J.D. Chapter 13 Principal Components Analysis: A Review of its Application on Molecular Dynamics Data. Annu. Rep. Comput. Chem. 2006, 2, 233–261. [Google Scholar] [CrossRef]
- Bode, C.; Kovacs, I.A.; Szalay, M.S.; Palotai, R.; Korcsmaros, T.; Csermely, P. Network analysis of protein dynamics. FEBS Lett. 2007, 581, 2776–2782. [Google Scholar] [CrossRef]
- Kasahara, K.; Fukuda, I.; Nakamura, H. A novel approach of dynamic cross correlation analysis on molecular dynamics simulations and its application to Ets1 dimer-DNA complex. PLoS ONE 2014, 9, e112419. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.R.; Bakan, A.; Shrivastava, I.H. Normal mode analysis of biomolecular structures: Functional mechanisms of membrane proteins. Chem. Rev. 2010, 110, 1463–1497. [Google Scholar] [CrossRef] [PubMed]
- Malmstrom, R.D.; Lee, C.T.; van Wart, A.; Amaro, R.E. On the Application of Molecular-Dynamics Based Markov State Models to Functional Proteins. J. Chem. Theory Comput. 2014, 10, 2648–2657. [Google Scholar] [CrossRef]
- Schütte, C.; Sarich, M. Metastability and Markov State Models in Molecular Dynamics: Modeling, Analysis, Algorithmic Approaches; American Mathematical Society: Providence, RI, USA, 2013; p. 128. [Google Scholar]
- Gao, K.; Wang, R.; Chen, J.; Cheng, L.; Frishcosy, J.; Huzumi, Y.; Qiu, Y.; Schluckbier, T.; Wei, X.; Wei, G.W. Methodology-Centered Review of Molecular Modeling, Simulation, and Prediction of SARS-CoV-2. Chem. Rev. 2022, 122, 11287–11368. [Google Scholar] [CrossRef]
- Zimmerman, M.I.; Hart, K.M.; Sibbald, C.A.; Frederick, T.E.; Jimah, J.R.; Knoverek, C.R.; Tolia, N.H.; Bowman, G.R. Prediction of New Stabilizing Mutations Based on Mechanistic Insights from Markov State Models. ACS Cent. Sci. 2017, 3, 1311–1321. [Google Scholar] [CrossRef]
- Cross, T.J.; Takahashi, G.R.; Diessner, E.M.; Crosby, M.G.; Farahmand, V.; Zhuang, S.; Butts, C.T.; Martin, R.W. Sequence Characterization and Molecular Modeling of Clinically Relevant Variants of the SARS-CoV-2 Main Protease. Biochemistry 2020, 59, 3741–3756. [Google Scholar] [CrossRef]
- Osuna, S. The challenge of predicting distal active site mutations in computational enzyme design. WIREs Comput. Mol. Sci. 2020, 11, e1502. [Google Scholar] [CrossRef]
- Sheik Amamuddy, O.; Afriyie Boateng, R.; Barozi, V.; Wavinya Nyamai, D.; Tastan Bishop, O. Novel dynamic residue network analysis approaches to study allosteric modulation: SARS-CoV-2 M(pro) and its evolutionary mutations as a case study. Comput. Struct. Biotechnol. J. 2021, 19, 6431–6455. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, N.; Baral, P.; Gerstman, B.S.; Chapagain, P.P. Structural and Dynamical Differences in the Spike Protein RBD in the SARS-CoV-2 Variants B.1.1.7 and B.1.351. J. Phys. Chem. B 2021, 125, 7101–7107. [Google Scholar] [CrossRef] [PubMed]
- Hantz, E.R.; Lindert, S. Adaptative Steered Molecular Dynamics Study of Mutagenesis Effects on Calcium Affinity in the Regulatory Domain of Cardiac Troponin C. J. Chem. Inf. Model. 2021, 61, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Hamelryck, T.; Borg, M.; Paluszewski, M.; Paulsen, J.; Frellsen, J.; Andreetta, C.; Boomsma, W.; Bottaro, S.; Ferkinghoff-Borg, J. Potentials of mean force for protein structure prediction vindicated, formalized and generalized. PLoS ONE 2010, 5, e13714. [Google Scholar] [CrossRef]
- Mukherjee, A.; Bhimalapuram, P.; Bagchi, B. Orientation-dependent potential of mean force for protein folding. J. Chem. Phys. 2005, 123, 014901. [Google Scholar] [CrossRef]
- Lee, T.S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; McGee, T.D., Jr.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; et al. Alchemical Binding Free Energy Calculations in AMBER20: Advances and Best Practices for Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5595–5623. [Google Scholar] [CrossRef]
- Meng, Y.; Dashti, D.S.; Roitberg, A.E. Computing Alchemical Free Energy Differences with Hamiltonian Replica Exchange Molecular Dynamics (H-REMD) Simulations. J. Chem. Theory Comput. 2011, 7, 2721–2727. [Google Scholar] [CrossRef]
- Clark, A.J.; Negron, C.; Hauser, K.; Sun, M.; Wang, L.; Abel, R.; Friesner, R.A. Relative Binding Affinity Prediction of Charge-Changing Sequence Mutations with FEP in Protein-Protein Interfaces. J. Mol. Biol. 2019, 431, 1481–1493. [Google Scholar] [CrossRef]
- Duan, J.; Lupyan, D.; Wang, L. Improving the Accuracy of Protein Thermostability Predictions for Single Point Mutations. Biophys. J. 2020, 119, 115–127. [Google Scholar] [CrossRef]
- Zou, J.; Simmerling, C.; Raleigh, D.P. Dissecting the Energetics of Intrinsically Disordered Proteins via a Hybrid Experimental and Computational Approach. J. Phys. Chem. B 2019, 123, 10394–10402. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Zhu, C.; Wang, L.; Abel, R.; Negron, C.; Pearlman, D.; Feyfant, E.; Duan, J.; Sherman, W. Predicting the Effect of Amino Acid Single-Point Mutations on Protein Stability-Large-Scale Validation of MD-Based Relative Free Energy Calculations. J. Mol. Biol. 2017, 429, 948–963. [Google Scholar] [CrossRef]
