
Potential COVID-19 Drug Candidates Based on Diazinyl-Thiazol-Imine Moieties: Synthesis and Greener Pastures Biological Study

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussions
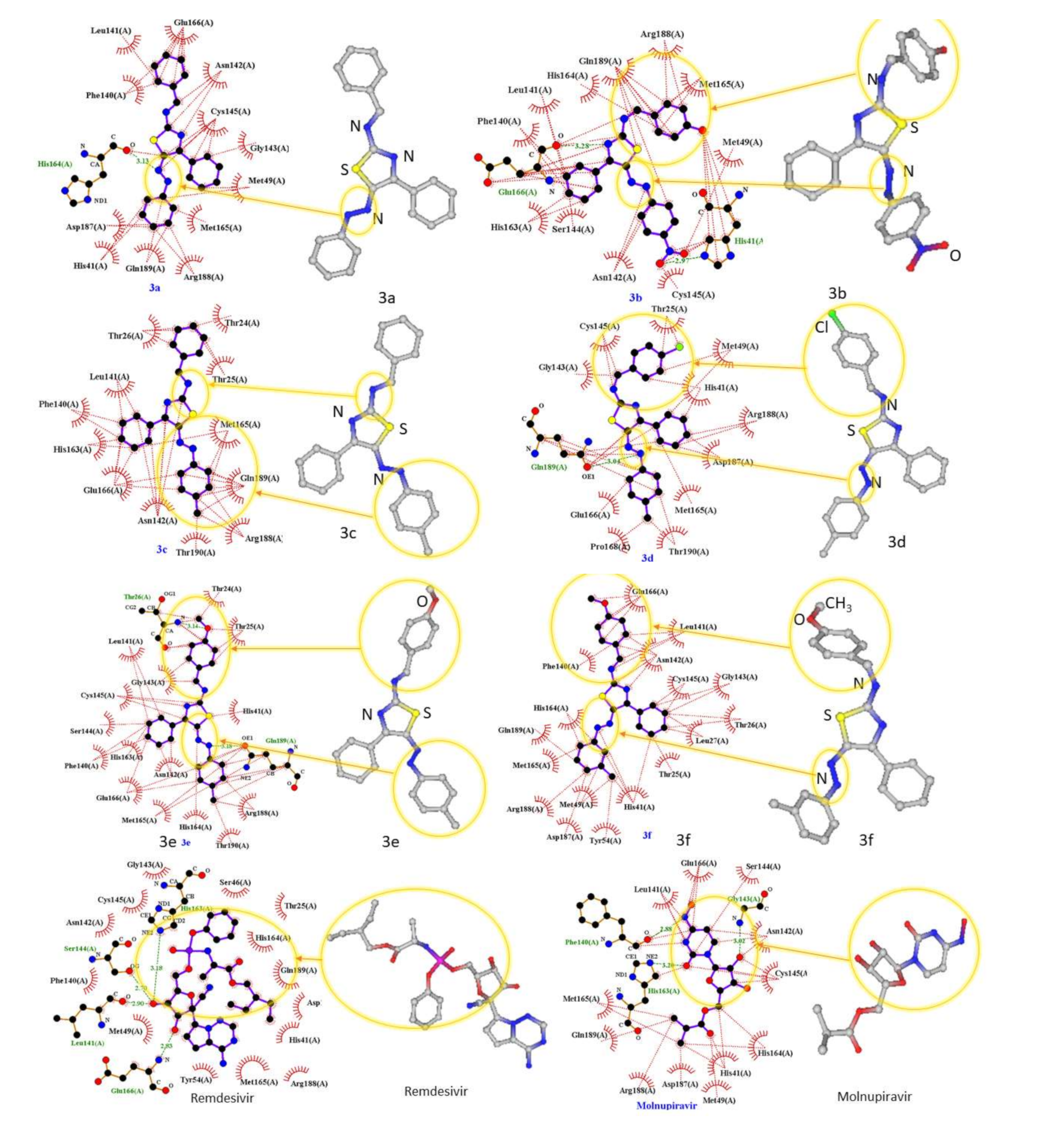
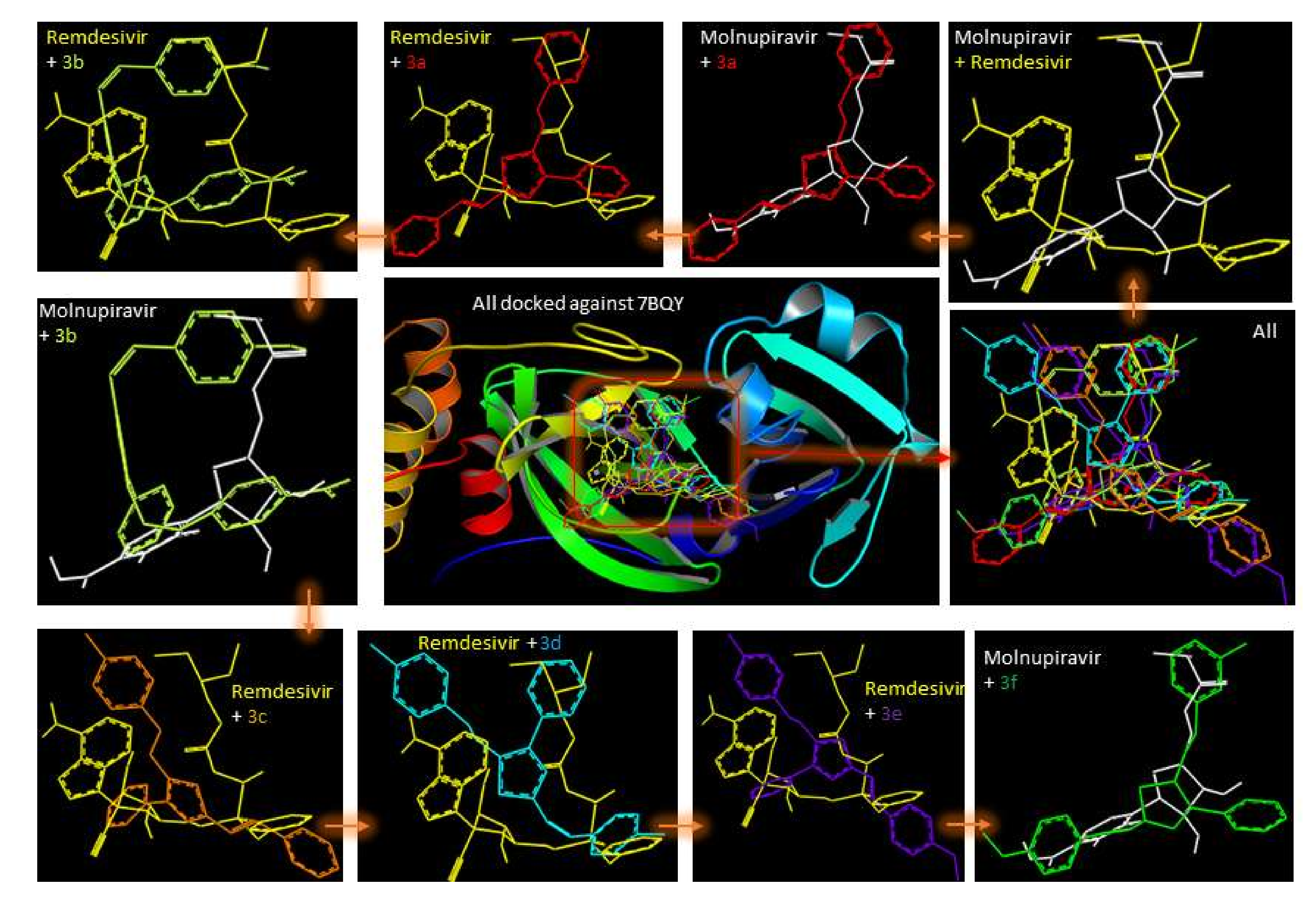
