
Advances in Fungal Phenaloenones—Natural Metabolites with Great Promise: Biosynthesis, Bioactivities, and an In Silico Evaluation of Their Potential as Human Glucose Transporter 1 Inhibitors

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Biosynthesis of Phenalenones
3. Bioactivities of Phenalenones
4. Human Glucose Transporter 1 (hGLUT1) Inhibitory Activity
4.1. Artificial Intelligence (AI)-Based Target Prediction for Phenalenone Derivatives
4.2. In Silico ADMET Properties of Selected Ligands
4.3. Ligands and Protein Preparation
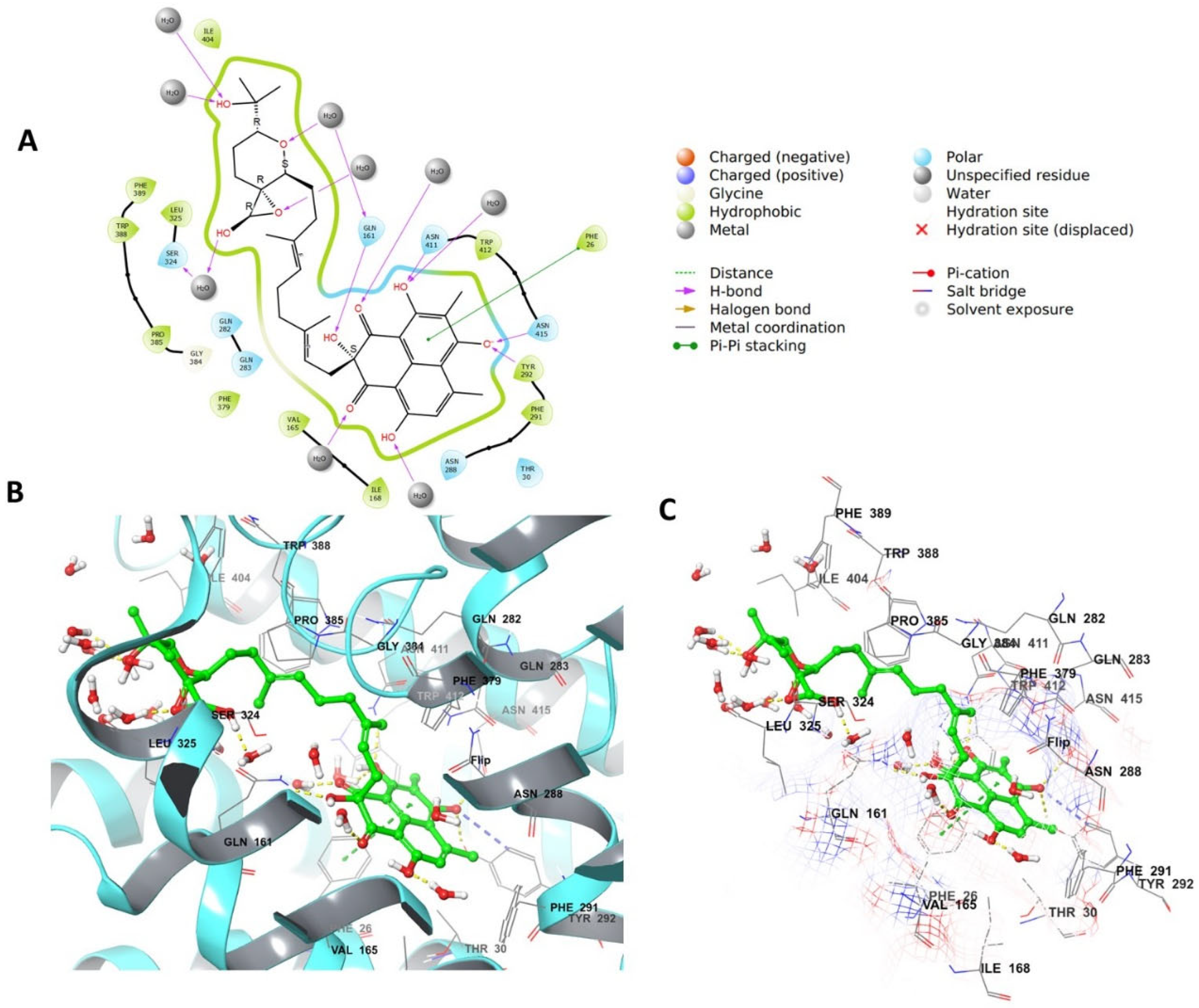
4.4. Grid Box Generation and Molecular Docking
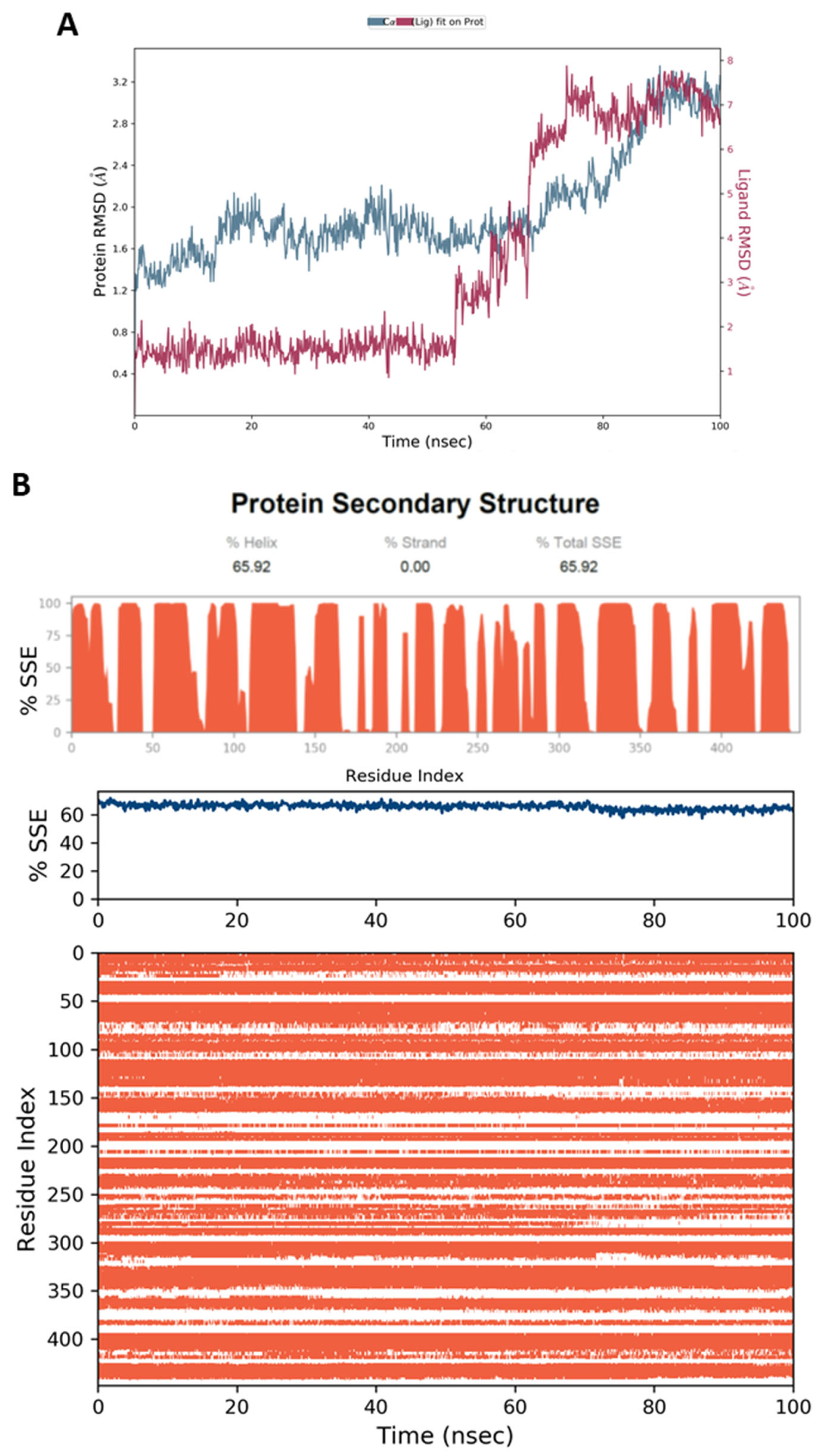
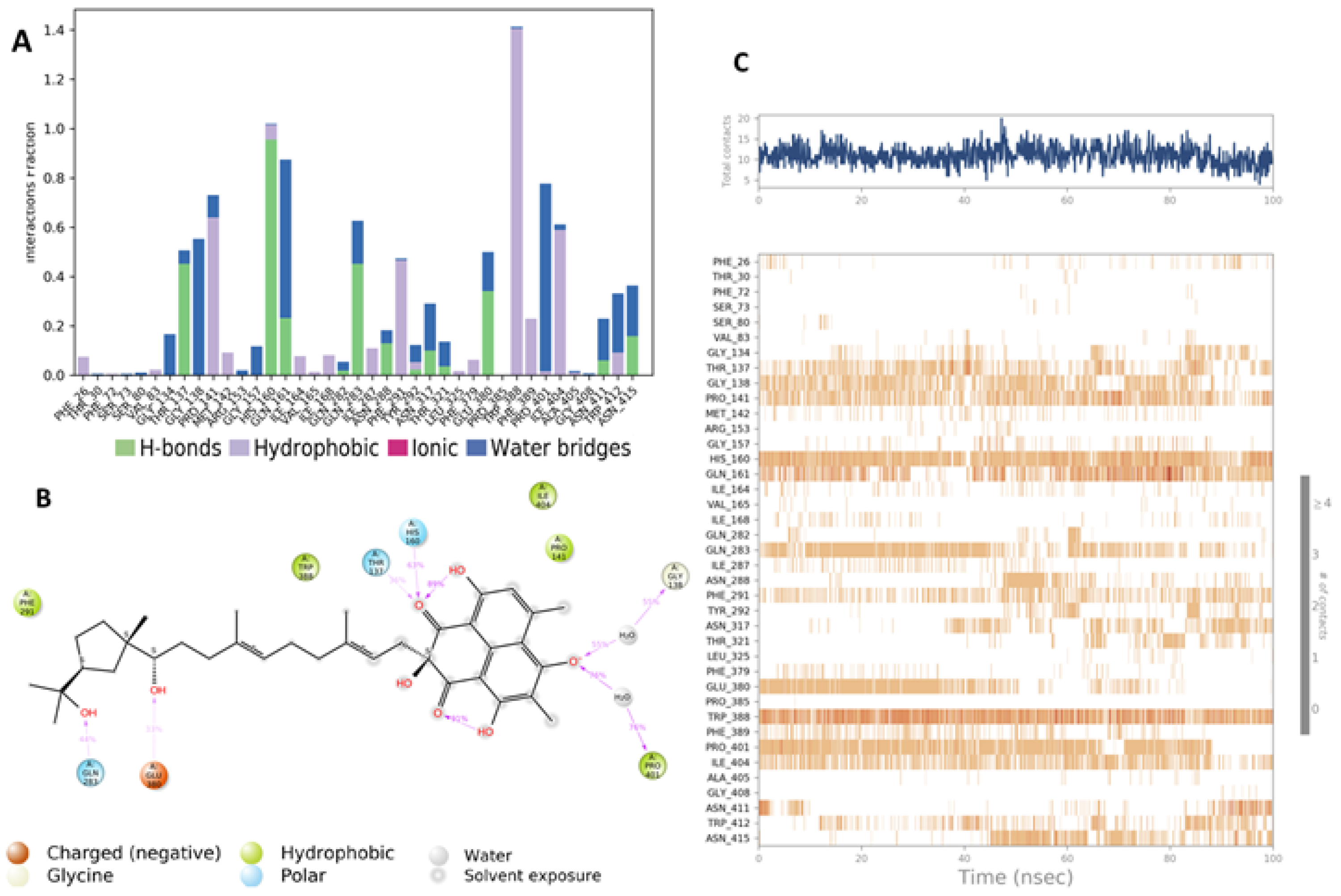
4.5. Molecular Dynamic Simulation (MD)
4.6. Material and Methods
4.6.1. ADMET Properties Prediction
4.6.2. Preparation of Protein and Ligands PDB Structures
4.6.3. Grid Generation and Docking
4.6.4. MD Simulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A-549 | Human lung carcinoma |
| ACE | Angiotensin-I-converting enzyme |
| AchE | Acetylcholinesterase |
| AMT | 2-Amino-5,6-dihydro-6-methyl-4H-1,3-thiazine hydrochloride |
| ASPC-1 | Human pancreas adenocarcinoma ascites metastasis |
| C6 | Rat glioma |
| CCK8 | Cell counting kit-8 |
| CD25 | Interleukin-2 receptor alpha chain |
| CD69 | Cluster of Differentiation 69 |
| CDC25B | Cell division cycle 25b |
| CFSE | Carboxyfluorescein succinimidyl ester |
| CYP2E1 | Cytochrome P450 2E1 |
| DFT | Density functional theory |
| DPPH | 1,1-Diphenyl-2-picrylhydrazyl |
| DPPH• | 1,1-diphenyl-2-picrylhydrazyl radical |
| EGFR | Epidermal growth factor receptor |
| ELISA | Enzyme-linked immunosorbent assay |
| GFP | Green fluorescent protein |
| GFPMA | Green fluorescent protein microplate assay |
| GIAO | Gauge invariant atomic orbital |
| H69AR | Human lung carcinoma |
| hBM-MSCs | Human bone marrow-mesenchymal stem cells |
| HeLa | Human cervix carcinoma |
| HepG2 | Human hepatocellular carcinoma |
| HL-60 | Human myeloid leukemia |
| HPLC | High-performance liquid chromatography |
| hPTP1B1-400 | Human PTP1B1-400 |
| HRESIMS | High resolution electrospray ionization mass spectrometry |
| HSCCC | High-speed counter-current chromatography |
| Huh7 | Human hepatoma |
| HUVECs | Human umbilical vascular endothelial cells |
| IC50 | Half inhibition concentration |
| IDO1 | Indoleamine 2,3-dioxygenase 1 |
| IKKα | Kappab kinase alpha |
| IL-2 | Interleukin 2 |
| K562 | Human chronic myelogenous leukemia |
| KB | Human oral carcinoma cell lines |
| L-02 | Human normal liver |
| LPS | Lipopolysaccharides |
| MAPK | Mitogen-activated protein kinase |
| MCF-7 | Human breast adenocarcinoma |
| MDA-MB-231 | Human breast cancer |
| MIC | Minimum inhibitory concentration |
| MRCNS | Methicillin-resistant coagulase-negative staphylococci |
| MRSA | Methicillin-resistant S. aureus |
| MTT | (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) |
| Na3VO4 | Sodium orthovanadate |
| NCI-H187 | Human small cell lung cancer |
| NF-κB | Nuclear factor kappa |
| NO | Nitric oxide |
| NS-1 | Mouse myeloma |
| O2•− | Superoxide anion |
| OH• | Hydroxyl radical |
| PC-12 | Rat a pheochromocytoma adrenal medulla |
| PMA/A23187 | Phorbol 12-myistrate 13-acetate |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PTKs | Protein tyrosine-kinases |
| PTPs | Protein tyrosine-phosphatases |
| REMA | Resazurin microplate assay |
| RKO | Human colorectal cancers |
| ROS | Reactive oxygen species |
| SKM1 | Myelodysplastic symdrom |
| SMMC-7721 | Hepatocellular carcinoma |
| SRB | Sulforhodamine B assay |
| SUP T1 | Human T cell lymphoblastic |
| SW480 | Hyman colon carcinoma |
| TAK1 | Transforming growth factor beta-activated kinase 1 |
| TCR | T cell receptor |
| U266 | Myeloma |
| U87MG | Human glioma |
| Vero | African green monkey kidney fibroblasts. |
References
- Ibrahim, S.R.M.; Altyar, A.E.; Mohamed, S.G.A.; Mohamed, G.A. Genus Thielavia: Phytochemicals, industrial importance and biological relevance. Nat. Prod. Res. 2021, 36, 5108–5123. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Mohamed, S.G.A.; Altyar, A.E.; Mohamed, G.A. Natural products of the fungal genus Humicola: Diversity, biological activity, and industrial importance. Curr. Microbiol. 2021, 78, 2488–2509. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Mohamed, S.G.A.; Sindi, I.A.; Mohamed, G.A. Biologically active secondary metabolites and biotechnological applications of species of the family Chaetomiaceae (Sordariales): An updated review from 2016 to 2021. Mycol. Prog. 2021, 20, 595–639. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Sirwi, A.; Eid, B.G.; Mohamed, S.G.A.; Mohamed, G.A. Bright Side of Fusarium oxysporum: Secondary Metabolites Bioactivities and Industrial Relevance in Biotechnology and Nanotechnology. J. Fungi. 2021, 7, 943. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.; Sirwi, A.; Eid, B.G.; Mohamed, S.; Mohamed, G.A. Fungal Depsides-Naturally Inspiring Molecules: Biosynthesis, Structural Characterization, and Biological Activities. Metabolites 2021, 11, 683. [Google Scholar] [CrossRef]
- Mohamed, G.A.; Ibrahim, S.R.M. Untapped Potential of Marine—Associated Cladosporium Species: An Overview on Secondary Metabolites, Biotechnological Relevance, and Biological Activities. Mar. Drugs. 2021, 19, 645. [Google Scholar] [CrossRef] [PubMed]
- Al-Rabia, M.W.; Mohamed, G.A.; Ibrahim, S.R.M.; Asfour, H.Z. Anti-inflammatory ergosterol derivatives from the endophytic fungus Fusarium chlamydosporium. Nat. Prod. Res. 2020, 35, 5011–5020. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, G.A.; Ibrahim, S.R.M.; Alhakamy, N.A.; Aljohani, O.S. Fusaroxazin, a novel cytotoxic and antimicrobial xanthone derivative from Fusarium oxysporum. Nat. Prod. Res. 2020, 36, 952–960. [Google Scholar] [CrossRef]
- Mohamed, G.A.; Ibrahim, S.R.M.; El-Agamy, D.S.; Elsaed, W.M.; Sirwi, A.; Asfour, H.Z.; Koshak, A.E.; Elhady, S.S. Terretonin as a New Protective Agent against Sepsis-Induced Acute Lung Injury: Impact on SIRT1/Nrf2/NF-κBp65/NLRP3 Signaling. Biology 2021, 10, 1219. [Google Scholar] [CrossRef] [PubMed]
- Khayat, M.T.; Ibrahim, S.R.M.; Mohamed, G.A.; Abdallah, H.M. Anti-inflammatory metabolites from endophytic fungus Fusarium sp. Phytochem Lett. 2019, 29, 104–109. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Bagalagel, A.A.; Diri, R.M.; Noor, A.O.; Bakhsh, H.T.; Muhammad, Y.A.; Mohamed, G.A.; Omar, A.M. Exploring the activity of fungal phenalenone derivatives as potential CK2 inhibitors using computational methods. J. Fungi 2022, 8, 443. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Mohamed, G.A.; Al Haidari, R.A.; El-Kholy, A.A.; Zayed, M.F.; Khayat, M.T. Biologically active fungal depsidones: Chemistry, biosynthesis, structural characterization, and bioactivities. Fitoterapia 2018, 129, 317–365. [Google Scholar] [CrossRef] [PubMed]
- Hareeri, R.H.; Aldurdunji, M.M.; Abdallah, H.M.; Alqarni, A.A.; Mohamed, S.G.A.; Mohamed, G.A.; Ibrahim, S.R.M. Aspergillus ochraceus: Metabolites, bioactivities, biosynthesis, and biotechnological potential. Molecules 2022, 27, 6759. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Mohamed, G.A.; Khedr, A.M.I. γ-Butyrolactones from Aspergillus species: Structures, biosynthesis, and biological activities. Nat. Prod. Commun. 2017, 12, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Abdallah, H.M.; Elkhayat, E.S.; Al Musayeib, N.M.; Asfour, H.Z.; Zayed, M.F.; Mohamed, G.A. Fusaripeptide A: New antifungal and anti-malarial cyclodepsipeptide from the endophytic fungus Fusarium sp. J. Asian Nat. Prod. Res. 2018, 20, 75–85. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Mohamed, G.A.; Al Haidari, R.A.; Zayed, M.F.; El-Kholy, A.A.; Elkhayat, E.S.; Ross, S.A. Fusarithioamide B, a new benzamide derivative from the endophytic fungus Fusarium chlamydosporium with potent cytotoxic and antimicrobial activities. Bioorg. Med. Chem. 2018, 26, 786–790. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Elkhayat, E.S.; Mohamed, G.A.; Fat’hi, S.M.; Ross, S.A. Fusarithioamide A, a new antimicrobial and cytotoxic benzamide derivative from the endophytic fungus Fusarium chlamydosporium. Biochem. Biophys. Res. Commun. 2016, 479, 211–216. [Google Scholar] [CrossRef]