- Markthaler, D.; Fleck, M.; Stankiewicz, B.; Hansen, N. Exploring the Effect of Enhanced Sampling on Protein Stability Prediction. J. Chem. Theory Comput. 2022, 18, 2569–2583. [Google Scholar] [CrossRef]
- Ramadoss, V.; Dehez, F.; Chipot, C. AlaScan: A Graphical User Interface for Alanine Scanning Free-Energy Calculations. J. Chem. Inf. Model. 2016, 56, 1122–1126. [Google Scholar] [CrossRef]
- Gapsys, V.; Perez-Benito, L.; Aldeghi, M.; Seeliger, D.; van Vlijmen, H.; Tresadern, G.; de Groot, B.L. Large scale relative protein ligand binding affinities using non-equilibrium alchemy. Chem. Sci. 2019, 11, 1140–1152. [Google Scholar] [CrossRef]
- Novichkova, D.A.; Lushchekina, S.V.; Dym, O.; Masson, P.; Silman, I.; Sussman, J.L. The four-helix bundle in cholinesterase dimers: Structural and energetic determinants of stability. Chem. Biol. Interact. 2019, 309, 108699. [Google Scholar] [CrossRef]
- Van der Kamp, M.W.; Mulholland, A.J. Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 2013, 52, 2708–2728. [Google Scholar] [CrossRef]
- Ryde, U. QM/MM Calculations on Proteins. Methods Enzymol 2016, 577, 119–158. [Google Scholar] [CrossRef]
- Zoi, I.; Suarez, J.; Antoniou, D.; Cameron, S.A.; Schramm, V.L.; Schwartz, S.D. Modulating Enzyme Catalysis through Mutations Designed to Alter Rapid Protein Dynamics. J. Am. Chem. Soc. 2016, 138, 3403–3409. [Google Scholar] [CrossRef]
- Kamerlin, S.C.; Haranczyk, M.; Warshel, A. Progress in ab initio QM/MM free-energy simulations of electrostatic energies in proteins: Accelerated QM/MM studies of pKa, redox reactions and solvation free energies. J. Phys. Chem. B 2009, 113, 1253–1272. [Google Scholar] [CrossRef]
- Boselt, L.; Thurlemann, M.; Riniker, S. Machine Learning in QM/MM Molecular Dynamics Simulations of Condensed-Phase Systems. J. Chem. Theory Comput. 2021, 17, 2641–2658. [Google Scholar] [CrossRef]
- Cui, Q.; Pal, T.; Xie, L. Biomolecular QM/MM Simulations: What Are Some of the “Burning Issues”? J. Phys. Chem. B 2021, 125, 689–702. [Google Scholar] [CrossRef]
- Yang, Z.; Hajlasz, N.; Kulik, H.J. Computational Modeling of Conformer Stability in Benenodin-1, a Thermally Actuated Lasso Peptide Switch. J. Phys. Chem. B 2022, 126, 3398–3406. [Google Scholar] [CrossRef]
- Ngo, K.; Bruno da Silva, F.; Leite, V.B.P.; Contessoto, V.G.; Onuchic, J.N. Improving the Thermostability of Xylanase A from Bacillus subtilis by Combining Bioinformatics and Electrostatic Interactions Optimization. J. Phys. Chem. B 2021, 125, 4359–4367. [Google Scholar] [CrossRef]
- Kim, S.B.; Palmer, J.C.; Debenedetti, P.G. Computational investigation of cold denaturation in the Trp-cage miniprotein. Proc. Natl. Acad. Sci. USA 2016, 113, 8991–8996. [Google Scholar] [CrossRef]
- Yang, C.; Jang, S.; Pak, Y. Computational Probing of Temperature-Dependent Unfolding of a Small Globular Protein: From Cold to Heat Denaturation. J. Chem. Theory Comput. 2021, 17, 515–524. [Google Scholar] [CrossRef]
- Uralcan, B.; Debenedetti, P.G. Computational Investigation of the Effect of Pressure on Protein Stability. J. Phys. Chem. Lett. 2019, 10, 1894–1899. [Google Scholar] [CrossRef]
- Arsiccio, A.; Shea, J.E. Pressure Unfolding of Proteins: New Insights into the Role of Bound Water. J. Phys. Chem. B 2021, 125, 8431–8442. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.D.; Mironov, V.A.; Chapagain, P.P.; Nemukhin, A.V.; Krylov, A.I. Exploring structural and optical properties of fluorescent proteins by squeezing: Modeling high-pressure effects on the mStrawberry and mCherry red fluorescent proteins. J. Phys. Chem. B 2012, 116, 12426–12440. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bandyopadhyay, D.; Bhatnagar, A.; Jain, S.; Pratyaksh, P. Selective Stabilization of Aspartic Acid Protonation State within a Given Protein Conformation Occurs via Specific “Molecular Association”. J. Phys. Chem. B 2020, 124, 5350–5361. [Google Scholar] [CrossRef]
- Reilley, D.J.; Wang, J.; Dokholyan, N.V.; Alexandrova, A.N. Titr-DMD-A Rapid, Coarse-Grained Quasi-All-Atom Constant pH Molecular Dynamics Framework. J. Chem. Theory Comput. 2021, 17, 4538–4549. [Google Scholar] [CrossRef]
- Deng, J.; Cui, Q. Reverse Protonation of Buried Ion-Pairs in Staphylococcal Nuclease Mutants. J. Chem. Theory Comput. 2021, 17, 4550–4563. [Google Scholar] [CrossRef]
- Mukherjee, S.; Schafer, L.V. Spatially Resolved Hydration Thermodynamics in Biomolecular Systems. J. Phys. Chem. B 2022, 126, 3619–3631. [Google Scholar] [CrossRef] [PubMed]
- Canchi, D.R.; Garcia, A.E. Cosolvent effects on protein stability. Annu. Rev. Phys. Chem. 2013, 64, 273–293. [Google Scholar] [CrossRef]