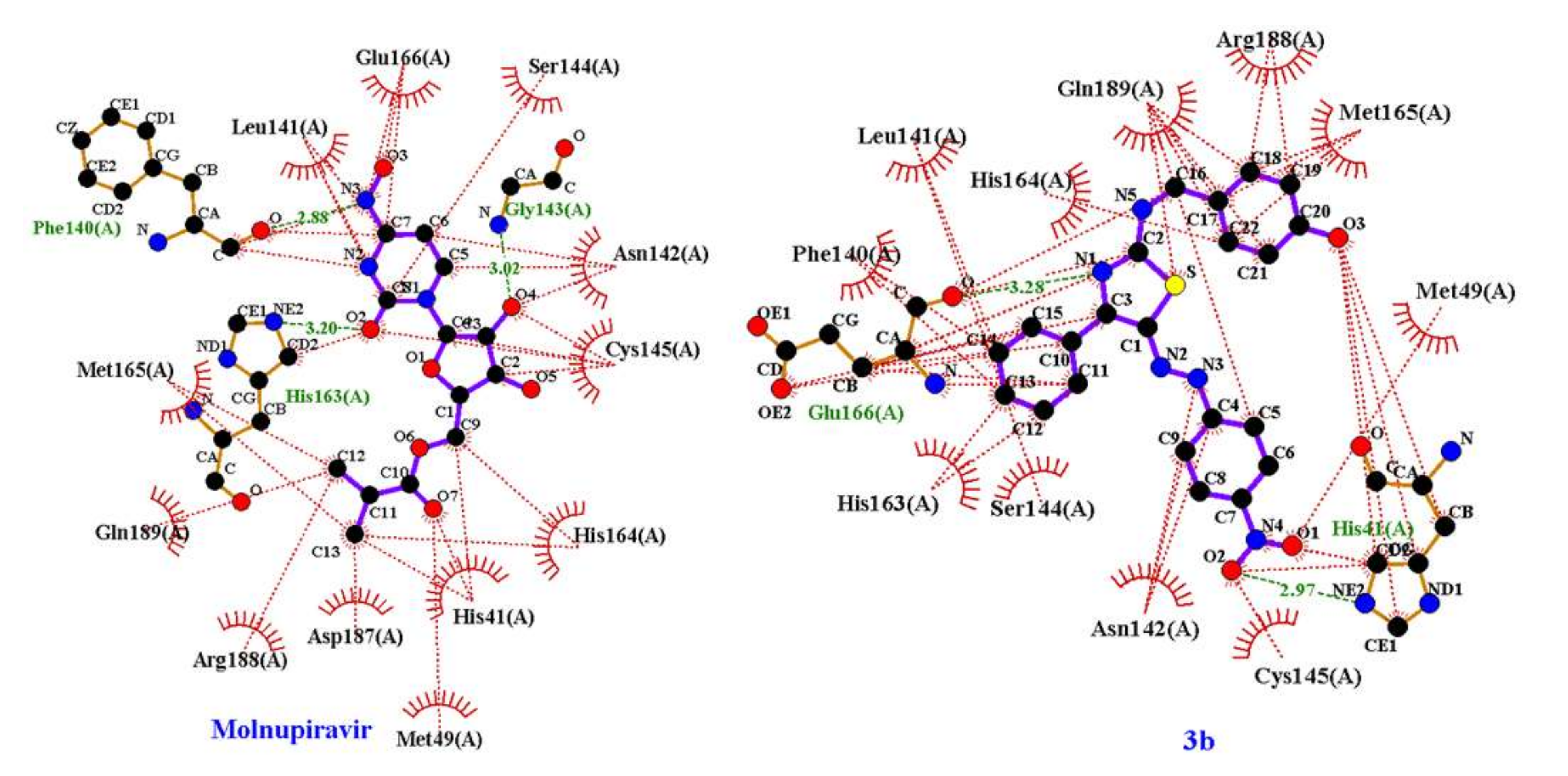
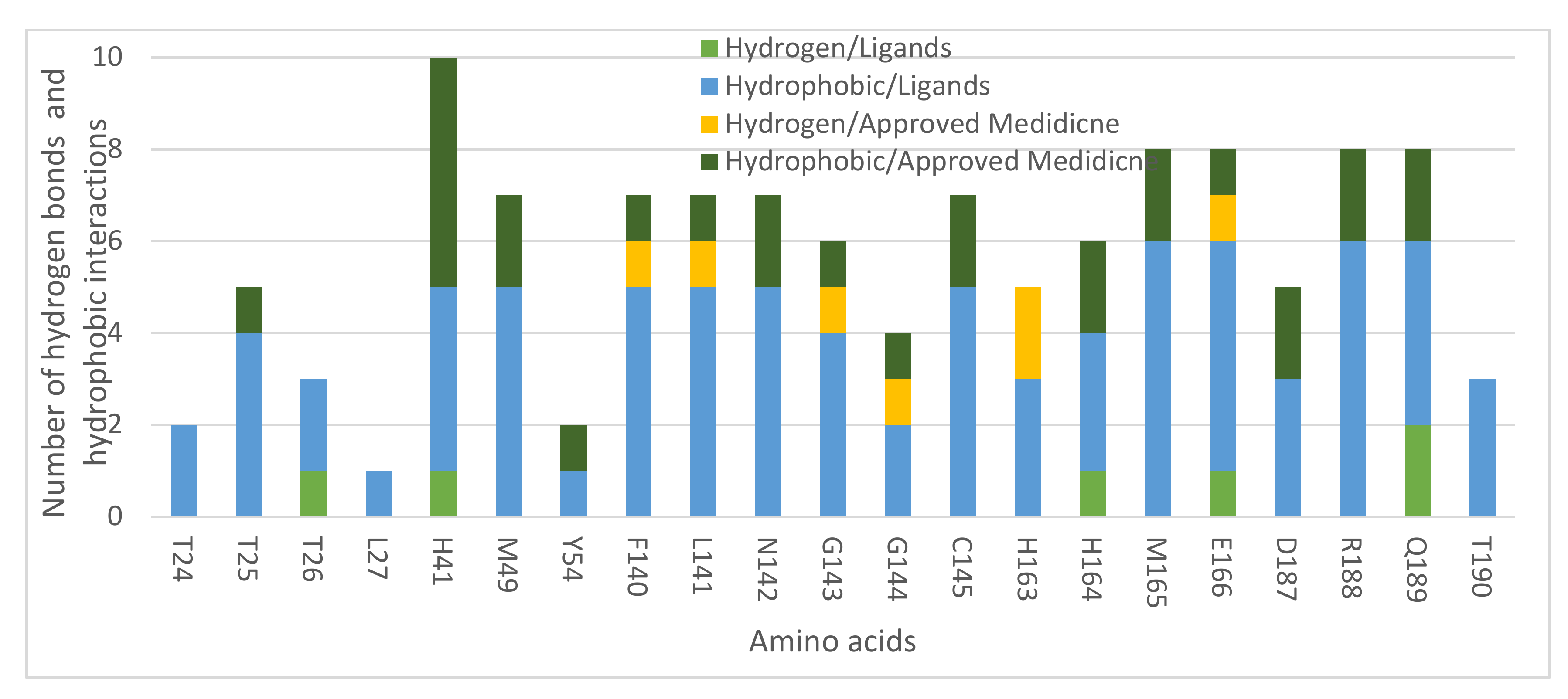