- Ibrahim, S.R.; Abdallah, H.M.; Mohamed, G.A.; Ross, S.A. Integracides H–J: New tetracyclic triterpenoids from the endophytic fungus Fusarium sp. Fitoterapia 2016, 112, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.; Mohamed, G.A.; Ross, S.A. Integracides F and G: New tetracyclic triterpenoids from the endophytic fungus Fusarium sp. Phytochem.Lett. 2016, 15, 125–130. [Google Scholar] [CrossRef]
- Nazir, M.; El Maddah, F.; Kehraus, S.; Egereva, E.; Piel, J.; Brachmann, A.O.; König, G.M. Phenalenones: Insight into the biosynthesis of polyketides from the marine alga-derived fungus Coniothyrium cereale. Org. Biomol. Chem. 2015, 13, 8071–8079. [Google Scholar] [CrossRef] [PubMed]
- Elsebai, M.F.; Saleem, M.; Tejesvi, M.V.; Kajula, M.; Mattila, S.; Mehiri, M.; Turpeinen, A.; Pirttilä, A.M. Fungal phenalenones: Chemistry, biology, biosynthesis and phylogeny. Nat. Prod. Rep. 2014, 31, 628–645. [Google Scholar] [CrossRef]
- Chooi, Y.H.; Tang, Y. Navigating the fungal polyketide chemical space: From genes to molecules. J. Org. Chem. 2012, 77, 9933–9953. [Google Scholar] [CrossRef]
- Song, R.; Feng, Y.; Wang, D.; Xu, Z.; Li, Z.; Shao, X. Phytoalexin phenalenone derivatives inactivate mosquito larvae and root-knot nematode as type-II photosensitizer. Sci. Rep. 2017, 7, 42058. [Google Scholar] [CrossRef]
- Hölscher, D.; Dhakshinamoorthy, S.; Alexandrov, T.; Becker, M.; Bretschneider, T.; Buerkert, A.; Crecelius, A.C.; De Waele, D.; Elsen, A.; Heckel, D.G.; et al. Phenalenone-type phytoalexins mediate resistance of banana plants (Musa spp.) to the burrowing nematode Radopholus similis. Proc. Natl. Acad. Sci. USA 2014, 111, 105–110. [Google Scholar] [CrossRef]
- Cooke, R.G.; Edwards, J.M. Naturally occurring phenalenones and related compounds. Fortschr. Chem. Org. Nat. 1981, 40, 153–190. [Google Scholar]
- Harman, R.E.; Cason, J.; Stodola, F.H.; Adkins, A.L. Structural features of herqueinone, a red pigment from Penicillium herquei. J. Org. Chem. 1955, 20, 1260. [Google Scholar] [CrossRef]
- Munde, T.; Brand, S.; Hidalgo, W.; Maddula, R.K.; Svatoš, A.; Schneider, B. Biosynthesis of tetraoxygenated phenylphenalenones in Wachendorfia thyrsiflora. Phytochemistry 2013, 91, 165–176. [Google Scholar] [CrossRef]
- Bucher, G.; Bresolí-Obach, R.; Brosa, C.; Flors, C.; Luis, J.G.; Grillo, T.A.; Nonell, S. β-Phenyl quenching of 9-phenylphenalenones: A novel photocyclisation reaction with biological implications. Phys. Chem. Chem. Phys. 2014, 16, 18813–18820. [Google Scholar] [CrossRef]
- Phatangare, K.R.; Lanke, S.K.; Sekar, N. Phenalenone fluorophores-synthesis.; photophysical properties and DFT study. J. Fluoresc. 2014, 24, 1827–1840. [Google Scholar] [CrossRef]
- Gao, S.S.; Duan, A.; Xu, W.; Yu, P.; Hang, L.; Houk, K.N.; Tang, Y. Phenalenone polyketide cyclization catalyzed by fungal polyketide synthase and flavin-dependent monooxygenase. J. Am. Chem. Soc. 2016, 138, 4249–4259. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Feng, B.-M.; Sun, Y.; Wu, H.-H.; Li, S.-G.; Liu, B.; Liu, F.; Zhang, W.-Y.; Chen, G.; Bai, J.; et al. Flaviphenalenones A.–C, three new phenalenone derivatives from the fungus Aspergillus flavipes PJ03-11. Tetrahedron Lett. 2016, 57, 645–649. [Google Scholar] [CrossRef]
- Lu, R.; Liu, X.; Gao, S.; Zhang, W.; Peng, F.; Hu, F.; Huang, B.; Chen, L.; Bao, G.; Li, C.; et al. New tyrosinase inhibitors from Paecilomyces gunnii. J. Agric. Food. Chem. 2014, 62, 11917–11923. [Google Scholar] [CrossRef] [PubMed]
- Rukachaisirikul, V.; Rungsaiwattana, N.; Klaiklay, S.; Phongpaichit, S.; Borwornwiriyapan, K.; Sakayaroj, J. γ-Butyrolactone.; cytochalasin.; cyclic carbonate.; eutypinic acid.; and phenalenone derivatives from the soil fungus Aspergillus sp. PSU-RSPG185. J. Nat. Prod. 2014, 77, 2375–2382. [Google Scholar] [CrossRef] [PubMed]
- Tansakul, C.; Rukachaisirikul, V.; Maha, A.; Kongprapan, T.; Phongpaichit, S.; Hutadilok-Towatana, N.; Borwornwiriyapan, K.; Sakayaroj, J. A new phenalenone derivative from the soil fungus Penicillium herquei PSU-RSPG93. Nat. Prod. Res. 2014, 28, 1718–1724. [Google Scholar] [CrossRef]
- Park, S.C.; Julianti, E.; Ahn, S.; Kim, D.; Lee, S.K.; Noh, M.; Oh, D.C.; Oh, K.B.; Shin, A.J. Phenalenones from a marine-derived fungus Penicillium sp. Mar. Drugs 2019, 17, 176. [Google Scholar] [CrossRef] [PubMed]
- Al Subeh, Z.Y.; Raja, H.A.; Burdette, J.E.; Falkinham, J.O.; Hemby, S.E.; Oberlies, N.H. Three diketomorpholines from a Penicillium sp. (strain G1071). Phytochemistry 2021, 189, 112830. [Google Scholar] [CrossRef]
- Elsebai, M.F.; Ghabbour, H.A.; Mehiri, M. Unusual nitrogenous phenalenone derivatives from the marine-derived fungus Coniothyrium cereale. Molecules 2016, 21, 178. [Google Scholar] [CrossRef]
- Lee, H.S.; Yu, J.S.; Kim, K.H.; Jeong, G.S. Diketoacetonylphenalenone, derived from Hawaiian volcanic soil-associated fungus Penicillium herquei FT729, regulates T Cell activation via nuclear factor-κB and mitogen-activated protein kinase pathway. Molecules 2020, 25, 5374. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhu, R.; Yi, W.; Chai, W.; Zhang, Z.; Lian, X.-Y. Peniciphenalenins A–F from the culture of a marine-associated fungus Penicillium sp. ZZ901. Phytochemistry 2018, 152, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, G.; Lopes, P.; Arcile, G.; Vlachou, P.; Van Elslande, E.; Retailleau, P.; Gallard, J.F.; Weis, M.; Benayahu, Y.; Fokialakis, N.; et al. Impact of the cultivation technique on the production of secondary metabolites by Chrysosporium lobatum TM-237-S5, Isolated from the sponge Acanthella cavernosa. Mar. Drugs 2019, 17, 678. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Shanmugam, K.; Kedike, B.; Narayanan, S.; Shanmugam, S.; Gopalasamudram Neelakantan, H. Trypethelone and phenalenone derivatives isolated from the mycobiont culture of Trypethelium eluteriae Spreng and their anti-mycobacterial properties. Nat. Prod. Res. 2020, 34, 3320–3327. [Google Scholar] [CrossRef] [PubMed]
- Intaraudom, C.; Nitthithanasilp, S.; Rachtawee, P.; Boonruangprapa, T.; Prabpai, S.; Kongsaeree, P.; Pittayakhajonwut, P. Phenalenone derivatives and the unusual tricyclic sesterterpene acid from the marine fungus Lophiostoma bipolare BCC25910. Phytochemistry 2015, 120, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Intaraudom, C.; Bunbamrung, N.; Dramae, A.; Boonyuen, N.; Choowong, W.; Rachtawee, P.; Pittayakhajonwut, P. Chromone derivatives, R- and S-taeniolin, from the marine-derived fungus Taeniolella sp. BCC31839. Nat. Prod. Res. 2021, 35, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Sun, C.; Li, C.; Zhang, G.; Zhu, T.; Li, D.; Che, Q. Antibacterial phenalenone derivatives from marine-derived fungus Pleosporales sp. HDN1811400. Tetrahedron Lett. 2021, 68, 152938. [Google Scholar] [CrossRef]
- Macabeo, A.P.G.; Pilapil, L.A.E.; Garcia, K.Y.M.; Quimque, M.T.J.; Phukhamsakda, C.; Cruz, A.J.C.; Hyde, K.D.; Stadler, M. Alpha-glucosidase- and lipase-inhibitory phenalenones from a new species of pseudolophiostoma originating from Thailand. Molecules 2020, 25, 965. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Q.; Mándi, A.; Li, X.M.; Liu, H.; Li, X.; Balázs Király, S.; Kurtán, T.; Wang, B.G. Separation and configurational assignment of stereoisomeric phenalenones from the marine mangrove-derived fungus Penicillium herquei MA-370. Bioorg. Chem. 2021, 106, 104477. [Google Scholar] [CrossRef]
- Zhang, Q.H.; Tian, L.; Sun, Z.L.; Fang, S.; Cai, G.L.; Wang, Y.J.; Pei, Y.H. Two new secondary metabolites from the marine-derived fungus Nigrospora sphaerica. J. Asian Nat. Prod. Res. 2015, 17, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yue, Q.; Jayanetti, D.R.; Swenson, D.C.; Bartholomeusz, G.A.; An, Z.; Gloer, J.B.; Bills, G.F. Anti-cryptococcus phenalenones and cyclic tetrapeptides from Auxarthron pseudauxarthron. J. Nat. Prod. 2017, 80, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.K.; Crombie, A.; Vuong, D.; Lacey, E.; Piggott, A.M.; Karuso, P. Talauxins: Hybrid phenalenone dimers from Talaromyces stipitatus. J. Nat. Prod. 2020, 83, 1051–1060. [Google Scholar] [CrossRef]