- Masson, P.; Lushchekina, S.; Schopfer, L.M.; Lockridge, O. Effects of viscosity and osmotic stress on the reaction of human butyrylcholinesterase with cresyl saligenin phosphate, a toxicant related to aerotoxic syndrome: Kinetic and molecular dynamics studies. Biochem. J. 2013, 454, 387–399. [Google Scholar] [CrossRef]
- Zhang, D.; Lazim, R. Application of conventional molecular dynamics simulation in evaluating the stability of apomyoglobin in urea solution. Sci. Rep. 2017, 7, 44651. [Google Scholar] [CrossRef]
- Zueva, I.V.; Lushchekina, S.V.; Masson, P. Water structure changes in oxime-mediated reactivation process of phosphorylated human acetylcholinesterase. BioSci. Rep. 2018, 38, BSR20180609. [Google Scholar] [CrossRef]
- Gomez, D.; Huber, K.; Klumpp, S. On Protein Folding in Crowded Conditions. J. Phys. Chem. Lett. 2019, 10, 7650–7656. [Google Scholar] [CrossRef]
- Lushchekina, S.V.; Inidjel, G.; Martinez, N.; Masson, P.; Trovaslet-Leroy, M.; Nachon, F.; Koza, M.M.; Seydel, T.; Peters, J. Impact of Sucrose as Osmolyte on Molecular Dynamics of Mouse Acetylcholinesterase. Biomolecules 2020, 10, 1664. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Sen, P. Shape-Dependent Macromolecular Crowding on the Thermodynamics and Microsecond Conformational Dynamics of Protein Unfolding Revealed at the Single-Molecule Level. J. Phys. Chem. B 2020, 124, 5858–5871. [Google Scholar] [CrossRef] [PubMed]
- Rickard, M.M.; Zhang, Y.; Pogorelov, T.V.; Gruebele, M. Crowding, Sticking, and Partial Folding of GTT WW Domain in a Small Cytoplasm Model. J. Phys. Chem. B 2020, 124, 4732–4740. [Google Scholar] [CrossRef]
- Mukherjee, M.; Mondal, J. Unifying the Contrasting Mechanisms of Protein-Stabilizing Osmolytes. J. Phys. Chem. B 2020, 124, 6565–6574. [Google Scholar] [CrossRef] [PubMed]
- Contessoto, V.G.; Ferreira, P.H.B.; Chahine, J.; Leite, V.B.P.; Oliveira, R.J. Small Neutral Crowding Solute Effects on Protein Folding Thermodynamic Stability and Kinetics. J. Phys. Chem. B 2021, 125, 11673–11686. [Google Scholar] [CrossRef] [PubMed]
- Katava, M.; Stirnemann, G.; Pachetti, M.; Capaccioli, S.; Paciaroni, A.; Sterpone, F. Specific Interactions and Environment Flexibility Tune Protein Stability under Extreme Crowding. J. Phys. Chem. B 2021, 125, 6103–6111. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.M.; Shanmugam, M.; Kutta, R.J.; Scrutton, N.S.; Lovett, J.E.; Hay, S. Combined Pulsed Electron Double Resonance EPR and Molecular Dynamics Investigations of Calmodulin Suggest Effects of Crowding Agents on Protein Structures. Biochemistry 2022, 61, 1735–1742. [Google Scholar] [CrossRef]
- Broom, A.; Jacobi, Z.; Trainor, K.; Meiering, E.M. Computational tools help improve protein stability but with a solubility tradeoff. J. Biol. Chem. 2017, 292, 14349–14361. [Google Scholar] [CrossRef] [PubMed]
- Qing, R.; Hao, S.; Smorodina, E.; Jin, D.; Zalevsky, A.; Zhang, S. Protein Design: From the Aspect of Water Solubility and Stability. Chem. Rev. 2022, 122, 14085–14179. [Google Scholar] [CrossRef]
- Geng, C.; Xue, L.C.; Roel-Touris, J.; Bonvin, A.M.J.J. Finding the ΔΔG spot: Are predictors of binding affinity changes upon mutations in protein–protein interactions ready for it? WIREs Comput. Mol. Sci. 2019, 9, e1410. [Google Scholar] [CrossRef]
- Piana, S.; Klepeis, J.L.; Shaw, D.E. Assessing the accuracy of physical models used in protein-folding simulations: Quantitative evidence from long molecular dynamics simulations. Curr. Opin. Struct. Biol. 2014, 24, 98–105. [Google Scholar] [CrossRef]
- Daggett, V. Molecular dynamics simulations of the protein unfolding/folding reaction. Acc. Chem. Res. 2002, 35, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Freddolino, P.L.; Harrison, C.B.; Liu, Y.; Schulten, K. Challenges in protein folding simulations: Timescale, representation, and analysis. Nat. Phys. 2010, 6, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Xiao, S.; Simmerling, C.; Raleigh, D.P. Quantitative Analysis of Protein Unfolded State Energetics: Experimental and Computational Studies Demonstrate That Non-Native Side-Chain Interactions Stabilize Local Native Backbone Structure. J. Phys. Chem. B 2021, 125, 3269–3277. [Google Scholar] [CrossRef]
- Klein, F.; Barrera, E.E.; Pantano, S. Assessing SIRAH’s Capability to Simulate Intrinsically Disordered Proteins and Peptides. J. Chem. Theory Comput. 2021, 17, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, E.H.; Leaver-Fay, A.; Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 2011, 79, 830–838. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Kassab, R.; Krishnan, K. Landscape-Based Protein Stability Analysis and Network Modeling of Multiple Conformational States of the SARS-CoV-2 Spike D614G Mutant: Conformational Plasticity and Frustration-Induced Allostery as Energetic Drivers of Highly Transmissible Spike Variants. J. Chem. Inf. Model. 2022, 62, 1956–1978. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, S.; Wang, W.; Pang, L.; Zhang, Q.; Liu, X. Mutation-Induced Impacts on the Switch Transformations of the GDP- and GTP-Bound K-Ras: Insights from Multiple Replica Gaussian Accelerated Molecular Dynamics and Free Energy Analysis. J. Chem. Inf. Model. 2021, 61, 1954–1969. [Google Scholar] [CrossRef]