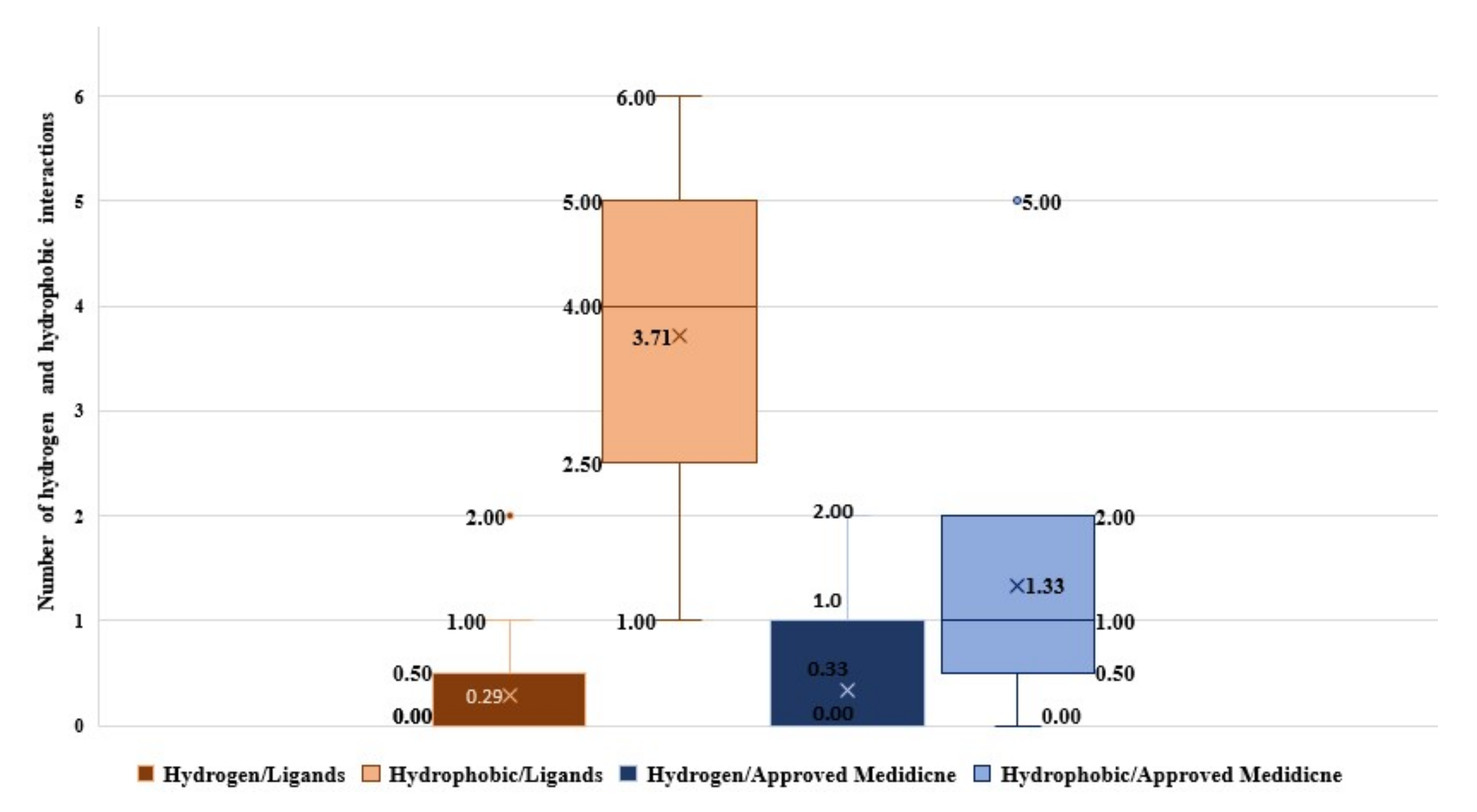
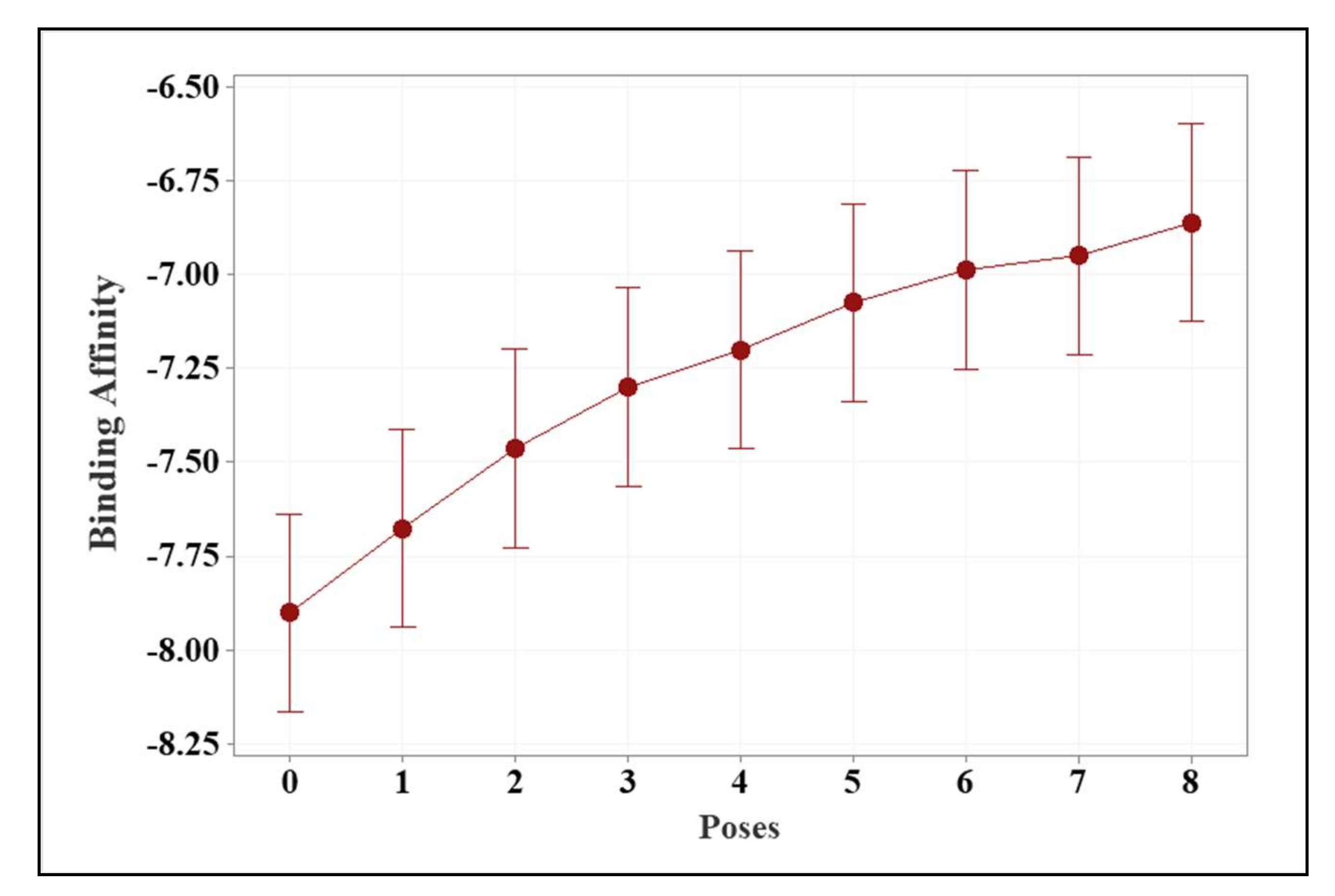
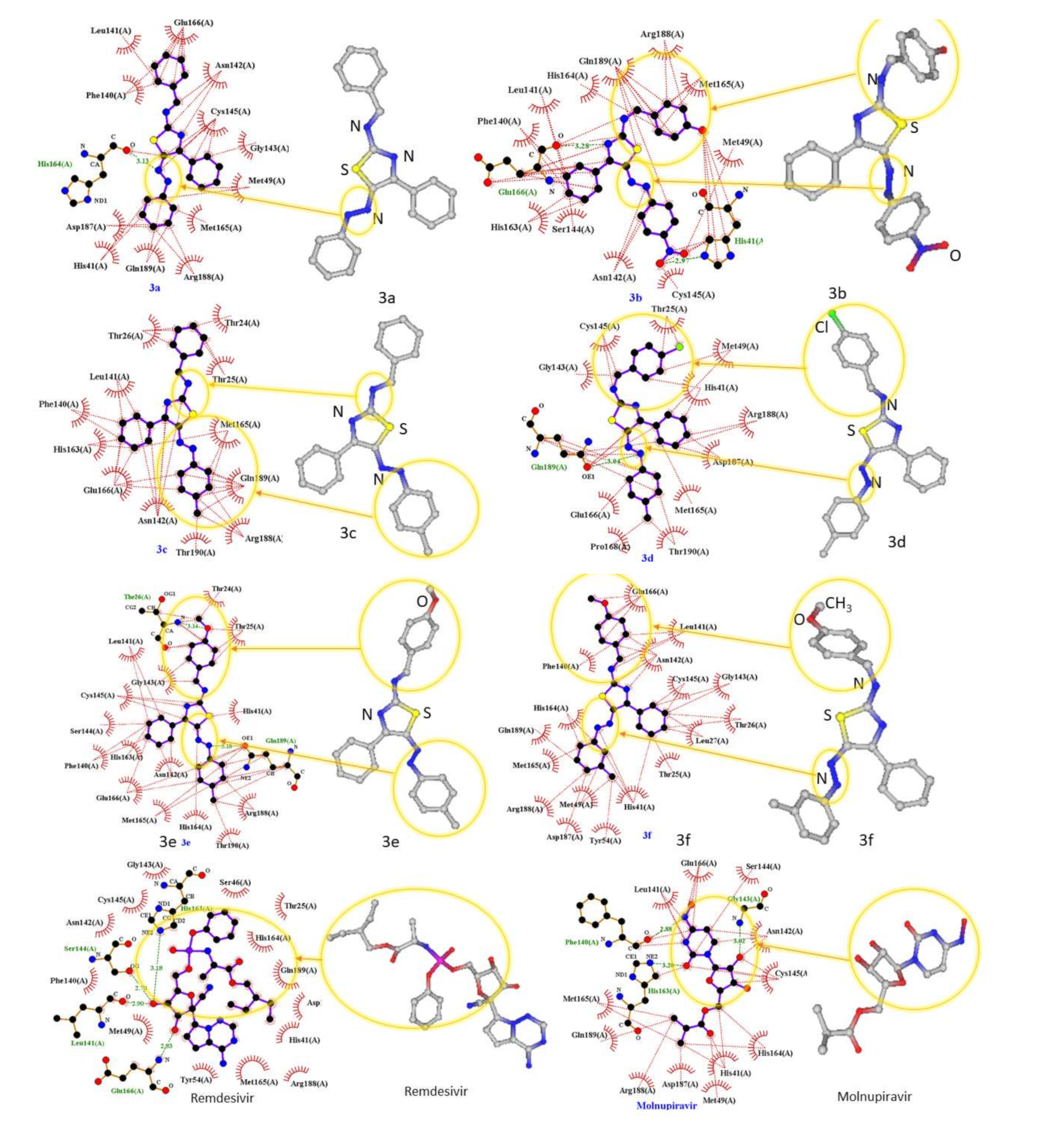