- Gombodorj, S.; Yang, M.-H.; Shang, Z.-C.; Liu, R.-H.; Li, T.-X.; Yin, G.-P.; Kong, L.-Y. New phenalenone derivatives from Pinellia ternata tubers derived Aspergillus sp. Fitoterapia 2017, 120, 72–78. [Google Scholar] [CrossRef]
- Pang, X.; Zhao, J.Y.; Fang, X.M.; Zhang, T.; Zhang, D.W.; Liu, H.Y.; Su, J.; Cen, S.; Yu, L.Y. Metabolites from the plant endophytic fungus Aspergillus sp. CPCC 400735 and their anti-HIV activities. J. Nat. Prod. 2017, 80, 2595–2601. [Google Scholar] [CrossRef] [PubMed]
- Basnet, B.B.; Liu, L.; Zhao, W.; Liu, R.; Ma, K.; Bao, L.; Ren, J.; Wei, X.; Yu, H.; Wei, J.; et al. New 1,2-naphthoquinone-derived pigments from the mycobiont of lichen Trypethelium eluteriae Sprengel. Nat. Prod. Res. 2019, 33, 2044–2050. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.C.; Minh Ha, T.; Sohn, J.H.; Yim, J.H.; Oh, H. Protein tyrosine phosphatase 1B inhibitors from a marine-derived fungal strain Aspergillus sp. SF-5929. Nat. Prod. Res. 2020, 34, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tan, X.; Li, Y.; Wang, Y.; Yu, M.; Qing, J.; Sun, B.; Niu, S.; Ding, G. Hispidulones A and B, two new phenalenone analogs from desert plant endophytic fungus Chaetosphaeronema hispidulum. J. Antibiot. 2020, 73, 56–59. [Google Scholar] [CrossRef]
- Yu, J.S.; Li, C.; Kwon, M.; Oh, T.; Lee, T.H.; Kim, D.H.; Ahn, J.S.; Ko, S.K.; Kim, C.S.; Cao, S.; et al. Herqueilenone, A, a unique rearranged benzoquinone-chromanone from the Hawaiian volcanic soil-associated fungal strain Penicillium herquei FT729. Bioorg. Chem. 2020, 105, 104397. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Yang, J.; Miao, C.P.; Yan, Y.; Ma, Y.T.; Li, X.N.; Zhao, L.X.; Huang, S.X. New duclauxamide from Penicillium manginii YIM PH30375 and structure revision of the duclauxin family. Org. Lett. 2015, 17, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, L.; Feng, L.; Hu, F.; Zhang, F.; Ren, J.; Qiu, Y.; Wang, Z. Verruculosins A-B; new oligophenalenone dimers from the soft coral-derived fungus Talaromyces verruculosus. Mar. Drugs 2019, 17, 516. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Arreola, B.S.; Aguilar-Ramírez, E.; Cano-Sánchez, P.; Morales-Jiménez, J.; González-Andrade, M.; Medina-Franco, J.L.; Rivera-Chávez, J. Dimeric phenalenones from Talaromyces sp. (IQ-313) inhibit hPTP1B1-400: Insights into mechanistic kinetics from in vitro and in silico studies. Bioorg. Chem. 2020, 101, 103893. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Ohlendorf, B.; Oesker, V.; Wiese, J.; Malien, S.; Schmaljohann, R.; Imhoff, J.F. Acetylcholinesterase inhibitors from a marine fungus Talaromyces sp. strain LF458. Mar. Biotechnol. 2015, 17, 110–119. [Google Scholar] [CrossRef]
- Zang, Y.; Genta-Jouve, G.; Escargueil, A.E.; Larsen, A.K.; Guedon, L.; Nay, B.; Prado, S. Antimicrobial oligophenalenone dimers from the soil fungus Talaromyces stipitatus. J. Nat. Prod. 2016, 79, 2991–2996. [Google Scholar] [CrossRef]
- Sankawa, U.; Taguchi, H.; Ogihara, Y.; Shibata, S. Biosynthesis of duclauxin. Tetrahedron Lett. 1966, 25, 2883–2886. [Google Scholar] [CrossRef]
- Aldemir, H.; Richarz, R.; Gulder, T.A. The biocatalytic repertoire of natural biaryl formation. Angew. Chem. Int. Ed. Engl. 2014, 53, 8286–8293. [Google Scholar] [CrossRef]
- Zhao, B.; Guengerich, F.P.; Bellamine, A.; Lamb, D.C.; Izumikawa, M.; Lei, L.; Podust, L.M.; Sundaramoorthy, M.; Kalaitzis, J.A.; Reddy, L.M.; et al. Binding of two flaviolin substrate molecules, oxidative coupling and crystal structure of Streptomyces coelicolor A3 (2) cytochrome P450 158A2. J. Biol. Chem. 2005, 280, 11599–11607. [Google Scholar] [CrossRef]
- Rontein, D.; Nishida, I.; Tashiro, G.; Yoshioka, K.; Wu, W.I.; Voelker, D.R.; Basset, G.; Hanson, A.D. Plants synthesize ethanolamine by direct decarboxylation of serine using a pyridoxal phosphate enzyme. J. Biol. Chem. 2001, 276, 35523–35529. [Google Scholar] [CrossRef]
- Elsebai, M.F.; Natesan, L.; Kehraus, S.; Mohamed, I.E.; Schnakenburg, G.; Sasse, F.; Shaaban, S.; Gütschow, M.; König, G.M. HLE-inhibitory alkaloids with a polyketide skeleton from the marine-derived fungus Coniothyrium cereale. J. Nat. Prod. 2011, 74, 2282–2285. [Google Scholar] [CrossRef]
- Wang, J.; Liu, P.; Wang, Y.; Wang, H.; Li, J.; Zhuang, Y.; Zhu, W. Antimicrobial aromatic polyketides from gorgonian-associated fungus; Penicillium commune 518. Chin. J. Chem. 2012, 30, 1236–1242. [Google Scholar] [CrossRef]
- Ernst-Russell, M.A.; Chai, C.L.L.; Elix, J.; McCarthy, P.M. Myeloconone A2, a new phenalenone from the lichen Myeloconis erumpens. Aust. J. Chem. 2000, 53, 1011–1013. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Uldry, M.; Ibberson, M.; Horisberger, J.D.; Chatton, J.Y.; Riederer, B.M.; Thorens, B. Identification of a mammalian H(+)-myo-inositol symporter expressed predominantly in the brain. EMBO J. 2001, 20, 4467–4477. [Google Scholar] [CrossRef] [PubMed]
- Yan, N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013, 38, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Almahmoud, S.; Wang, X.; Vennerstrom, J.L.; Zhong, H.A. Conformational studies of glucose transporter 1 (GLUT1) as an anticancer drug target. Molecules 2019, 24, 2159. [Google Scholar] [CrossRef]
- Mueckler, M.; Caruso, C.; Baldwin, S.A.; Panico, M.; Blench, I.; Morris, H.R.; Allard, W.J.; Lienhard, G.E.; Lodish, H.F. Sequence and structure of a human glucose transporter. Science 1985, 229, 941–945. [Google Scholar] [CrossRef]
- Olson, A.L.; Pessin, J.E. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu. Rev. Nutr. 1996, 16, 235–256. [Google Scholar] [CrossRef]
- Santer, R.; Klepper, J. Disorders of Glucose Transport. In Inborn Metabolic Diseases: Diagnosis and Treatment; Saudubray, J.M., van den Berghe, G., Walter, J.H., Eds.; Springer: New York, NY, USA, 2011; pp. 175–181. [Google Scholar]
- Kapoor, K.; Finer-Moore, J.S.; Pedersen, B.P.; Caboni, L.; Waight, A.; Hillig, R.C.; Bringmann, P.; Heisler, I.; Müller, T.; Siebeneicher, H.; et al. Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. USA 2016, 113, 4711–4716. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Akt, hexokinase, mTOR: Targeting cellular energy metabolism for cancer therapy. Drug Discov. Today Dis. Mech. 2005, 2, 239–246. [Google Scholar] [CrossRef]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Omar, A.M.; Bagalagel, A.A.; Diri, R.M.; Noor, A.O.; Almasri, D.M.; Mohamed, S.G.A.; Mohamed, G.A. Thiophenes-Naturally Occurring Plant Metabolites: Biological Activities and In Silico Evaluation of Their Potential as Cathepsin D Inhibitors. Plants 2022, 11, 539. [Google Scholar] [CrossRef]
- Yang, S.Q.; Ye, Q.; Ding, J.J.; Yin, M.Z.; Lu, A.P.; Chen, X.; Hou, T.J.; Cao, D.S. Current advances in ligand-based target prediction. WIREs Comput. Mol. Sci. 2021, 11, e1504. [Google Scholar] [CrossRef]
- Nickel, J.; Gohlke, B.O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Dunkel, M.; Preissner, R. SuperPred: Update on drug classification and target prediction. Nucleic Acids Res. 2014, 42, W26–W31. [Google Scholar] [CrossRef]
- Omar, A.M.; Mohamed, G.A.; Ibrahim, S.R.M. Chaetomugilins and chaetoviridins-promising natural metabolites: Structures, separation, characterization, biosynthesis, bioactivities, molecular docking, and molecular dynamics. J. Fungi. 2022, 8, 127. [Google Scholar] [CrossRef]
- Schrödinger, LLC. Schrödinger Release 2021-4: LigPrep; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger, LLC. Schrödinger Release 2021-4: QikProp; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger, LLC. Schrödinger Release 2021-4: Glide; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Baier, A.; Szyszka, R. Compounds from Natural Sources as Protein Kinase Inhibitors. Biomolecules 2020, 10, 1546. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. Schrödinger Release 2021-4: Desmond Molecular Dynamics System; D.E. Shaw Research: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger. Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2021. [Google Scholar]
- RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 15 March 2022).