- Yang, Y.I.; Shao, Q.; Zhang, J.; Yang, L.; Gao, Y.Q. Enhanced sampling in molecular dynamics. J. Chem. Phys. 2019, 151, 070902. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta 2015, 1850, 872–877. [Google Scholar] [CrossRef]
- Hénin, J.; Lelièvre, T.; Shirts, M.R.; Valsson, O.; Delemotte, L. Enhanced sampling methods for molecular dynamics simulations. arXiv 2022, arXiv:2202.04164. [Google Scholar] [CrossRef]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef]
- Smith, Z.; Ravindra, P.; Wang, Y.; Cooley, R.; Tiwary, P. Discovering Protein Conformational Flexibility through Artificial-Intelligence-Aided Molecular Dynamics. J. Phys. Chem. B 2020, 124, 8221–8229. [Google Scholar] [CrossRef]
- Leidner, F.; Kurt Yilmaz, N.; Schiffer, C.A. Deciphering Complex Mechanisms of Resistance and Loss of Potency through Coupled Molecular Dynamics and Machine Learning. J. Chem. Theory Comput. 2021, 17, 2054–2064. [Google Scholar] [CrossRef]
- Kleiman, D.E.; Shukla, D. Multiagent Reinforcement Learning-Based Adaptive Sampling for Conformational Dynamics of Proteins. J. Chem. Theory Comput. 2022, 18, 5422–5434. [Google Scholar] [CrossRef]
- Khan, S.; Vihinen, M. Performance of protein stability predictors. Hum. Mutat. 2010, 31, 675–684. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011, 12, 151. [Google Scholar] [CrossRef]
- Montanucci, L.; Capriotti, E.; Birolo, G.; Benevenuta, S.; Pancotti, C.; Lal, D.; Fariselli, P. DDGun: An untrained predictor of protein stability changes upon amino acid variants. Nucleic Acids Res. 2022, 50, 222–227. [Google Scholar] [CrossRef]
- Garcia-Cebollada, H.; Lopez, A.; Sancho, J. Protposer: The web server that readily proposes protein stabilizing mutations with high PPV. Comput. Struct. Biotechnol. J. 2022, 20, 2415–2433. [Google Scholar] [CrossRef]
- Pan, Q.; Nguyen, T.B.; Ascher, D.B.; Pires, D.E.V. Systematic evaluation of computational tools to predict the effects of mutations on protein stability in the absence of experimental structures. Brief. Bioinform. 2022, 23, bbac025. [Google Scholar] [CrossRef]
- Parlade, E.; Volta-Duran, E.; Cano-Garrido, O.; Sanchez, J.M.; Unzueta, U.; Lopez-Laguna, H.; Serna, N.; Cano, M.; Rodriguez-Mariscal, M.; Vazquez, E.; et al. An In Silico Methodology That Facilitates Decision Making in the Engineering of Nanoscale Protein Materials. Int. J. Mol. Sci. 2022, 23, 4958. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef]
- Simons, K.T.; Bonneau, R.; Ruczinski, I.; Baker, D. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins Struct. Funct. Bioinform. 1999, 37, 171–176. [Google Scholar] [CrossRef]
- Ibarra, A.A.; Bartlett, G.J.; Hegedus, Z.; Dutt, S.; Hobor, F.; Horner, K.A.; Hetherington, K.; Spence, K.; Nelson, A.; Edwards, T.A.; et al. Predicting and Experimentally Validating Hot-Spot Residues at Protein-Protein Interfaces. ACS Chem. Biol. 2019, 14, 2252–2263. [Google Scholar] [CrossRef]
- Shorthouse, D.; Hall, M.W.J.; Hall, B.A. Computational Saturation Screen Reveals the Landscape of Mutations in Human Fumarate Hydratase. J. Chem. Inf. Model. 2021, 61, 1970–1980. [Google Scholar] [CrossRef]
- Polyakov, I.V.; Kniga, A.E.; Grigorenko, B.L.; Nemukhin, A.V. Structure of the Brain N-Acetylaspartate Biosynthetic Enzyme NAT8L Revealed by Computer Modeling. ACS Chem. Neurosci. 2020, 11, 2296–2302. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, R.; Shao, W.; Wang, J.; Silman, I.; Sussman, J.L. Do Newly Born Orphan Proteins Resemble Never Born Proteins? A Study using Deep Learning Algorithms. Proteins 2022, in press. [CrossRef]
- Wei, G.W.; Zhu, F.; Merz, K.M. Editorial on Machine Learning. J. Chem. Inf. Model. 2022, 62, 3941. [Google Scholar] [CrossRef]
- Mulnaes, D.; Porta, N.; Clemens, R.; Apanasenko, I.; Reiners, J.; Gremer, L.; Neudecker, P.; Smits, S.H.J.; Gohlke, H. TopModel: Template-Based Protein Structure Prediction at Low Sequence Identity Using Top-Down Consensus and Deep Neural Networks. J. Chem. Theory Comput. 2020, 16, 1953–1967. [Google Scholar] [CrossRef]
- Shamsi, Z.; Chan, M.; Shukla, D. TLmutation: Predicting the Effects of Mutations Using Transfer Learning. J. Phys. Chem. B 2020, 124, 3845–3854. [Google Scholar] [CrossRef]
- Casadio, R.; Savojardo, C.; Fariselli, P.; Capriotti, E.; Martelli, P.L. Turning Failures into Applications: The Problem of Protein DeltaDeltaG Prediction. Methods Mol. Biol. 2022, 2449, 169–185. [Google Scholar] [CrossRef]