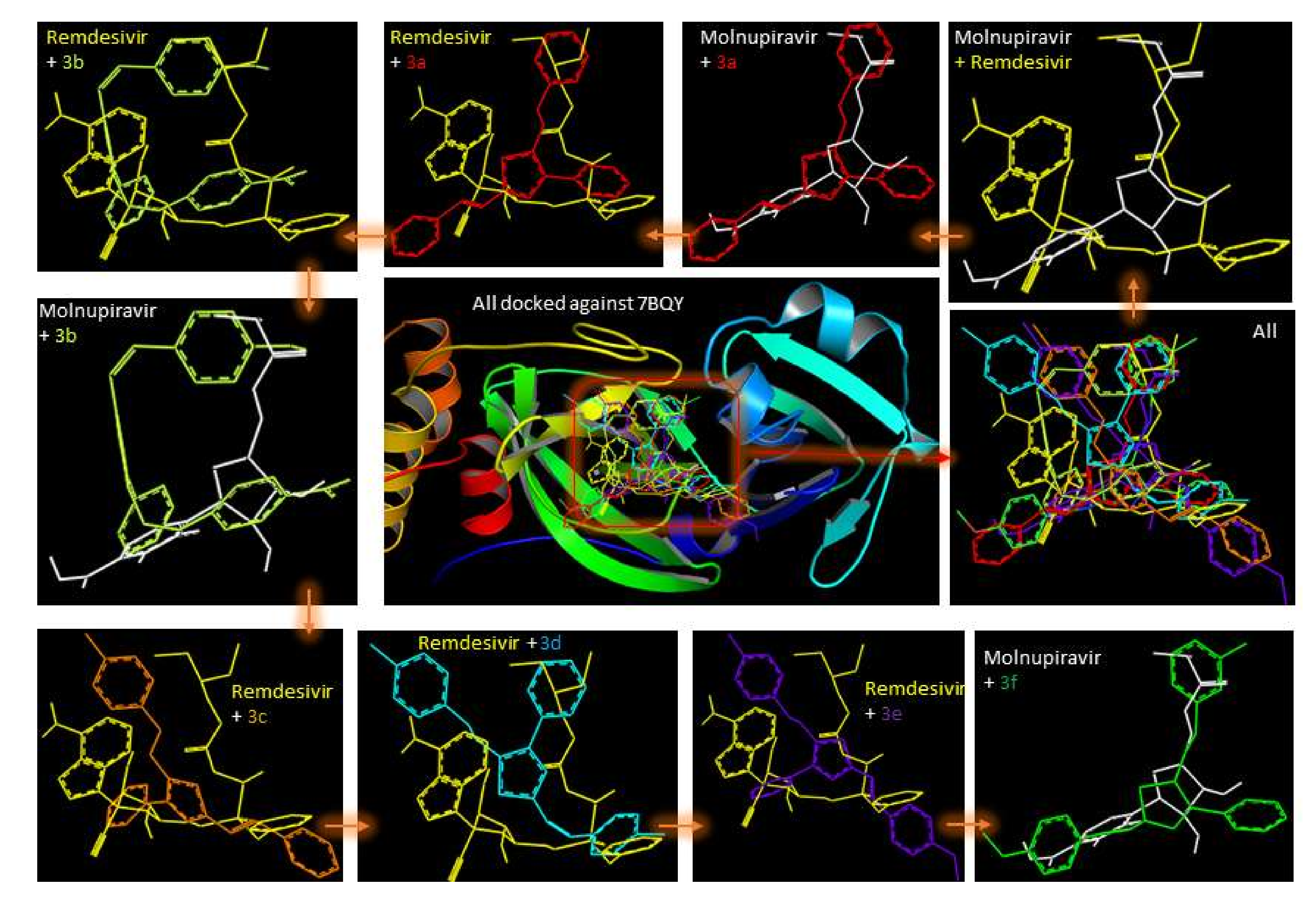
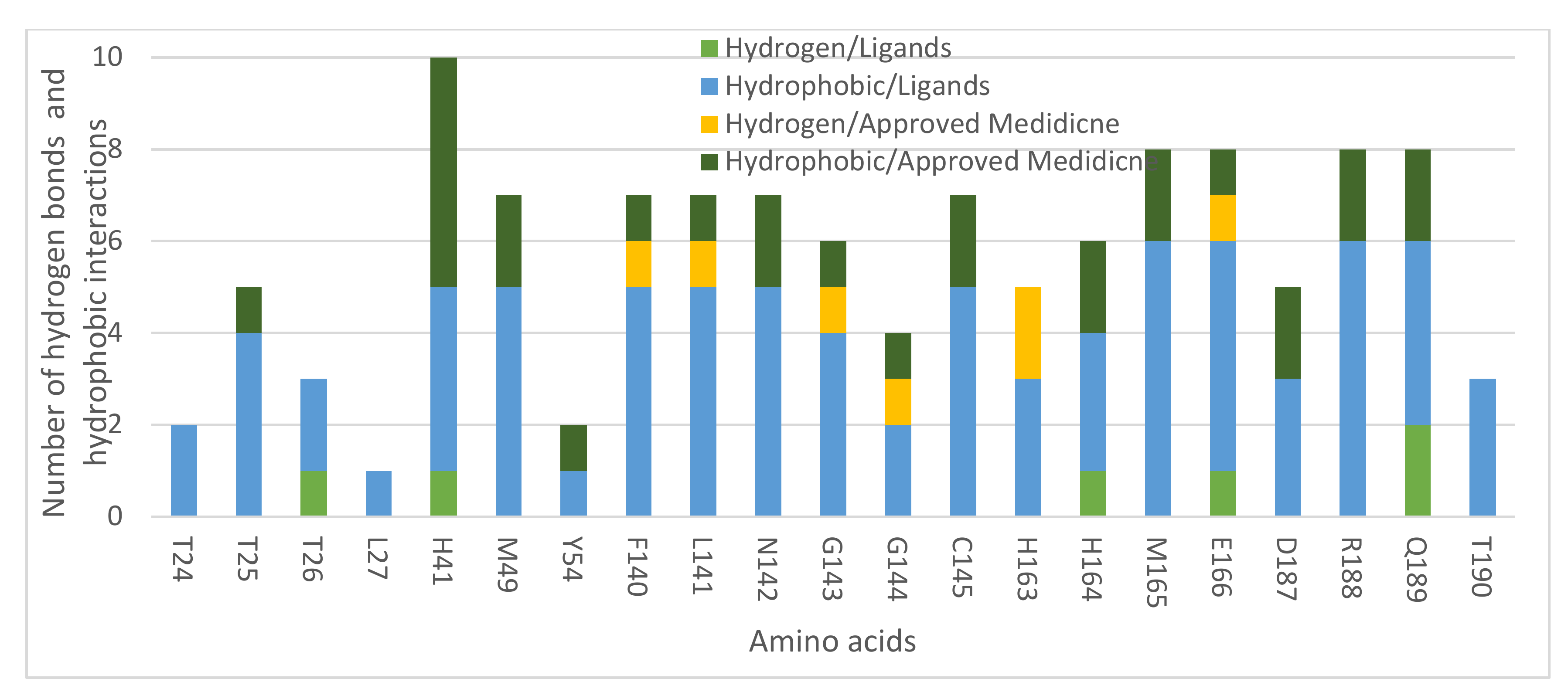