- Houštecká, R.; Hadzima, M.; Fanfrlík, J.; Brynda, J.; Pallová, L.; Hánová, I.; Mertlíková-Kaiserová, H.; Lepšík, M.; Horn, M.; Smrčina, M.; et al. Biomimetic macrocyclic inhibitors of human cathepsin D: Structure-activity relationship and binding mode analysis. J. Med. Chem. 2020, 63, 1576–1596. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Harvey, C.J.B.; Tang, M.; Schlechtm, U.; Horecka, J.; Fischer, C.R.; Lin, H.C.; Li, J.; Naughton, B.; Cherry, J.; Miranda, M.; et al. HEx: A heterologous expression platform for the discovery of fungal natural products. Sci. Adv. 2018, 4, eaar5459. [Google Scholar] [CrossRef]
- Hautbergue, T.; Jamin, E.L.; Debrauwer, L.; Puel, O.; Oswald, I.P. From genomics to metabolomics, moving toward an integrated strategy for the discovery of fungal secondary metabolites. Nat. Prod. Rep. 2018, 35, 147–173. [Google Scholar] [CrossRef] [PubMed]
- Raschka, S.; Kaufman, B. Machine learning and AI-based approaches for bioactive ligand discovery and GPCR-ligand recognition. Methods 2020, 180, 89–110. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A deep learning approach to antibiotic discovery. Cell 2020, 180, 688–702. [Google Scholar] [CrossRef]































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Mol. Wt. | Mol. Formula | Fungal Source | Host (Part, Family) | Place | Ref. |
|---|---|---|---|---|---|---|
| Paecilomycone A (1) | 288 | C15H12O6 | Paecilomyces gunnii | Culture | China | [32] |
| Paecilomycone B (2) | 286 | C15H10O6 | Paecilomyces gunnii | Culture | China | [32] |
| Paecilomycone C (3) | 287 | C15H13NO5 | Paecilomyces gunnii | Culture | China | [32] |
| Myeloconone A (4) | 286 | C16H14O5 | Paecilomyces gunnii | Culture | China | [32] |
| Aspergillussanone A (5) | 540 | C31H40O8 | Aspergillus sp. PSU-RSPG185 | Soil | Surat Thani, Thailand | [33] |
| Aspergillussanone B (6) | 540 | C31H40O8 | Aspergillus sp. PSU-RSPG185 | Soil | Surat Thani, Thailand | [33] |
| Peniciherqueinone (7) | 388 | C20H20O8 | Penicillium herquei PSU-RSPG93 | Soil | Surat Thani, Thailand | [34] |
| ent-Peniciherqueinone (8) | 388 | C20H20O8 | Penicillium sp. | Marine sediment | Gagudo, Korea | [35] |
| Herqueinone (9) | 372 | C20H20O7 | Penicillium herquei PSU-RSPG93 | Soil | Surat Thani, Thailand | [34] |
| Penicillium sp. | Marine sediment | Gagudo, Korea | [35] | |||
| Penicillium sp. G1071 | Mushroom fruiting body | Freshwater stream, Hebron, Connecticut, USA | [36] | |||
| Norherqueinone (10) | 358 | C19H18O7 | Penicillium sp. G1071 | Mushroom fruiting body | Freshwater stream, Hebron, Connecticut, USA | [36] |
| 12-Hydroxynorherqueinone (11) | 374 | C19H18O8 | Penicillium sp. | Marine sediment | Gagudo, Korea | [35] |
| Isoherqueinone (12) | 372 | C20H20O7 | Penicillium sp. | Marine sediment | Gagudo, Korea | [35] |
| ent-Isoherqueinone (13) | 372 | C20H20O7 | Penicillium sp. | Marine sediment | Gagudo, Korea | [35] |
| Deoxyherqueinone (14) | 356 | C20H20O6 | Penicillium herquei PSU-RSPG93 | Soil | Surat Thani, Thailand | [34] |
| Oxopropylisoherqueinone A (15) | 414 | C22H22O8 | Penicillium sp. | Marine sediment | Gagudo, Korea | [35] |
| Oxopropylisoherqueinone B (16) | 414 | C22H22O8 | Penicillium sp. | Marine sediment | Gagudo, Korea | [35] |
| Diketoacetonylphenalenone = Acetone adduct of a triketone (17) | 398 | C22H22O7 | Penicillium sp. | Marine sediment | Gagudo, Korea | [35] |
| Acetone adduct of antatrovenetinone (18) | 398 | C22H22O7 | Penicillium herquei PSU-RSPG93 | Soil | Surat Thani, Thailand | [34] |
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [37] | |||
| Penicillium herquei FT729 | Volcanic soil sample | Big Island, Hawaii | [38] | |||
| Antatrovenetinone (19) | 340 | C19H16O6 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [37] |
| Sclerodin (20) | 328 | C18H16O6 | Penicillium herquei PSU-RSPG93 | Soil | Surat Thani, Thailand | [34] |
| (+)-Sclerodin (21) | 328 | C18H16O6 | Penicillium sp. ZZ901 | Scapharca broughtonii (Schrenck blood clam, Arcidae) | Sea shoal, East China, Zhoushan Archipelago, China | [39] |
| Chrysosporium lobatum TM-237-S5 | Acanthella cavernosa (Sponge, Dictyonellidae) | Mesophotic reef in Eilat, Dekel Beach, Gulf of Aqaba, Israel | [40] | |||
| Trypethelium eluteriae Sprengl | Anacardium occidentale (Bark, Anacardiaceae) | Vadanemmeli, Kanchipuram, Tamil Nadu, India | [41] | |||
| (−)-Sclerodin (22) | 328 | C18H16O6 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| Penicillium sp. | Marine sediment | Gagudo, Korea | [35] | |||
| Taeniolella sp. BCC31839 | Wood of mangrove forest (Poaceae) | Bangkok, Thailand | [43] | |||
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] | |||
| (+)-8-Hydroxyslerodin (23) | 344 | C18H16O7 | Chrysosporium lobatum TM-237-S5 | Acanthella cavernosa (Sponge, Dictyonellidae) | Mesophotic reef in Eilat, Dekel Beach, Gulf of Aqaba, Israel | [40] |
| (−)-Sclerodinol (24) | 344 | C18H16O7 | Pleosporales sp. HDN1811400 | Sediment sample | Fildes Peninsula | [44] |
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] | |||
| (−)-Bipolaride D (25) | 342 | C19H18O6 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| Taeniolella sp. BCC31839 | Wood of mangrove forest (Poaceae) | Bangkok, Thailand | [43] | |||
| Bipolarol C (26) | 344 | C18H16O7 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| 4-Hydroxysclerodin (27) | 346 | C18H18O7 | Penicillium sp. | Marine sediment | Gagudo, Korea | [35] |
| (+)-Sclerodione (28) | 312 | C18H16O5 | Penicillium sp. ZZ901 | Scapharca broughtonii (Schrenck blood clam, Arcidae) | Sea shoal, East China, Zhoushan Archipelago, China | [39] |
| (−)-Sclerodione (29) | 312 | C18H16O5 | Pseudolophiostoma sp. MFLUCC 17-2081 | Clematis fulvicoma (Ranunculaceae) | Chiang Rai, Thailand | [45] |
| Taeniolella sp. BCC31839 | Wood of mangrove forest (Poaceae) | Bangkok, Thailand | [43] | |||
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] | |||
| (−)-Bipolaride B (30) | 326 | C19H18O5 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| Taeniolella sp. BCC31839 | Wood of mangrove forest (Poaceae) | Bangkok, Thailand | [43] | |||
| Peniciphenalenin H (31) | 328 | C18H16O6 | Pleosporales sp. HDN1811400 | Sediment sample | Fildes Peninsula | [44] |
| Bipolarol A (32) | 342 | C19H18O6 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| (+)-Scleroderolide (33) | 328 | C18H16O6 | Penicillium sp. ZZ901 | Scapharca broughtonii (Schrenck blood clam, Arcidae) | Sea shoal, East China, Zhoushan Archipelago, China | [39] |
| Chrysosporium lobatum TM-237-S5 | Acanthella cavernosa (Sponge, Dictyonellidae) | Mesophotic reef in Eilat, Dekel Beach, Gulf of Aqaba, Israel | [40] | |||
| Penicillium herquei MA-370 | Rhizospheric soil of the mangrove plant Rhizophora mucronata (Rhizophoraceae) | Hainan Island, China | [46] | |||
| (−)-Scleroderolide (34) | 328 | C18H16O6 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| Penicillium sp. | Marine sediments | Gagudo, Korea | [35] | |||
| Pseudolophiostoma sp. MFLUCC 17-2081 | Clematis fulvicoma (Ranunculaceae) | Chiang Rai, Thailand | [45] | |||
| Taeniolella sp. BCC31839 | Wood of mangrove forest (Poaceae) | Bangkok, Thailand | [43] | |||
| Penicillium herquei MA-370 | Rhizospheric soil of the mangrove plant Rhizophora mucronata (Rhizophoraceae) | Hainan Island, China | [46] | |||
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] | |||
| (+)-8-Hydroxyscleroderolide (35) | 344 | C18H16O7 | Chrysosporium lobatum TM-237-S5 | Acanthella cavernosa (Sponge, Dictyonellidae) | Mesophotic reef in Eilat, Dekel Beach, Gulf of Aqaba, Israel | [40] |
| (−)-Bipolaride A (36) | 342 | C19H18O6 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| (−)-Cereolactone = (−)-7,8-Dihydro-3,6-dihydroxy-1,7,7,8-tetramethyl-5H-furo-[2′,3′:5,6]naphtho-[1,8-bc]furan-5-one (37) | 300 | C17H16O5 | Penicillium herquei PSU-RSPG93 | Soil | Surat Thani, Thailand | [34] |
| Chrysosporium lobatum TM-237-S5 | Acanthella cavernosa (Sponge, Dictyonellidae) | Mesophotic reef in Eilat, Dekel Beach, Gulf of Aqaba, Israel | [40] | |||
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] | |||
| (R)-6-Hydroxy-3-methoxy-1,7,7,8-tetramethyl-7,8-dihydro-5H-naphtho[1,2-b:5,4-b′c′]difuran-5-one (38) | 314 | C18H18O5 | Trypethelium eluteriae | Anacardium occidentale (Bark, Anacardiaceae) | Vadanemmeli, Kanchipuram, Tamil Nadu, India | [41] |
| (−)-Bipolaride E (39) | 314 | C18H18O5 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| Bipolarol B (40) | 344 | C19H20O6 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| Bipolarol D (41) | 330 | C18H18O6 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| Bipolaride C (42) | 328 | C19H20O5 | Lophiostoma bipolare BCC25910 | Mangrove wood | Beach in Haad Wanakorn National Park, Prachuap Khiri Khan, Thailand | [42] |
| 2,5,7-Trihydroxy-4-(3′-methylbut-2′-en-1′-yl) oxy-2H-naphtho [1,8-bc] furan-9-one (43) | 302 | C16H14O6 | Nigrospora sphaerica | Xylocarpus granatum (Mangrove plant, Meliaceae) | Intertidal zone, China Sea, Huanghai, China | [47] |
| Rousselianone A = 4,9-Dihydroxy-6-methyl-7-((3-methylbut-2-en-1-yl)oxy) -1H-phenalene-1,2,3-trione (44) | 340 | C19H16O6 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20,37] |
| Flaviphenalenone A (45) | 442 | C24H26O8 | Aspergillus flavipes PJ03-11 | Wetland mud | Panjin Red Beach National Nature Reserve, Liaoning, China | [31] |
| Flaviphenalenone B (46) | 372 | C20H20O7 | Aspergillus flavipes PJ03-11 | Wetland mud | Panjin Red Beach National Nature Reserve, Liaoning, China | [31] |
| Flaviphenalenone C (47) | 372 | C20H20O7 | Aspergillus flavipes PJ03-11 | Wetland mud | Panjin Red Beach National Nature Reserve, Liaoning, China | [31] |