- Pucci, F.; Schwersensky, M.; Rooman, M. Artificial intelligence challenges for predicting the impact of mutations on protein stability. Curr. Opin. Struct. Biol. 2022, 72, 161–168. [Google Scholar] [CrossRef]
- Pancotti, C.; Benevenuta, S.; Birolo, G.; Alberini, V.; Repetto, V.; Sanavia, T.; Capriotti, E.; Fariselli, P. Predicting protein stability changes upon single-point mutation: A thorough comparison of the available tools on a new dataset. Brief. Bioinform. 2022, 23, bbab555. [Google Scholar] [CrossRef]
- Vila, J.A. Proteins’ Evolution upon Point Mutations. ACS Omega 2022, 7, 14371–14376. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Cramer, P. AlphaFold2 and the future of structural biology. Nat. Struct. Mol. Biol. 2021, 28, 704–705. [Google Scholar] [CrossRef]
- Serpell, L.C.; Radford, S.E.; Otzen, D.E. AlphaFold: A Special Issue and A Special Time for Protein Science. J. Mol. Biol. 2021, 433, 167231. [Google Scholar] [CrossRef]
- Buel, G.R.; Walters, K.J. Can AlphaFold2 predict the impact of missense mutations on structure? Nat. Struct. Mol. Biol. 2022, 29, 1–2. [Google Scholar] [CrossRef]
- Diwan, G.D.; Gonzalez-Sanchez, J.C.; Apic, G.; Russell, R.B. Next Generation Protein Structure Predictions and Genetic Variant Interpretation. J. Mol. Biol. 2021, 433, 167180. [Google Scholar] [CrossRef]
- Sen, N.; Anishchenko, I.; Bordin, N.; Sillitoe, I.; Velankar, S.; Baker, D.; Orengo, C. Characterizing and explaining the impact of disease-associated mutations in proteins without known structures or structural homologs. Brief. Bioinform. 2022, 23, bbac187. [Google Scholar] [CrossRef]
- Iqbal, S.; Ge, F.; Li, F.; Akutsu, T.; Zheng, Y.; Gasser, R.B.; Yu, D.J.; Webb, G.I.; Song, J. PROST: AlphaFold2-aware Sequence-Based Predictor to Estimate Protein Stability Changes upon Missense Mutations. J. Chem. Inf. Model. 2022, 62, 4270–4282. [Google Scholar] [CrossRef]
- Creighton, T.E. Counting integral numbers of amino acid residues per polypeptide chain. Nature 1980, 284, 487–489. [Google Scholar] [CrossRef]
- Sculley, M.J.; Treacy, O.B.; Jeffrey, P.D. A new theoretical approach to the investigation of the symmetry of protein oligomers with bifunctional reagents. Biophys. Chem. 1984, 19, 39–47. [Google Scholar] [CrossRef]
- Goldenberg, D.P.; Creighton, T.E. Gel electrophoresis in studies of protein conformation and folding. Anal. Biochem. 1984, 138, 1–18. [Google Scholar] [CrossRef]
- Gianazza, E.; Eberini, I.; Santi, O.; Vignati, M. Denaturant-gradient gel electrophoresis: Technical aspects and practical applications. Anal. Chim. Acta 1998, 372, 99–120. [Google Scholar] [CrossRef]
- Rodbard, D.; Chrambach, A. Estimation of molecular radius, free mobility, and valence using polyacrylamide gel electrophoresis. Anal. Biochem. 1971, 40, 95–134. [Google Scholar] [CrossRef]
- Chung, M.; Kim, D.; Herr, A.E. Polymer sieving matrices in microanalytical electrophoresis. Analyst 2014, 139, 5635–5654. [Google Scholar] [CrossRef]
- Cléry, C.; Renault, F.; Masson, P. Pressure-induced molten globule state of cholinesterase. FEBS Lett. 1995, 370, 212–214. [Google Scholar] [CrossRef]
- Masson, P. Electrophoresis of proteins under high hydrostatic pressure. In High-Pressure Techniques in Chemistry and Physics: A Practical Approach; Holzapfel, W.B., Isaacs, N.S., Eds.; Oxford University Press: Oxford, UK, 1997; pp. 353–373. [Google Scholar]
- Chen, B.; Chrambach, A.; Rodbard, D. Continuous optical scanning in polyacrylamide gel electrophoresis: Estimation of the apparent diffusion coefficient of β-lactoglobulin B. Anal. Biochem. 1979, 97, 120–130. [Google Scholar] [CrossRef]
- Nyström, B.; Roots, J. Dynamic light scattering studies of protein solutions under high pressure. J. Chem. Phys. 1983, 78, 2833–2837. [Google Scholar] [CrossRef]
- Hawley, S.A. Electrophoretic separation of conformational states of α-chymotrypsinogen A at high pressures. Biochim. Biophys. Acta (BBA)-Protein Struct. 1973, 317, 236–239. [Google Scholar] [CrossRef]
- Hawley, S.A.; Mitchell, R.M. An electrophoretic study of reversible protein denaturation: Chymotrypsinogen at high pressures. Biochemistry 1975, 14, 3257–3264. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Reybaud, J. Hydrophobic interaction electrophoresis under high hydrostatic pressure: Study of the effects of pressure upon the interaction of serum albumin with a long-chain aliphatic ligand. Electrophoresis 1988, 9, 157–161. [Google Scholar] [CrossRef]
- Paladini, A.A.; Weber, G.; Erijman, L. Analysis of dissociation and unfolding of oligomeric proteins using a flat bed gel electrophoresis at high pressure. Anal. Biochem. 1994, 218, 364–369. [Google Scholar] [CrossRef]
- Paladini, A.A.; Silva, J.L.; Weber, G. Slab gel electrophoresis of oligomeric proteins under high hydrostatic pressure. Anal. Biochem. 1987, 161, 358–364. [Google Scholar] [CrossRef]