| Rousselianone A′ = Acetone adduct of 4,9-dihydroxy-6-methyl-7-((3-methylbut-2-en-1-yl)oxy)-1H-phenalene-1,2,3-trione (48) | 398 | C22H22O7 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [37] |
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] | |||
| Auxarthrone A (49) | 358 | C19H18O7 | Auxarthron pseudauxarthron TTI-0363 | Sylvilagus audubonii (Desert rabbit dung, Leporidae) | U.S. Route 385, Marathon, and the north entrance of Big Bend National Park, Brewster, Texas, USA | [48] |
| Auxarthrone B (50) | 358 | C19H18O7 | Auxarthron pseudauxarthron TTI-0363 | Sylvilagus audubonii (Desert rabbit dung, Leporidae) | U.S. Route 385, Marathon, and the north entrance of Big Bend National Park, Brewster, Texas, USA | [48] |
| Auxarthrone D (51) | 372 | C20H20O7 | Auxarthron pseudauxarthron TTI-0363 | Sylvilagus audubonii (Desert rabbit dung, Leporidae) | U.S. Route 385, Marathon, and the north entrance of Big Bend National Park, Brewster, Texas, USA | [48] |
| Auxarthrone C (52) | 340 | C19H16O6 | Auxarthron pseudauxarthron TTI-0363 | Sylvilagus audubonii (Desert rabbit dung, Leporidae) | U.S. Route 385, Marathon, and the north entrance of Big Bend National Park, Brewster, Texas. | [48] |
| Auxarthrone E (53) | 354 | C20H18O6 | Auxarthron pseudauxarthron TTI-0363 | Sylvilagus audubonii (Desert rabbit dung, Leporidae) | U.S. Route 385, Marathon, and the north entrance of Big Bend National Park, Brewster, Texas, USA | [48] |
| FR-901235 (54) | 344 | C18H16O7 | Auxarthron pseudauxarthron TTI-0363 | Sylvilagus audubonii (Desert rabbit dung, Leporidae) | U.S. Route 385, Marathon, and the north entrance of Big Bend National Park, Brewster, Texas, USA | [48] |
| 9-Demethyl FR-901235 (55) | 330 | C17H14O7 | Talaromyces stipitatus | Soil | Collaroy, New South Wales, Australia | [49] |
| (10S)-6-amino-3,6,8-trihydroxy-1,9,9,10-tetramethyl-9,10-dihydro-5H-furo[2′,3′:5,6]naphtho[1,8-bc]oxepine-5,7(6H)-dione (56) | 373 | C19H19NO7 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [37] |
| (S)-3,8-Dihydroxy-6-imino-1,9,9,10-tetramethyl-9,10-dihydro-5H-furo[2′,3′:5,6]naphtho[1,8-bc]oxepine -5,7(6H)-dione (57) | 355 | C19H17NO6 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [37] |
| S-Dehydroazasirosterol (58) | C47H59NO6 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [37] | |
| Conio-azasterol (59) | 733 | C47H59NO6 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [37] |
| Aspergillussanone C (60) | 624 | C35H44O10 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
| Aspergillussanone D (61) | 592 | C35H44O8 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
| Aspergillussanone E (62) | 592 | C35H44O8 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
| Aspergillussanone F (63) | 594 | C35H46O8 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
| Aspergillussanone G (64) | 610 | C35H46O9 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
| Aspergillussanone H (65) | 606 | C35H42O9 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
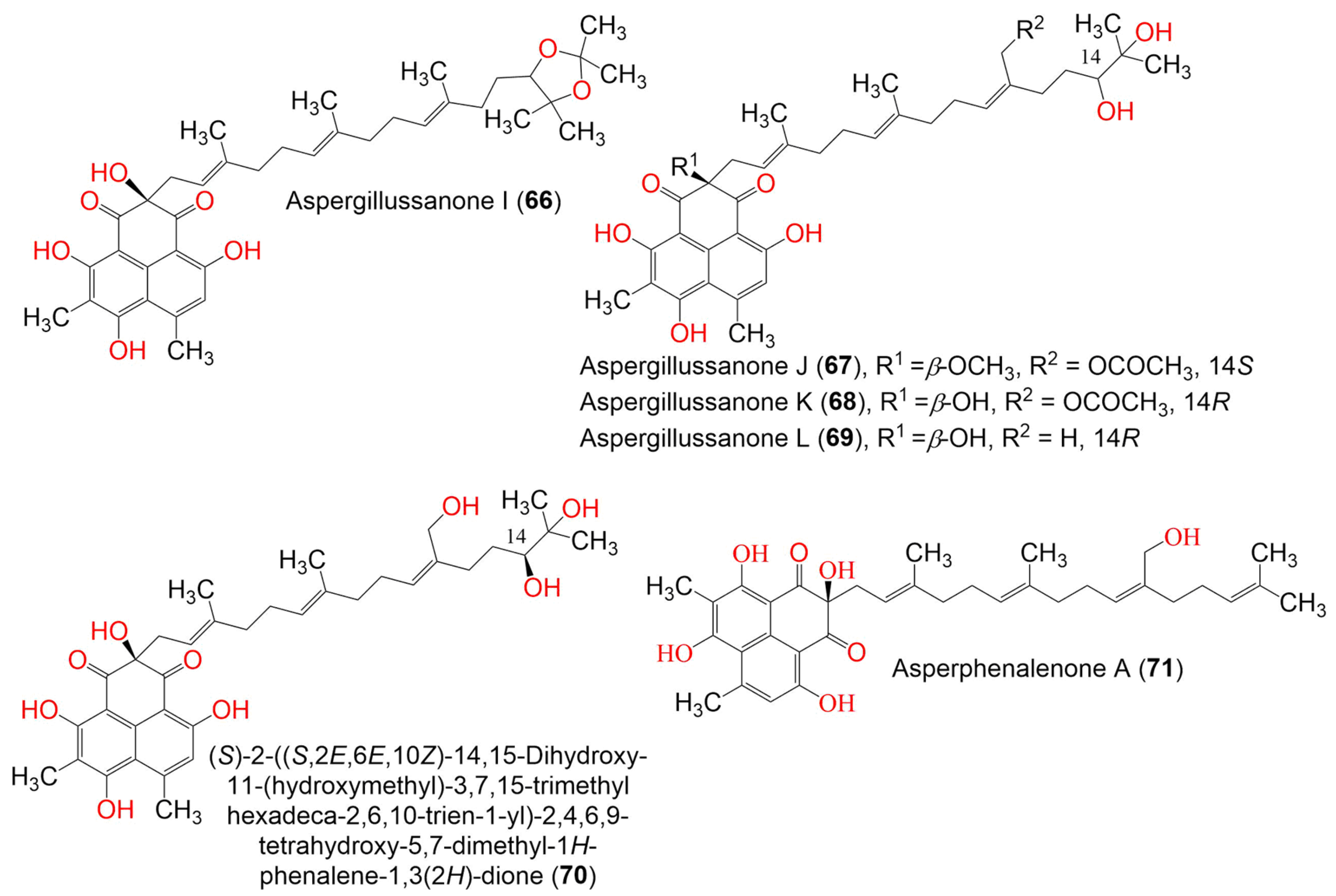
| Aspergillussanone I (66) | 634 | C38H50O8 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
| Aspergillussanone J (67) | 666 | C38H50O10 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
| Aspergillussanone K (68) | 652 | C37H48O10 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
| Aspergillussanone L (69) | 594 | C35H46O8 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
| (S)-2-((S,2E,6E,10Z)-14,15-Dihydroxy-11-(hydroxymethyl)-3,7,15-trimethylhexadeca-2,6,10-trien-1-yl)-2,4,6,9-tetrahydroxy- 5,7-dimethyl-1H-phenalene-1,3(2H)-dione (70) | 610 | C35H46O9 | Aspergillus sp. | Pinellia ternate (Tuber, Araceae) | Suburb of Nanjing, Jiangsu, China | [50] |
| Asperphenalenone A (71) | 576 | C35H44O7 | Aspergillus sp. CPCC 400735 | Kadsura longipedunculata (Schisandraceae) | Leishan, in Guizhou province, China | [51] |
| Asperphenalenone B (72) | 624 | C35H44O10 | Aspergillus sp. CPCC 400735 | Kadsura longipedunculata (Schisandraceae) | Leishan, in Guizhou province, China | [51] |
| Asperphenalenone C (73) | 624 | C35H44O10 | Aspergillus sp. CPCC 400735 | Kadsura longipedunculata (Schisandraceae) | Leishan, in Guizhou province, China | [51] |
| Asperphenalenone D (74) | 654 | C37H50O10 | Aspergillus sp. CPCC 400735 | Kadsura longipedunculata (Schisandraceae) | Leishan, in Guizhou province, China | [51] |
| Asperphenalenone E (75) | 610 | C35H46O9 | Aspergillus sp. CPCC 400735 | Kadsura longipedunculata (Schisandraceae) | Leishan, in Guizhou province, China | [51] |
| Peniciphenalenin A (76) | 328 | C18H16O6 | Penicillium sp. ZZ901 | Scapharca broughtonii (Schrenck blood clam, Arcidae) | Sea shoal, East China, Zhoushan Archipelago, China | [39] |
| Peniciphenalenin B (77) | 404 | C20H20O9 | Penicillium sp. ZZ901 | Scapharca broughtonii (Schrenck blood clam, Arcidae) | Sea shoal, East China, Zhoushan Archipelago, China | [39] |
| Peniciphenalenin C (78) | 418 | C21H22O9 | Penicillium sp. ZZ901 | Scapharca broughtonii (Schrenck blood clam, Arcidae) | Sea shoal, East China, Zhoushan Archipelago, China | [39] |
| Peniciphenalenin D (79) | 316 | C17H16O6 | Penicillium sp. ZZ901 | Scapharca broughtonii (Schrenck blood clam, Arcidae) | Sea shoal, East China, Zhoushan Archipelago, China | [39] |
| Chrysosporium lobatum TM-237-S5 | Acanthella cavernosa (Sponge, Dictyonellidae) | Mesophotic reef in Eilat, Dekel Beach, Gulf of Aqaba, Israel | [40] | |||
| Peniciphenalenin E, Peniciphenalenin Ea (80) | 300 | C17H16O5 | Penicillium sp. ZZ901 | Scapharca broughtonii (Schrenck blood clam, Arcidae) | Sea shoal, East China, Zhoushan Archipelago, China | [39] |
| Peniciphenalenin F (81) | 300 | C17H16O5 | Penicillium sp. ZZ901 | Scapharca broughtonii (Schrenck blood clam, Arcidae) | Sea shoal, East China, Zhoushan Archipelago, China | [39] |
| (−)-Peniciphenalenin F (82) | 300 | C17H16O5 | Chrysosporium lobatum TM-237-S5 | Acanthella cavernosa (Sponge, Dictyonellidae) | Mesophotic reef in Eilat, Dekel Beach, Gulf of Aqaba, Israel | [40] |
| Peniciphenalenin G (83) | 364 | C18H17ClO6 | Pleosporales sp. HDN1811400 | Sediment | Fildes Peninsula | [44] |
| Peniciphenalenin I (84) | 328 | C18H16O6 | Pleosporales sp. HDN1811400 | Sediment | Fildes Peninsula | [44] |
| Coniosclerodione (85) | 312 | C18H16O5 | Pleosporales sp. HDN1811400 | Sediment | Fildes Peninsula | [44] |
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] | |||
| Trypethelonamide A (86) | 399 | C23H29NO5 | Trypethelium eluteriae Sprengl | - | Hainan, China | [52] |
| 5′-Hydroxytrypethelone (87) | 288 | C16H16O5 | Trypethelium eluteriae Sprengl | - | Hainan, China | [52] |
| (+)-8-Hydroxy-7-methoxytrypethelone (88) | 302 | C17H18O5 | Trypethelium eluteriae Sprengl | - | Hainan, China | [52] |
| (+)-Trypethelone (89) | 272 | C16H16O4 | Trypethelium eluteriae Sprengl | - | Hainan, China | [52] |
| Trypethelium eluteriae Sprengl | Anacardium occidentale (Bark, Anacardiaceae) | Vadanemmeli, Kanchipuram, Tamil Nadu, India | [41] | |||
| (−)-Trypethelone (90) | 272 | C16H16O4 | Trypethelium eluteriae Sprengl | - | Hainan, China | [52] |
| Pseudolophiostoma sp. MFLUCC 17-2081 | Clematis fulvicoma (Ranunculaceae) | Chiang Rai, Thailand | [45] | |||
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] | |||
| Trypethelone methyl ether (91) | 286 | C17H18O4 | Trypethelium eluteriae Sprengl | Anacardium occidentale (Bark, Anacardiaceae) | Vadanemmeli, Kanchipuram, Tamil Nadu, India | [41] |
| 8-Hydroxytrypethelone methyl ether (92) | 302 | C17H18O5 | Trypethelium eluteriae Sprengl | Anacardium occidentale (Bark, Anacardiaceae) | Vadanemmeli, Kanchipuram, Tamil Nadu, India | [41] |
| 8-Methoxytrypethelone (93) | 302 | C17H18O5 | Trypethelium eluteriae Sprengl | Anacardium occidentale (Bark, Anacardiaceae) | Vadanemmeli, Kanchipuram, Tamil Nadu, India | [41] |
| 4′-Hydroxy-8-methoxytrypethelone methyl ether (94) | 332 | C18H20O6 | Trypethelium eluteriae Sprengl | Anacardium occidentale (Bark, Anacardiaceae) | Vadanemmeli, Kanchipuram, Tamil Nadu, India | [41] |
| 5′-Hydroxy-8-ethoxytrypethelone (95) | 332 | C18H20O6 | Trypethelium eluteriae Sprengl | Anacardium occidentale (Bark, Anacardiaceae) | Vadanemmeli, Kanchipuram, Tamil Nadu, India | [41] |
| 8-Methoxytrypethelone methyl ether (96) | 316 | C18H20O5 | Trypethelium eluteriae Sprengl | Anacardium occidentale (Bark, Anacardiaceae) | Vadanemmeli, Kanchipuram, Tamil Nadu, India | [41] |
| Isoconiolactone (97) | 300 | C17H16O5 | Chrysosporium lobatum TM-237-S5 | Acanthella cavernosa (Sponge, Dictyonellidae) | Mesophotic reef in Eilat, Dekel Beach, Gulf of Aqaba, Israel | [40] |
| Coniolactone (98) | 300 | C17H16O5 | Chrysosporium lobatum TM-237-S5 | Acanthella cavernosa (Sponge, Dictyonellidae) | Mesophotic reef in Eilat, Dekel Beach, Gulf of Aqaba, Israel | [40] |
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] | |||
| Coniosclerodin (99) | 328 | C18H16O6 | Chrysosporium lobatum TM-237-S5 | Acanthella cavernosa (Sponge, Dictyonellidae) | Mesophotic reef in Eilat, Dekel Beach, Gulf of Aqaba, Israel | [40] |
| Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] | |||
| O-Desmethylfunalenone (100) | 274 | C14H10O6 | Talaromyces stipitatus | Soil | Collaroy, New South Wales, Australia | [49] |
| Funalenone (101) | 288 | C15H12O6 | Aspergillus sp. SF-5929. | Coral | Ross Sea, Antarctica | [53] |
| Hispidulone A (102) | 276 | C14H12O6 | Chaetosphaeronema hispidulum TS-8-1 | Desert plant | Desert, arid, and grassland areas, China | [54] |
| Hispidulone B (103) | 290 | C15H14O6 | Chaetosphaeronema hispidulum TS-8-1 | Desert plant | Desert, arid, and grassland areas, China | [54] |
| Aceneoherqueinone A (104) | 414 | C22H22O8 | Penicillium herquei MA-370 | Rhizospheric soil of the mangrove plant Rhizophora mucronata (Rhizophoraceae) | Hainan Island, China | [46] |
| Aceneoherqueinone B (105) | 414 | C22H22O8 | Penicillium herquei MA-370 | Rhizospheric soil of the mangrove plant Rhizophora mucronata (Rhizophoraceae) | Hainan Island, China | [46] |
| (+)-Aceatrovenetinone A (106) | 398 | C22H22O7 | Penicillium herquei MA-370 | Rhizospheric soil of the mangrove plant Rhizophora mucronata (Rhizophoraceae) | Hainan Island, China | [46] |
| (−)-Aceatrovenetinone B (107) | 398 | C22H22O7 | Penicillium herquei MA-370 | Rhizospheric soil of the mangrove plant Rhizophora mucronata (Rhizophoraceae) | Hainan Island, China | [46] |
| (−)-aceatrovenetinone A (108) | 398 | C22H22O7 | Penicillium herquei MA-370 | Rhizospheric soil of the mangrove plant Rhizophora mucronata (Rhizophoraceae) | Hainan Island, China | [46] |
| (+)-Aceatrovenetinone B (109) | 398 | C22H22O7 | Penicillium herquei MA-370 | Rhizospheric soil of the mangrove plant Rhizophora mucronata (Rhizophoraceae) | Hainan Island, China | [46] |
| Lamellicolic anhydride (110) | 260. | C13H8O6 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] |
| (−)-Cereoaldomine (111) | 301 | C16H15NO5 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] |
| Z-Coniosclerodinol (112) | 344 | C18H16O7 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] |
| E-Coniosclerodinol (113) | 344 | C18H16O7 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] |
| Conioscleroderolide (114) | 328 | C18H16O6 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] |
| Cereolactam (115) | 299 | C17H17NO4 | Coniothyrium cereale | Enteromorpha sp. (Algae, Ulvaceae) | Fehmarn, Baltic Sea, Germany | [20] |
| Herqueilenone A (116) | 581 | C30H31NO11 | Penicillium herquei FT729 | Volcanic soil sample | Big Island, Hawaii | [55] |
| Erabulenol B (117) | 550 | C30H30O10 | Penicillium herquei FT729 | Volcanic soil sample | Big Island, Hawaii | [55] |
| Erabulenol C (118) | 550 | C30H30O10 | Penicillium herquei FT729 | Volcanic soil | Big Island, Hawaii | [55] |
| Duclauxamide A1 (119) | 557 | C30H23NO10 | Penicillium manginii YIM PH30375 | Panax notoginseng (Root, Araliaceae) | Wenshan, Yunnan, China | [56] |
| Duclauxin (120) | 546 | C29H22O11 | Penicillium manginii YIM PH30375 | Panax notoginseng (Root, Araliaceae) | Wenshan, Yunnan, China | [56] |
| Talaromyces verruculosus | Goniopora sp. (Soft coral, Poritidae) | Sanya, Hainan Island, South China Sea, China | [57] | |||
| Talaromyces stipitatus | Soil | Collaroy, New South Wales, Australia | [49] | |||
| Talaromyces sp. IQ-313 | Anthill soil | Huasteca Hidalguense, Hidalgo State, Mexico | [58] | |||
| Talaromycesone A (121) | 548 | C29H24O11 | Talaromyces sp. LF458 | Axinellaverrucosa (Sponge, Axinellidae) | Punta di Fetovaia, Isle of Elba, Mediterranean Sea, Italy | [59] |
| Talaromycesone B (122) | 502 | C27H18O10 | Talaromyces sp. LF458 | Axinellaverrucosa (Sponge, Axinellidae) | Punta di Fetovaia, Isle of Elba, Mediterranean Sea, Italy | [59] |
| Talaromyces sp. IQ-313 | Anthill soil | Huasteca Hidalguense, Hidalgo State, Mexico. | [58] | |||
| Bacillisporin A (123) | 516 | C28H20O10 | Talaromyces stipitatus ATCC 10500 | Culture | France | [60] |
| 9a-Epi-bacillisporin E (124) | 532 | C28H20O11 | Talaromyces stipitatus ATCC 10500 | Culture | France | [60] |
| Bacillisporin F (125) | 546 | C29H22O11 | Talaromyces stipitatus ATCC 10500 | Culture | France | [60] |
| Talaromyces verruculosus | Goniopora sp. (Soft coral, Poritidae) | Sanya, Hainan Island, South China Sea, China | [57] | |||
| 1- Epi-bacillisporin F (126) | 546 | C29H22O11 | Talaromyces stipitatus ATCC 10500 | Culture | France | [60] |
| Bacillisporin G (127) | 488 | C27H20O9 | Talaromyces stipitatus ATCC 10500 | Culture | France | [60] |
| Talaromyces sp. IQ-313 | Anthill soil | Huasteca Hidalguense, Hidalgo State, Mexico. | [58] | |||
| Bacillisporin H (128) | 545 | C29H23NO10 | Talaromyces stipitatus ATCC 10500 | Culture | France | [60] |
| Verruculosin A (129) | 604 | C32H28O12 | Talaromyces verruculosus | Goniopora sp. (Soft coral, Poritidae) | Sanya, Hainan Island, South China Sea, China | [57] |
| Verruculosin B (130) | 620 | C32H28O13 | Talaromyces verruculosus | Goniopora sp. (Soft coral, Poritidae) | Sanya, Hainan Island, South China Sea, China | [57] |
| Xenoclauxin (131) | 562 | C29H22O12 | Talaromyces verruculosus | Goniopora sp. (Soft coral, Poritidae) | Sanya, Hainan Island, South China Sea, China | [57] |
| Talaromyces sp. IQ-313 | Anthill soil | Huasteca Hidalguense, Hidalgo State, Mexico. | [58] | |||
| Talauxin E (132) | 675 | C34H29NO14 | Talaromyces stipitatus | Soil | Collaroy, New South Wales, Australia | [49] |
| Talauxin I (133) | 659 | C35H33NO12 | Talaromyces stipitatus | Soil | Collaroy, New South Wales, Australia | [49] |
| Talauxin L (134) | 659 | C35H33NO12 | Talaromyces stipitatus | Soil | Collaroy, New South Wales, Australia | [49] |
| Talauxin Q (135) | 674 | C34H30N2O13 | Talaromyces stipitatus | Soil | Collaroy, New South Wales, Australia | [49] |
| Talauxin V (136) | 645 | C34H31NO12 | Talaromyces stipitatus | Soil | Collaroy, New South Wales, Australia | [49] |
| Epitalauxin I (137) | 659 | C35H33NO12 | Talaromyces stipitatus | Soil | Collaroy, New South Wales, Australia | [49] |
| Epitalauxin L (138) | 659 | C35H33NO12 | Talaromyces stipitatus | Soil | Collaroy, New South Wales, Australia | [49] |
| Pseudolophiostoma sp. MFLUCC 17-2081 | Clematis fulvicoma (Ranunculaceae) | Chiang Rai, Thailand | [45] | |||
| Desacetyldesmethyltalauxin V (139) | 571 | C31H25NO10 | Talaromyces stipitatus | Soil sample | Collaroy, New South Wales, Australia | [49] |
| Compound Name | Biological Activity | Assay, Organism, or Cell Line | Biological Results | Positive Control | Ref. |
|---|---|---|---|---|---|
| Paecilomycone A (1) | Tyrosinase inhibition | Colorimetric-microtiter plates/Tyrosinase enzyme | 0.11 mM (IC50) | Kojic acid 0.10 mM (IC50) Arbutin 0.20 mM (IC50) | [32] |
| Paecilomycone B (2) | Tyrosinase inhibition | Colorimetric-microtiter plates/Tyrosinase enzyme | 0.17 mM (IC50) | Kojic acid 0.10 mM (IC50) Arbutin 0.20 mM (IC50) | [32] |
| Paecilomycone C (3) | Tyrosinase inhibition | Colorimetric-microtiter plates/Tyrosinase enzyme | 0.14 mM (IC50) | Kojic acid 0.10 mM (IC50) Arbutin 0.20 mM (IC50) | [32] |
| Aspergillussanone A (5) | Cytotoxicity | Resazurin microplate/KB | 48.4 μM (IC50) | Ellipticine 4.1 μM (IC50) | [33] |
| Resazurin microplate/Vero | 34.2 μM (IC50) | Ellipticine 4.5 μM (IC50) | [33] | ||
| ent-Peniciherqueinone (8) | Adipogenesis induction | Adiponectin production assay/hBM-MSC(B7) | 57.5 µM (IC50) | Pioglitazone 0.69 µM (IC50) | [35] |
| Herqueinone (9) | Antioxidant | DPPH/DPPH• | 0.48 mM (IC50) | Butylated hydroxytoluene 0.11 mM (IC50) | [34] |
| Hydroxyl radical scavenging/OH• | 6.34 mM (IC50) | Tannic acid 0.26 mM (IC50) | [34] | ||
| Superoxide radical scavenging/O2•− | 4.11 mM (IC50) | Trolox 0.96 mM (IC50) | [34] | ||
| Isoherqueinone (12) | Adipogenesis induction | Adiponectin production assay/hBM-MSC(B7) | 39.7 µM (IC50) | Pioglitazone 0.69 µM (IC50) | [35] |
| Acetone adduct of a triketone (17) | Anti-inflammatory | Nitric oxide synthase/RAW 264.7 | 3.2 µM (IC50) | AMT 0.2 µM (IC50) | [35] |
| (+)-Sclerodin (21) | Cytotoxicity | SRB/U87MG | 55.99 µM (IC50) | Doxorubicin 1.2 µM (IC50) | [39] |
| SRB/C6 | 44.65 µM (IC50) | Doxorubicin 0.47 µM (IC50) | [39] | ||
| (−)-Sclerodinol (24) | Antimicrobial | Agar dilution/B. cereus | 25.0 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] |
| Agar dilution/Proteus species | 50.0 µM (MIC) | Ciprofloxacin 0.78 µM (MIC) | [44] | ||
| Agar dilution/M. Phlei | 25.0 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/B. subtilis | 12.5 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/V. parahemolyticus | 12.5 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/MRCNS | 50.0 µM (MIC) | Ciprofloxacin 25.0 µM (MIC) | [44] | ||
| Agar dilution/MRSA | 50.0 µM (MIC) | Ciprofloxacin > 50 µM (MIC) | [44] | ||
| Bipolarol C (26) | Antibacterial | REMA/B. cereus | 25.0 µg/mL (MIC) | Vancomycin 1.0 µg/mL (MIC) | [42] |
| 4-Hydroxysclerodin (27) | Anti-angiogenetic | Tube formation assay/HUVECs | 20.9 µM (IC50) | Sunitinib 1.5 µM (IC50) | [35] |
| (+)-Sclerodione (28) | Cytotoxicity | SRB/U87MG | 60.93 µM (IC50) | Doxorubicin 1.2 µM (IC50) | [39] |
| SRB/C6 | 60.81 µM (IC50) | Doxorubicin 0.47 µM (IC50) | [39] | ||
| Antibacterial | Micro-broth dilution/MRSA | 23.0 µg/mL (MIC) | Gentamicin 0.5 µg/mL (MIC) | [39] | |
| Micro-broth dilution/E. coli | 35.0 µg/mL (MIC) | Gentamicin 1.0 µg/mL (MIC) | [39] | ||
| (−)–Sclerodione (29) | α-Glucosidase inhibition | Colorimetric/α-Glucosidase | 120 µM (IC50) | N-deoxynojirimycin 130.5 µM (IC50) | [45] |
| Porcine-lipase inhibition | Colorimetric/Porcine lipase | 1.0 µM (IC50) | Orlistat 9.4 µM (IC50) | [45] | |
| (−)-Bipolaride B (30) | Antibacterial | REMA/B. cereus | 25.0 µg/mL (MIC) | Vancomycin 1.0 µg/mL (MIC) | [42] |
| Cytotoxicity | REMA/MCF-7 | 79.4 µM (IC50) | Doxorubicin 12.99 µM (IC50) Tamoxifen 19.39 µM (IC50) | [42] | |
| REMA/KB | 96.8 µM (IC50) | Ellipticine 8.32 µM (IC50) Doxorubicin 1.15 µM (IC50) | [42] | ||
| REMA/NCI-H187 | 56.5 µM (IC50) | Ellipticine 9.74 µM (IC50) Doxorubicin 0.19 µM (IC50) | [42] | ||
| Peniciphenalenin H (31) | Antimicrobial | Agar dilution/Proteus species | 50.0 µM (MIC) | Ciprofloxacin 0.78 µM (MIC) | [44] |
| Agar dilution/B. subtilis | 25.0 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/V. parahemolyticus | 25.0 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/MRCNS | 25.0 µM (MIC) | Ciprofloxacin 25.0 µM (MIC) | [44] | ||