- Masson, P.; Arciero, D.M.; Hooper, A.B.; Balny, C. Electrophoresis at elevated hydrostatic pressure of the multiheme hydroxylamine oxidoreductase. Electrophoresis 1990, 11, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Clery-Barraud, C.; Renault, F.; Leva, J.; El Bakdouri, N.; Masson, P.; Rochu, D. Exploring the structural and functional stabilities of different paraoxonase-1 formulations through electrophoretic mobilities and enzyme activity parameters under hydrostatic pressure. Biochim. Biophys. Acta 2009, 1794, 680–688. [Google Scholar] [CrossRef]
- Erijman, L.; Clegg, R.M. High pressure electrophoresis in narrow bore glass tubes: One- and two-dimensional separations of protein subunits. Rev. Sci. Instrum. 1996, 67, 813–817. [Google Scholar] [CrossRef]
- Masson, P.; Cléry, C. Pressure-induced molten globule states of proteins. In Proceedings of the High Pressure Bioscience and Biotechnology, Kyoto, Japan, 5–9 November 1995; Elsevier: Amsterdam, The Netherlands, 1996; pp. 117–126. [Google Scholar]
- Clery-Barraud, C.; Ordentlich, A.; Grosfeld, H.; Shafferman, A.; Masson, P. Pressure and heat inactivation of recombinant human acetylcholinesterase. Importance of residue E202 for enzyme stability. Eur. J. Biochem. 2002, 269, 4297–4307. [Google Scholar] [CrossRef]
- Marion, J.; Trovaslet, M.; Martinez, N.; Masson, P.; Schweins, R.; Nachon, F.; Trapp, M.; Peters, J. Pressure-induced molten globule state of human acetylcholinesterase: Structural and dynamical changes monitored by neutron scattering. Phys. Chem. Chem. Phys. 2015, 17, 3157–3163. [Google Scholar] [CrossRef]
- Gentile, F.; Veneziani, B.M.; Sellitto, C. Polyacrylamide gel electrophoresis in discontinuous transverse urea-gradient gels. Anal. Biochem. 1997, 244, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Creighton, T.E.; Pain, R.H. Unfolding and refolding of Staphylococcus aureus penicillinase by urea-gradient electrophoresis. J. Mol. Biol. 1980, 137, 431–436. [Google Scholar] [CrossRef]
- Creighton, T.E. Kinetic study of protein unfolding and refolding using urea gradient electrophoresis. J. Mol. Biol. 1980, 137, 61–80. [Google Scholar] [CrossRef]
- van den Oetelaar, P.J.M.; de Man, B.M.; Hoenders, H.J. Protein folding and aggregation studied by isoelectric focusing across a urea gradient and isoelectric focusing in two dimensions. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1989, 995, 82–90. [Google Scholar] [CrossRef]
- Beringhelli, T.; Eberini, I.; Galliano, M.; Pedoto, A.; Perduca, M.; Sportiello, A.; Fontana, E.; Monaco, H.L.; Gianazza, E. pH and ionic strength dependence of protein (un)folding and ligand binding to bovine beta-lactoglobulins A and B. Biochemistry 2002, 41, 15415–15422. [Google Scholar] [CrossRef]
- Creighton, T.E. Electrophoretic analysis of the unfolding of proteins by urea. J. Mol. Biol. 1979, 129, 235–264. [Google Scholar] [CrossRef]
- Masson, P.; Goasdoue, J.-L. Evidence that the conformational stability of ‘aged’ organophosphate-inhibited cholinesterase is altered. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1986, 869, 304–313. [Google Scholar] [CrossRef]
- Masson, P.; Lockridge, O. Butyrylcholinesterase for protection from organophosphorus poisons: Catalytic complexities and hysteretic behavior. Arch. Biochem. Biophys. 2010, 494, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Juul, P. Human plasma cholinesterase isoenzymes. Clin. Chim. Acta 1968, 19, 205–213. [Google Scholar] [CrossRef]
- Laurents, D.V.; Huyghues-Despointes, B.M.P.; Bruix, M.; Thurlkill, R.L.; Schell, D.; Newsom, S.; Grimsley, G.R.; Shaw, K.L.; Treviño, S.; Rico, M.; et al. Charge–Charge Interactions are Key Determinants of the pK Values of Ionizable Groups in Ribonuclease Sa (pI = 3.5) and a Basic Variant (pI = 10.2). J. Mol. Biol. 2003, 325, 1077–1092. [Google Scholar] [CrossRef]
- Ewbank, J.J.; Creighton, T.E. Structural characterization of the disulfide folding intermediates of bovine alpha-lactalbumin. Biochemistry 1993, 32, 3694–3707. [Google Scholar] [CrossRef]
- Klemm, J.D.; Wozniak, J.A.; Alber, T.; Goldenberg, D.P. Correlation between mutational destabilization of phage T4 lysozyme and increased unfolding rates. Biochemistry 1991, 30, 589–594. [Google Scholar] [CrossRef]
- Creighton, T.E.; Shortle, D. Electrophoretic characterization of the denatured states of staphylococcal nuclease. J. Mol. Biol. 1994, 242, 670–682. [Google Scholar] [CrossRef]
- Darby, N.J.; Kemmink, J.; Creighton, T.E. Identifying and characterizing a structural domain of protein disulfide isomerase. Biochemistry 1996, 35, 10517–10528. [Google Scholar] [CrossRef]
- Hollecker, M.; Creighton, T.E. Effect on protein stability of reversing the charge on amino groups. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1982, 701, 395–404. [Google Scholar] [CrossRef]