| Agar dilution/MRSA | 50.0 µM (MIC) | Ciprofloxacin > 50 µM (MIC) | [44] | ||
| Bipolarol A (32) | Cytotoxicity | REMA/MCF-7 | 110.4 µM (IC50) | Doxorubicin 12.99 µM (IC50) Tamoxifen 19.39 µM (IC50) | [42] |
| (+)-Scleroderolide (33) | Cytotoxicity | SRB/U87MG | 37.26 µM (IC50) | Doxorubicin 1.2 µM (IC50) | [39] |
| SRB/C6 | 23.24 µM (IC50) | Doxorubicin 0.47 µM (IC50) | [39] | ||
| Antibacterial | Micro-broth dilution/MRSA | 7.0 µg/mL (MIC) | Gentamicin 0.5 µg/mL (MIC) | [39] | |
| Micro-broth dilution/E. coli | 9.0 µg/mL (MIC) | Gentamicin 1.0 µg/mL (MIC) | [39] | ||
| (−)–Scleroderolide (34) | α-Glucosidase inhibition | Colorimetric/α-Glucosidase | 48.7 µM (IC50) | N-Deoxynojirimycin 130.5 µM (IC50) | [45] |
| porcine-lipase inhibition | Colorimetric/Porcine lipase | 3.4 µM (IC50) | Orlistat 9.4 µM (IC50) | [45] | |
| (−)-Bipolaride A (36) | Antibacterial | REMA/B. cereus | 12.5 µg/mL (MIC) | Vancomycin 1.0 µg/mL (MIC) | [42] |
| Cytotoxicity | REMA/NCI-H187 | 60.2 µM (IC50) | Ellipticine 9.74 µM (IC50) Doxorubicin 0.19 µM (IC50) | [42] | |
| (−)-Bipolaride E (39) | Antibacterial | REMA/B. cereus | 12.5 µg/mL (MIC) | Vancomycin 1.0 µg/mL (MIC) | [42] |
| Cytotoxicity | REMA/MCF-7 | 65.1 µM (IC50) | Doxorubicin 12.99 µM (IC50) Tamoxifen 19.39 µM (IC50) | [42] | |
| REMA/KB | 94.4 µM (IC50) | Ellipticine 8.32 µM (IC50) Doxorubicin 1.15 µM (IC50) | [42] | ||
| REMA/NCI-H187 | 86.9 µM (IC50) | Ellipticine 9.74 µM (IC50) Doxorubicin 0.19 µM (IC50) | [42] | ||
| Bipolarol B (40) | Cytotoxicity | REMA/MCF-7 | 65.3 µM (IC50) | Doxorubicin 12.99 µM (IC50) Tamoxifen 19.39 µM (IC50) | [42] |
| REMA/KB | 52.5 µM (IC50) | Ellipticine 8.32 µM (IC50) Doxorubicin 1.15 µM (IC50) | [42] | ||
| REMA/NCI-H187 | 48.3 µM (IC50) | Ellipticine 9.74 µM (IC50) Doxorubicin 0.19 µM (IC50) | [42] | ||
| Bipolarol D (41) | REMA/MCF-7 | REMA/MCF-7 | 108.7 µM (IC50) | Doxorubicin 12.99 µM (IC50) Tamoxifen 19.39 µM (IC50) | [42] |
| (−)-Bipolaride C (42) | Antibacterial | REMA/B. cereus | 12.5 µg/mL (MIC) | Vancomycin 1.0 µg/mL (MIC) | [42] |
| Cytotoxicity | REMA/MCF-7 | 48.9 µM (IC50) | Doxorubicin 12.99 µM (IC50) Tamoxifen 19.39 µM (IC50) | [42] | |
| REMA/KB | 34.4 µM (IC50) | Ellipticine 8.32 µM (IC50) Doxorubicin 1.15 µM (IC50) | [42] | ||
| REMA/NCI-H187 | 59.8 µM (IC50) | Ellipticine 9.74 µM (IC50) Doxorubicin 0.19 µM (IC50) | [42] | ||
| REMA/Vero | 53.1 µM (IC50) | Ellipticine 2.13 µM (IC50) | [42] | ||
| Flaviphenalenone A (45) | Cytotoxicity | MTT/A549 | 6.6 µg/mL (IC50) | Doxorubicin HCl 0.2 µg/mL (IC50) | [31] |
| MTT/MCF-7 | 10.0 µg/mL (IC50) | Doxorubicin HCl 0.4 µg/mL (IC50) | [31] | ||
| Flaviphenalenone B (46) | α-Glucosidase inhibition | Colorimetrically/α-Glucosidase | 94.95 µM (IC50) | Acarbose 685.36 µM (IC50) | [31] |
| Flaviphenalenone C (47) | α-Glucosidase inhibition | Colorimetrically/α-Glucosidase | 78.96 µM (IC50) | Acarbose 685.36 µM (IC50) | [31] |
| Cytotoxicity | MTT/A549 | 28.5 µg/mL (IC50) | Doxorubicin HCl 0.2 µg/mL (IC50) | [31] | |
| MTT/MCF-7 | 50.0 µg/mL (IC50) | Doxorubicin HCl 0.4 µg/mL (IC50) | [31] | ||
| Auxarthrone A (49) | Antifungal | Serial dilution/C. neoformans | 3.2 µg/mL (MIC) | Amphotericin B 0.8 µg/mL (MIC) | [48] |
| Serial dilution/C. albicans | 3.2 µg/mL (MIC) | Amphotericin B 0.8 µg/mL (MIC) | [48] | ||
| Auxarthrone B (50) | Antifungal | Serial dilution/C. neoformans | 12.8 µg/mL (MIC) | Amphotericin B 0.8 µg/mL (MIC) | [48] |
| Serial dilution/C. albicans | 25.6 µg/mL (MIC) | Amphotericin B 0.8 µg/mL (MIC) | [48] | ||
| Auxarthrone D (51) | Antifungal | Serial dilution/C. neoformans | 6.4 µg/mL (MIC) | Amphotericin B 0.8 µg/mL (MIC) | [48] |
| Serial dilution/C. albicans | 6.4 µg/mL (MIC) | Amphotericin B 0.8 µg/mL (MIC) | [48] | ||
| Auxarthrone C (52) | Antifungal | Serial dilution/C. neoformans | 25.6 µg/mL (MIC) | Amphotericin B 0.8 µg/mL (MIC) | [48] |
| Serial dilution/C. albicans | 51.2 µg/mL (MIC) | Amphotericin B 0.8 µg/mL (MIC) | [48] | ||
| FR-901235 (54) | Antifungal | Serial dilution/C. neoformans | 51.2 µg/mL (MIC) | Amphotericin B 0.8 µg/mL (MIC) | [48] |
| Serial dilution/C. albicans | 51.2 µg/mL (MIC) | Amphotericin B 0.8 µg/mL (MIC) | [48] | ||
| Aspergillussanone D (61) | Antibacterial | Broth microdilution/P. aeruginosa | 38.47 µg/mL (MIC50) | Streptomycin 0.34 µg/mL (MIC50) | [50] |
| Broth microdilution/S. aureus | 29.91 µg/mL (MIC50) | Penicillin 0.063 µg/mL (MIC50) | [50] | ||
| Aspergillussanone E (62) | Antibacterial | Broth microdilution/E. coli | 7.83 µg/mL (MIC50) | Streptomycin 0.34 µg/mL (MIC50) | [50] |
| Aspergillussanone F (63) | Antibacterial | Broth microdilution/P. aeruginosa | 26.56 µg/mL (MIC50) | Streptomycin 0.34 µg/mL (MIC50) | [50] |
| Broth microdilution/E. coli | 3.93 µg/mL (MIC50) | Streptomycin 0.25 µg/mL (MIC50) | [50] | ||
| Broth microdilution/S. aureus | 16.48 µg/mL (MIC50) | Penicillin 0.063 µg/mL (MIC50) | [50] | ||
| Aspergillussanone G (64) | Antibacterial | Broth microdilution/P. aeruginosa | 24.46 µg/mL (MIC50) | Streptomycin 0.34 µg/mL (MIC50) | [50] |
| Broth microdilution/S. aureus | 34.66 µg/mL (MIC50) | Penicillin 0.063 µg/mL (MIC50) | [50] | ||
| Aspergillussanone H (65) | Antibacterial | Broth microdilution/P. aeruginosa | 8.59 µg/mL (MIC50) | Streptomycin 0.34 µg/mL (MIC50) | [50] |
| Broth microdilution/E. coli | 5.87 µg/mL (MIC50) | Streptomycin 0.25 µg/mL (MIC50) | [50] | ||
| Aspergillussanone I (66) | Antibacterial | Broth microdilution/P. aeruginosa | 12.00 µg/mL (MIC50) | Streptomycin 0.34 µg/mL (MIC50) | [50] |
| Aspergillussanone J (67) | Antibacterial | Broth microdilution/P. aeruginosa | 28.50 µg/mL (MIC50) | Streptomycin 0.34 µg/mL (MIC50) | [50] |
| Broth microdilution/E. coli | 5.34 µg/mL (MIC50) | Streptomycin 0.25 µg/mL (MIC50) | [50] | ||
| Broth microdilution/S. aureus | 29.87 µg/mL (MIC50) | Penicillin 0.063 µg/mL (MIC50) | [50] | ||
| Aspergillussanone K (68) | Antibacterial | Broth microdilution/P. aeruginosa | 6.55 µg/mL (MIC50) | Streptomycin 0.34 µg/mL (MIC50) | [50] |
| Broth microdilution/S. aureus | 21.02 µg/mL (MIC50) | Penicillin 0.063 µg/mL (MIC50) | [50] | ||
| Aspergillussanone L (69) | Antibacterial | Broth microdilution/P. aeruginosa | 1.87 µg/mL (MIC50) | Streptomycin 0.34 µg/mL (MIC50) | [50] |
| Broth microdilution/S. aureus | 2.77 µg/mL (MIC50) | Penicillin 0.063 µg/mL (MIC50) | [50] | ||
| Broth microdilution/B. subtilis | 4.80 µg/mL (MIC50) | Penicillin 0.063 µg/mL (MIC50) | [50] | ||
| (S)-2-((S,2E,6E,10Z)-14,15-Dihydroxy-11-(hydroxymethyl)-3,7,15-trimethylhexadeca-2,6,10-trien-1-yl)-2,4,6,9-tetrahydroxy-5,7-dimethyl-1H-phenalene-1,3(2H)-dione (70) | Antibacterial | Broth microdilution/P. aeruginosa | 19.07 µg/mL (MIC50) | Streptomycin 0.34 µg/mL (MIC50) | [50] |
| Broth microdilution/E. coli | 1.88 µg/mL (MIC50) | Streptomycin 0.25 µg/mL (MIC50) | [50] | ||
| Asperphenalenone A (71) | Anti-HIV-1 | Luciferase assay/SupT1 cells | 4.2 µM (IC50) | Lamivudine 0.1 µM (IC50) Efavirenz 0.0004 µM (IC50) | [51] |
| Asperphenalenone B (72) | Anti-HIV-1 | Luciferase assay/SupT1 cells | 32.6 µM (IC50) | Lamivudine 0.1 µM (IC50) Efavirenz 0.0004 µM (IC50) | [51] |
| Asperphenalenone D (74) | Anti-HIV-1 | Luciferase assay/SupT1 cells | 2.4 µM (IC50) | Lamivudine 0.1 µM (IC50) Efavirenz 0.0004 µM (IC50) | [51] |
| Anti-HIV-1 | Luciferase assay/SupT1 cells | 22.1 µM (IC50) | Lamivudine 0.1 µM (IC50) Efavirenz 0.0004 µM (IC50) | [51] | |
| Peniciphenalenin G (83) | Antimicrobial | Agar dilution/B. cereus | 25.0 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] |
| Agar dilution/Proteus species | 50.0 µM (MIC) | Ciprofloxacin 0.78 µM (MIC) | [44] | ||
| Agar dilution/M. Phlei | 50.0 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/B. subtilis | 25.0 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/V. Parahemolyticus | 50.0 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/MRCNS | 25.0 µM (MIC) | Ciprofloxacin 25.0 µM (MIC) | [44] | ||
| Agar dilution/MRSA | 12.5 µM (MIC) | Ciprofloxacin > 50 µM (MIC) | [44] | ||
| Coniosclerodione (85) | Antimicrobial | Agar dilution/B. cereus | 25.0 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] |
| Agar dilution/Proteus sp. | 25.0 µM (MIC) | Ciprofloxacin 0.78 µM (MIC) | [44] | ||
| Agar dilution/M. Phlei | 25.0 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/B. subtilis | 12.5 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/V. parahemolyticus | 12.5 µM (MIC) | Ciprofloxacin 0.39 µM (MIC) | [44] | ||
| Agar dilution/MRCNS | 12.5 µM (MIC) | Ciprofloxacin 25.0 µM (MIC) | [44] | ||
| Agar dilution/MRSA | 6.25 µM (MIC) | Ciprofloxacin > 50 µM (MIC) | [44] | ||
| Trypethelonamide A (86) | Cytotoxicity | CCK8/RKO | 63.6 µM (IC50) | Taxol 0.05 µM (IC50) | [52] |
| 5′-Hydroxytrypethelone (87) | Cytotoxicity | CCK8/RKO | 22.6 µM (IC50) | Taxol 0.05 µM (IC50) | [52] |
| (+)-8-Hydroxy-7-methoxytrypethelone (88) | Cytotoxicity | CCK8/RKO | 113.5 µM (IC50) | Taxol 0.05 µM (IC50) | [52] |
| CCK8/HepG2 | 183.2 µM (IC50) | Taxol 1.0 µM (IC50) | [52] | ||
| (+)-Trypethelone (89) | Cytotoxicity | CCK8/RKO | 49.3 µM (IC50) | Taxol 0.05 µM (IC50) | [52] |
| (−)-Trypethelone (90) | Cytotoxicity | CCK8/RKO | 30.3 µM (IC50) | Taxol 0.05 µM (IC50) | [52] |
| O-Desmethylfunalenone (100) | Antibacterial | REMA/B. subtilis | 265 µM (IC50) | Clotrimazole 0.4 µM (IC50) | [49] |
| Cytotoxicity | Resazurin microplate/NS-1 | 70 µM (IC50) | 5-Fluorouracil 4.6 µM (IC50) | [49] | |
| Funalenone (101) | hPTP1B1-400 inhibition | Photocolorimetric/hPTP1B1-400 | 6.1 µM (IC50) | Ursolic acid 4.3 µM (IC50) | [53] |
| Hispidulone B (103) | Cytotoxicity | MTT/A-549 | 2.71 µM (IC50) | cis-Platinum 8.73 µM (IC50) | [54] |
| MTT/Huh7 | 22.93 µM (IC50) | cis-Platinum 5.89 µM (IC50) | [54] | ||
| MTT/HeLa | 23.94 µM (IC50) | cis-Platinum 14.68 µM (IC50) | [54] | ||
| Aceneoherqueinone A (104) | Angiotensin-I-converting enzyme inhibition | Spectrophotometric/Hippuryl-L-histidyl-L-leucine | 3.10 µM (IC50) | Captopril 9.23 nM (IC50) | [46] |
| Aceneoherqueinone B (105) | Angiotensin-I-converting enzyme inhibition | Spectrophotometric/Hippuryl-L-histidyl-L-leucine | 11.28 µM (IC50) | Captopril 9.23 nM (IC50) | [46] |
| Erabulenol B (117) | Indoleamine dioxygenase 1 inhibition | ELISA/Indoleamine 2,3-dioxygenase 1 | 13.69 µM (IC50) | Epacadostat 0.015 µM (IC50) | [55] |
| Erabulenol C (118) | Indoleamine dioxygenase 1 inhibition | ELISA/Indoleamine 2,3-dioxygenase 1 | 14.38 µM (IC50) | Epacadostat 0.015 µM (IC50) | [55] |
| Duclauxin (120) | Antitumor | ELISA/EGFR | 0.95 µM (IC50) | Afatinib 0.0005 µM (IC50) | [57] |
| ELISA/CDC25B | 0.75 µM (IC50) | Na3VO4 0.52 µM (IC50) | [57] | ||
| Cytotoxicity | Resazurin microplate/NS-1 | 140 µM (IC50) | 5-Fluorouracil 4.6 µM (IC50) | [49] | |
| hPTP1B1-400 inhibition | Photocolorimetric/hPTP1B1-400 | 12.7 µM (IC50) | Ursolic acid 26.6 µM (IC50) | [58] | |
| Talaromycesone A (121) | Antibacterial | REMA/S. epidermidis | 3.70 µM (IC50) | Chloramphenicol 1.81 µM (IC50) | [59] |
| Antibacterial | REMA/MRSA | 5.48 µM (IC50) | Chloramphenicol 2.46 µM (IC50) | [59] | |
| AchE inhibition | Modified Ellman’s enzyme/Immunosorbent assay | 7.49 µM (IC50) | Huperzine 11.60 µM (IC50) | [59] | |
| Talaromycesone B (122) | Antibacterial | REMA/S. epidermidis | 17.36 µM (IC50) | Chloramphenicol 1.81 µM (IC50) | [59] |
| REMA/MRSA | 19.50 µM (IC50) | Chloramphenicol 2.46 µM (IC50) | [59] | ||
| hPTP1B1-400 inhibition | Photocolorimetric/hPTP1B1-400 | 82.1 µM (IC50) | Ursolic acid 26.6 µM (IC50) | [58] | |
| Bacillisporin A (123) | Antibacterial | Microtiter plate/S. aureus | 5.2 µg/mL (MIC) | Tetracycline 0.05 µg/mL (MIC) | [60] |
| Microtiter plate/S. hemolyticus | 9.5 µg/mL (MIC) | Tetracycline 29.2 µg/mL (MIC) | [60] | ||
| Microtiter plate/E. faecalis | 2.4 µg/mL (MIC) | Tetracycline 0.4 µg/mL (MIC) | [60] | ||
| 9a-Epi-bacillisporin E (124) | Antibacterial | Microtiter plate/S. aureus | 29.3 µg/mL (MIC) | Tetracycline 0.05 µg/mL (MIC) | [60] |
| Bacillisporin F (125) | Antibacterial | Microtiter plate/S. aureus | 15.6 µg/mL (MIC) | Tetracycline 0.05 µg/mL (MIC) | [60] |
| Antitumor | ELISA/EGFR | 4.41 µM (IC50) | Afatinib 0.0005 µM (IC50) | [57] | |
| ELISA/CDC25B | 0.40 µM (IC50) | Na3VO4 0.52 µM (IC50) | [57] | ||
| Antitumor | ELISA/EGFR | 4.41 µM (IC50) | Afatinib 0.0005 µM (IC50) | [57] | |
| ELISA/CDC25B | 0.40 µM (IC50) | Na3VO4 0.52 µM (IC50) | [57] | ||
| Bacillisporin G (127) | hPTP1B1-400 inhibition | Photocolorimetric/hPTP1B1-400 | 13.5 µM (IC50) | Ursolic acid 26.6 µM (IC50) | [58] |
| Bacillisporin H (128) | Cytotoxicity | MTT/HeLa | 49.5 µM (IC50) | Cisplatin 10.6 µM (IC50) | [60] |
| Antibacterial | Microtiter plate/S. aureus | 5.0 µg/mL (MIC) | Tetracycline 0.05 µg/mL (MIC) | [60] | |
| Microtiter plate/S. hemolyticus | 20.4 µg/mL (MIC) | Tetracycline 29.2 µg/mL (MIC) | [60] | ||
| Verruculosin A (129) | Antitumor | ELISA/EGFR | 0.92 µM (IC50) | Afatinib 0.0005 µM (IC50) | [57] |
| Antitumor | ELISA/CDC25B | 0.38 µM (IC50) | Na3VO4 0.52 µM (IC50) | [57] | |
| Verruculosin B (130) | Antitumor | ELISA/EGFR | 1.22 µM (IC50) | Afatinib 0.0005 µM (IC50) | [57] |
| Xenoclauxin (131) | Antitumor | ELISA/EGFR | 0.24 µM (IC50) | Afatinib 0.0005 µM (IC50) | [57] |
| ELISA/CDC25B | 0.26 µM (IC50) | Na3VO4 0.52 µM (IC50) | [57] | ||
| hPTP1B1-400 inhibition | Photocolorimetric/hPTP1B1-400 | 21.8 µM (IC50) | Ursolic acid 26.6 µM (IC50) | [58] |
| Compound # | Probability * | Model Accuracy ** |
|---|---|---|
| 7 | 63.54% | 98.75% |
| 118 | 78.92% | 98.75% |
| 71 | 64.66% | 98.75% |
| 69 | 68% | 99% |
| 70 | 66% | 99% |
| 72 | 67.3% | 99% |
| 75 | 66% | 99% |
| 73 | 67.3% | 99% |
| 68 | 78% | 99% |
| 5 | 68% | 99% |
| 67 | 71.95% | 98.75% |
| 6 | 75% | 99% |
| 66 | NA | NA |
| 62 | 53% | 99% |
| 61 | 53% | 99% |
| 63 | 72% | 99% |
| 64 | 76% | 99% |
| 60 | 52.61% | 98.75% |
| 74 | 66% | 99% |
| 65 | 74% | 99% |
| Title | Mol_MW | # Stars | Dipole | SASA | HBD | HBA | QPlogPo/w | QPlogS | QPlogKhsa | # Metab | QPlogBB | %Human Oral Absorption | QPlogHERG | CNS | # RtvFG |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Recommended range | (130–725) | (0.0–5.0) | (1–12.50) | (300–1000) | (0–6) | (2.0–20.0) | (−2–6.5) | (−6.5–0.5) | (−1.5–1.5) | (1–8) | (−3–1.2) | (<25% poor; >80% high) | concern below −5 | (−2 inactive) (+2 active) | (0–2) |
| 7 | 388.373 | 0 | 3.013 | 566.818 | 2 | 6.5 | 1.851 | −3.559 | 0.097 | 6 | −1.437 | 74.987 | −3.478 | −2 | 0 |
| 118 | 550.561 | 1 | 7.485 | 794.384 | 2 | 8.25 | 3.782 | −6.151 | 0.715 | 9 | −2.297 | 69.345 | −5.022 | −2 | 0 |
| 71 | 576.728 | 3 | 8.802 | 910.147 | 3 | 6.7 | 6.314 | −7.258 | 1.316 | 18 | −2.368 | 79.595 | −5.491 | −2 | 0 |
| 69 | 594.744 | 2 | 9.484 | 789.214 | 4 | 7.45 | 5.322 | −4.622 | 0.981 | 17 | −2.174 | 57.778 | −3.532 | −2 | 0 |
| 70 | 610.743 | 7 | 9.975 | 1043.134 | 5 | 9.15 | 5.066 | −7.89 | 0.907 | 17 | −4.262 | 41.015 | −6.435 | −2 | 0 |
| 72 | 624.727 | 8 | 8.459 | 1026.611 | 5 | 9.45 | 5.31 | −7.68 | 0.735 | 16 | −4.35 | 28.338 | −4.482 | −2 | 1 |
| 75 | 610.743 | 7 | 9.879 | 1030.596 | 5 | 9.15 | 4.911 | −7.677 | 0.918 | 17 | −4.46 | 49.169 | −6.255 | −2 | 0 |
| 73 | 624.727 | 8 | 8.459 | 1026.616 | 5 | 9.45 | 5.31 | −7.68 | 0.735 | 16 | −4.351 | 28.338 | −4.482 | −2 | 1 |
| 68 | 652.78 | 8 | 12.335 | 1096.494 | 4 | 9.45 | 6.114 | −8.908 | 1.288 | 17 | −4.061 | 50.421 | −6.561 | −2 | 1 |
| 5 | 540.652 | 3 | 14.173 | 889.502 | 3 | 7.45 | 5.255 | −6.976 | 0.997 | 13 | −2.485 | 70.591 | −5.556 | −2 | 0 |
| 67 | 666.807 | 8 | 10.067 | 1027.334 | 3 | 9.45 | 6.512 | −7.869 | 1.359 | 16 | −3.158 | 73.463 | −5.735 | −2 | 1 |
| 6 | 540.652 | 2 | 7.928 | 919.785 | 3 | 7.45 | 5.282 | −7.502 | 1.064 | 13 | −2.834 | 67.029 | −5.832 | −2 | 0 |
| 66 | 634.808 | 6 | 10.74 | 970.033 | 2 | 6.5 | 7.963 | −8.898 | 1.935 | 15 | −1.591 | 100 | −5.533 | −2 | 1 |
| 62 | 592.728 | 6 | 12.392 | 1005.377 | 3 | 7.45 | 6.358 | −9.167 | 1.54 | 17 | −2.88 | 73.816 | −6.238 | −2 | 0 |
| 61 | 592.728 | 6 | 11.434 | 1011.989 | 3 | 7.45 | 6.379 | −9.284 | 1.561 | 17 | −2.954 | 73.171 | −6.288 | −2 | 0 |
| 63 | 592.771 | 5 | 11.447 | 968.98 | 3 | 5.75 | 7.264 | −8.949 | 1.821 | 13 | −2.355 | 85.225 | −5.777 | −2 | 0 |
| 64 | 608.77 | 5 | 12.352 | 986.08 | 4 | 7.45 | 5.988 | −8.302 | 1.398 | 14 | −3.232 | 53.649 | −5.838 | −2 | 0 |
| 60 | 624.727 | 4 | 11.95 | 974.752 | 4 | 11.15 | 4.249 | −7.089 | 0.71 | 14 | −3.346 | 53.716 | −5.828 | −2 | 3 |
| 74 | 654.796 | 7 | 10.567 | 1030.237 | 3 | 10.85 | 5.564 | −7.91 | 1.034 | 14 | −3.042 | 68.666 | −6.017 | −2 | 1 |
| 65 | 606.711 | 5 | 9.825 | 992.894 | 3 | 8.75 | 5.513 | −8.33 | 1.235 | 15 | −3.349 | 62.369 | −6.083 | −2 | 1 |
| Title | Docking Score | XP GScore | Glide Emodel | XP GScore |
|---|---|---|---|---|
| 60 | −15.082 | −15.777 | −112.586 | −14.777 |
| 64 | −14.829 | −15.239 | −101.024 | −14.239 |
| 66 | −13.511 | −15.227 | −88.992 | −14.227 |
| 71 | −13.348 | −15.063 | −96.987 | −14.063 |
| 68 | −14.276 | −14.973 | −130.242 | −13.973 |
| 61 | −14.073 | −14.77 | −93.445 | −13.77 |
| 70 | −14.248 | −14.658 | −98.707 | −13.658 |
| 65 | −12.689 | −14.404 | −96.631 | −13.404 |
| 69 | −12.733 | −14.277 | −85.473 | −13.277 |
| 75 | −13.343 | −14.04 | −108.477 | −13.04 |
| 74 | −12.284 | −14.002 | −99.09 | −13.002 |
| 63 | −13.061 | −13.757 | −104.496 | −12.757 |
| 72 | −11.996 | −13.711 | −78.259 | −12.711 |
| 67 | −12.07 | −13.571 | −105.73 | −12.571 |
| 5 | −12.01 | −13.511 | −94.603 | −12.511 |
| 6 | −13.162 | −13.466 | −88.095 | −12.466 |
| 73 | −11.712 | −13.361 | −96.264 | −12.361 |
| 62 | −12.586 | −13.283 | −98.251 | −12.283 |
| 7 | −11.849 | −12.42 | −78.277 | −11.42 |
| 5RE | −11.206 | −11.207 | −88.062 | −10.207 |
| 118 | −9.901 | −11.187 | −84.952 | −10.187 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, S.R.M.; Omar, A.M.; Muhammad, Y.A.; Alqarni, A.A.; Alshehri, A.M.; Mohamed, S.G.A.; Abdallah, H.M.; Elfaky, M.A.; Mohamed, G.A.; Xiao, J. Advances in Fungal Phenaloenones—Natural Metabolites with Great Promise: Biosynthesis, Bioactivities, and an In Silico Evaluation of Their Potential as Human Glucose Transporter 1 Inhibitors. Molecules 2022, 27, 6797. https://doi.org/10.3390/molecules27206797
Ibrahim SRM, Omar AM, Muhammad YA, Alqarni AA, Alshehri AM, Mohamed SGA, Abdallah HM, Elfaky MA, Mohamed GA, Xiao J. Advances in Fungal Phenaloenones—Natural Metabolites with Great Promise: Biosynthesis, Bioactivities, and an In Silico Evaluation of Their Potential as Human Glucose Transporter 1 Inhibitors. Molecules. 2022; 27(20):6797. https://doi.org/10.3390/molecules27206797
Chicago/Turabian StyleIbrahim, Sabrin R. M., Abdelsattar M. Omar, Yosra A. Muhammad, Ali A. Alqarni, Abdullah M. Alshehri, Shaimaa G. A. Mohamed, Hossam M. Abdallah, Mahmoud A. Elfaky, Gamal A. Mohamed, and Jianbo Xiao. 2022. "Advances in Fungal Phenaloenones—Natural Metabolites with Great Promise: Biosynthesis, Bioactivities, and an In Silico Evaluation of Their Potential as Human Glucose Transporter 1 Inhibitors" Molecules 27, no. 20: 6797. https://doi.org/10.3390/molecules27206797
APA StyleIbrahim, S. R. M., Omar, A. M., Muhammad, Y. A., Alqarni, A. A., Alshehri, A. M., Mohamed, S. G. A., Abdallah, H. M., Elfaky, M. A., Mohamed, G. A., & Xiao, J. (2022). Advances in Fungal Phenaloenones—Natural Metabolites with Great Promise: Biosynthesis, Bioactivities, and an In Silico Evaluation of Their Potential as Human Glucose Transporter 1 Inhibitors. Molecules, 27(20), 6797. https://doi.org/10.3390/molecules27206797