- Matthews, C.R.; Crisanti, M.M. Urea-induced unfolding of the alpha subunit of tryptophan synthase: Evidence for a multistate process. Biochemistry 1981, 20, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.W.; Williams, J. The electrophoresis of transferrins in urea/polyacrylamide gels. Biochem. J. 1980, 189, 541–546. [Google Scholar] [CrossRef]
- Gianazza, E.; Calabresi, L.; Santi, O.; Sirtori, C.R.; Franceschini, G. Denaturation and self-association of apolipoprotein A-I investigated by electrophoretic techniques. Biochemistry 1997, 36, 7898–7905. [Google Scholar] [CrossRef]
- Carra, J.H.; Privalov, P.L. Energetics of denaturation and m values of staphylococcal nuclease mutants. Biochemistry 1995, 34, 2034–2041. [Google Scholar] [CrossRef]
- Masson, P.; Gouet, P.; Clery, C. Pressure and propylene carbonate denaturation of native and “aged” phosphorylated cholinesterase. J. Mol. Biol. 1994, 238, 466–478. [Google Scholar] [CrossRef]
- Pace, C.N.; Marshall, H.F. A comparison of the effectiveness of protein denaturants for β-lactoglobulin and ribonuclease. Arch. Biochem. Biophys. 1980, 199, 270–276. [Google Scholar] [CrossRef]
- Artoni, G.; Gianazza, E.; Zanoni, M.; Gelfi, C.; Tanzi, M.C.; Barozzi, C.; Ferruti, P.; Righetti, P.G. Fractionation techniques in a hydro-organic environment: II. Acryloyl-morpholine polymers as a matrix for electrophoresis in hydro-organic solvents. Anal. Biochem. 1984, 137, 420–428. [Google Scholar] [CrossRef]
- Zazra, J.J.; Szklaruk, B. Tetramethylurea as a protein denaturing agent in gel electrophoresis. Electrophoresis 1987, 8, 331–332. [Google Scholar] [CrossRef]
- Vecchio, G.; Righetti, P.G.; Zanoni, M.; Artoni, G.; Gianazza, E. Fractionation techniques in a hydro-organic environment: I. Sulfolane as a solvent for hydrophobic proteins. Anal. Biochem. 1984, 137, 410–419. [Google Scholar] [CrossRef]
- Thatcher, D.R.; Hodson, B. Denaturation of proteins and nucleic acids by thermal-gradient electrophoresis. Biochem. J. 1981, 197, 105–109. [Google Scholar] [CrossRef]
- Thatcher, D.R.; Sheikh, R. The relative conformational stability of the alcohol dehydrogenase alleloenzymes of the fruitfly Drosophila melanogaster. Biochem. J. 1981, 197, 111–117. [Google Scholar] [CrossRef]
- Rosenbaum, V.; Riesner, D. Temperature-gradient gel electrophoresis. Biophys. Chem. 1987, 26, 235–246. [Google Scholar] [CrossRef]
- Riesner, D.; Henco, K.; Steger, G. Temperature-Gradient Gel Electrophoresis: A method for the analysis of conformational transitions and mutations in nucleic acids and proteins. In Advances in Electrophoresis; Chrambach, A., Dunn, M.J., Radola, B.J., Eds.; VCH: Weinheim, Germany, 1991; Volume 4, pp. 169–250. [Google Scholar]
- Birmes, A.; Sattler, A.; Maurer, K.H.; Riesner, D. Analysis of the conformational transitions of proteins by temperature-gradient gel electrophoresis. Electrophoresis 1990, 11, 795–801. [Google Scholar] [CrossRef]
- Arakawa, T.; Hung, L.; Pan, V.; Horan, T.P.; Kolvenbach, C.G.; Narhi, L.O. Analysis of the heat-induced denaturation of proteins using temperature gradient gel electrophoresis. Anal. Biochem. 1993, 208, 255–259. [Google Scholar] [CrossRef]
- Perrella, M.; Heyda, A.; Mosca, A.; Rossi-Bernardi, L. Isoelectric focusing and electrophoresis at subzero temperatures. Anal. Biochem. 1978, 88, 212–224. [Google Scholar] [CrossRef]
- Perrella, M.; Cremonesi, L.; Benazzi, L.; Rossi-Bernardi, L. Isolation of intermediate valence hybrids between ferrous and methemoglobin at subzero temperatures. J. Biol. Chem. 1981, 256, 11098–11103. [Google Scholar] [CrossRef]
- Franks, F.; Eagland, D. The role of solvent interactions in protein conformation. CRC Crit. Rev. Biochem. 1975, 3, 165–219. [Google Scholar] [CrossRef]
- Perrella, M.; Samaja, M.; Rossi-Bernardi, L. Hybrid formation for liganded hemoglobins A and C at subzero temperatures. J. Biol. Chem. 1979, 254, 8748–8750. [Google Scholar] [CrossRef]
- Perrella, M.; Benazzi, L.; Cremonesi, L.; Vesely, S.; Viggiano, G.; Berger, R.L. Subzero temperature quenching and electrophoretic methods for the isolation of protein reaction intermediates. J. Biochem. Biophys. Methods 1983, 7, 187–197. [Google Scholar] [CrossRef]
- Curtil, C.; Channac, L.; Ebel, C.; Masson, P. Cold-induced conformational changes of ribonuclease A as investigated by subzero transverse temperature gradient gel electrophoresis. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1994, 1208, 1–7. [Google Scholar] [CrossRef]
- Rochu, D.; Masson, P. Multiple advantages of capillary zone electrophoresis for exploring protein conformational stability. Electrophoresis 2002, 23, 189–202. [Google Scholar] [CrossRef]
- Rochu, D.; Ducret, G.; Ribes, F.; Vanin, S.; Masson, P. Capillary zone electrophoresis with optimized temperature control for studying thermal denaturation of proteins at various pH. Electrophoresis 1999, 20, 1586–1594. [Google Scholar] [CrossRef]
- Rochu, D.; Georges, C.; Répiton, J.; Viguié, N.; Saliou, B.; Bon, C.; Masson, P. Thermal stability of acetylcholinesterase from Bungarus fasciatus venom as investigated by capillary electrophoresis. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 2001, 1545, 216–226. [Google Scholar] [CrossRef]
- Rochu, D.; Pernet, T.; Renault, F.; Bon, C.; Masson, P. Dual effect of high electric field in capillary electrophoresis study of the conformational stability of Bungarus fasciatus acetylcholinesterase. J. Chromatogr. A 2001, 910, 347–357. [Google Scholar] [CrossRef]
- Rochu, D.; Renault, F.; Clery-Barraud, C.; Chabriere, E.; Masson, P. Stability of highly purified human paraoxonase (PON1): Association with human phosphate binding protein (HPBP) is essential for preserving its active conformation(s). Biochim. Biophys. Acta 2007, 1774, 874–883. [Google Scholar] [CrossRef]
- Rochu, D.; Renault, F.; Masson, P. Detection of unwanted protein-bound ligands by capillary zone electrophoresis: The case of hidden ligands that stabilize cholinesterase conformation. Electrophoresis 2002, 23, 930–937. [Google Scholar] [CrossRef]
- Rochu, D.; Clery-Barraud, C.; Renault, F.; Chevalier, A.; Bon, C.; Masson, P. Capillary electrophoresis versus differential scanning calorimetry for the analysis of free enzyme versus enzyme-ligand complexes: In the search of the ligand-free status of cholinesterases. Electrophoresis 2006, 27, 442–451. [Google Scholar] [CrossRef]
- Rochu, D.; Ducret, G.; Masson, P. Measuring conformational stability of proteins using an optimized temperature-controlled capillary electrophoresis approach. J. Chromatogr. A 1999, 838, 157–165. [Google Scholar] [CrossRef]
- Creighton, T.E. Detection of folding intermediates using urea-gradient electrophoresis. Methods Enzymol. 1986, 131, 156–172. [Google Scholar] [CrossRef]
- Hawley, S.A.; Macleod, R.M. Electrophoretic separation of molecular species associated with the thermal transition of chymotrypsinogen A. J. Mol. Biol. 1976, 103, 655–657. [Google Scholar] [CrossRef]
- Weber, G. Dynamics of oligomeric proteins. J. Mol. Liq. 1989, 42, 255–268. [Google Scholar] [CrossRef]
- Weingand-Ziade, A.; Ribes, F.; Renault, F.; Masson, P. Pressure- and heat-induced inactivation of butyrylcholinesterase: Evidence for multiple intermediates and the remnant inactivation process. Biochem. J. 2001, 356, 487–493. [Google Scholar] [CrossRef]
- Kornblatt, M.J.; Hui Bon Hoa, G. The pressure-induced inactivation of mammalian enolases is accompanied by dissociation of the dimeric enzyme. Arch. Biochem. Biophys. 1987, 252, 277–283. [Google Scholar] [CrossRef]
- Froment, M.-T.; Lockridge, O.; Masson, P. Resistance of butyrylcholinesterase to inactivation by ultrasound: Effects of ultrasound on catalytic activity and subunit association. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1998, 1387, 53–64. [Google Scholar] [CrossRef]
- Aoki, K.; Hiramatsu, K.; Tanaka, M.; Kaneshina, S. Bovine serum albumin exposed to high pressure. Biochim. Biophys. Acta (BBA)-Protein Struct. 1968, 160, 368–377. [Google Scholar] [CrossRef]
- Narhi, L.O.; Arakawa, T. Sodium dodecyl sulfate Polyacrylamide gel electrophoresis as a method for studying the stability of subtilisin. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1989, 990, 144–149. [Google Scholar] [CrossRef]
- Boukil, A.; Marciniak, A.; Mezdour, S.; Pouliot, Y.; Doyen, A. Effect of High Hydrostatic Pressure Intensity on Structural Modifications in Mealworm (Tenebrio molitor) Proteins. Foods 2022, 11, 956. [Google Scholar] [CrossRef] [PubMed]
- Bartosz, G.; Soszynski, M.; Retelewska, W. Aging of the erythrocyte. X. Immunoelectrophoretic studies on the denaturation of superoxide dismutase. Mech. Ageing Dev. 1981, 17, 237–251. [Google Scholar] [CrossRef]
- Manjrekar, J.; Krishnan, K.S. Thermal gels: A procedure for determination of heat-inactivation temperature of enzymes. Anal. Biochem. 1987, 160, 409–413. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masson, P.; Lushchekina, S. Conformational Stability and Denaturation Processes of Proteins Investigated by Electrophoresis under Extreme Conditions. Molecules 2022, 27, 6861. https://doi.org/10.3390/molecules27206861
Masson P, Lushchekina S. Conformational Stability and Denaturation Processes of Proteins Investigated by Electrophoresis under Extreme Conditions. Molecules. 2022; 27(20):6861. https://doi.org/10.3390/molecules27206861
Chicago/Turabian StyleMasson, Patrick, and Sofya Lushchekina. 2022. "Conformational Stability and Denaturation Processes of Proteins Investigated by Electrophoresis under Extreme Conditions" Molecules 27, no. 20: 6861. https://doi.org/10.3390/molecules27206861
APA StyleMasson, P., & Lushchekina, S. (2022). Conformational Stability and Denaturation Processes of Proteins Investigated by Electrophoresis under Extreme Conditions. Molecules, 27(20), 6861. https://doi.org/10.3390/molecules27206861