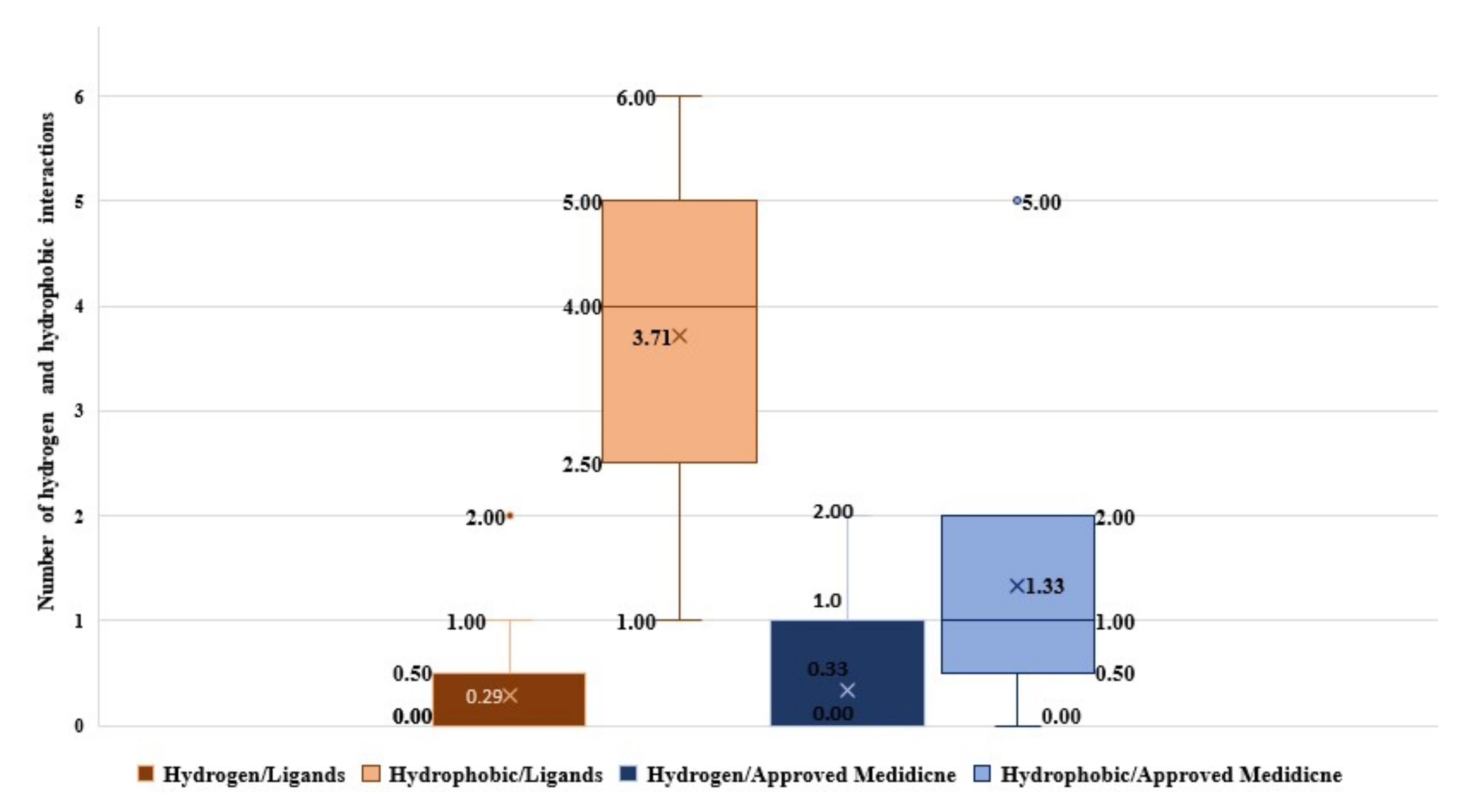
2.1. Molecular Modeling against 7BQY
2.2. In Silico Toxicity Prediction for Compounds 3a–f
2.3. Drug Likeness Evaluation
3. Materials and Methods
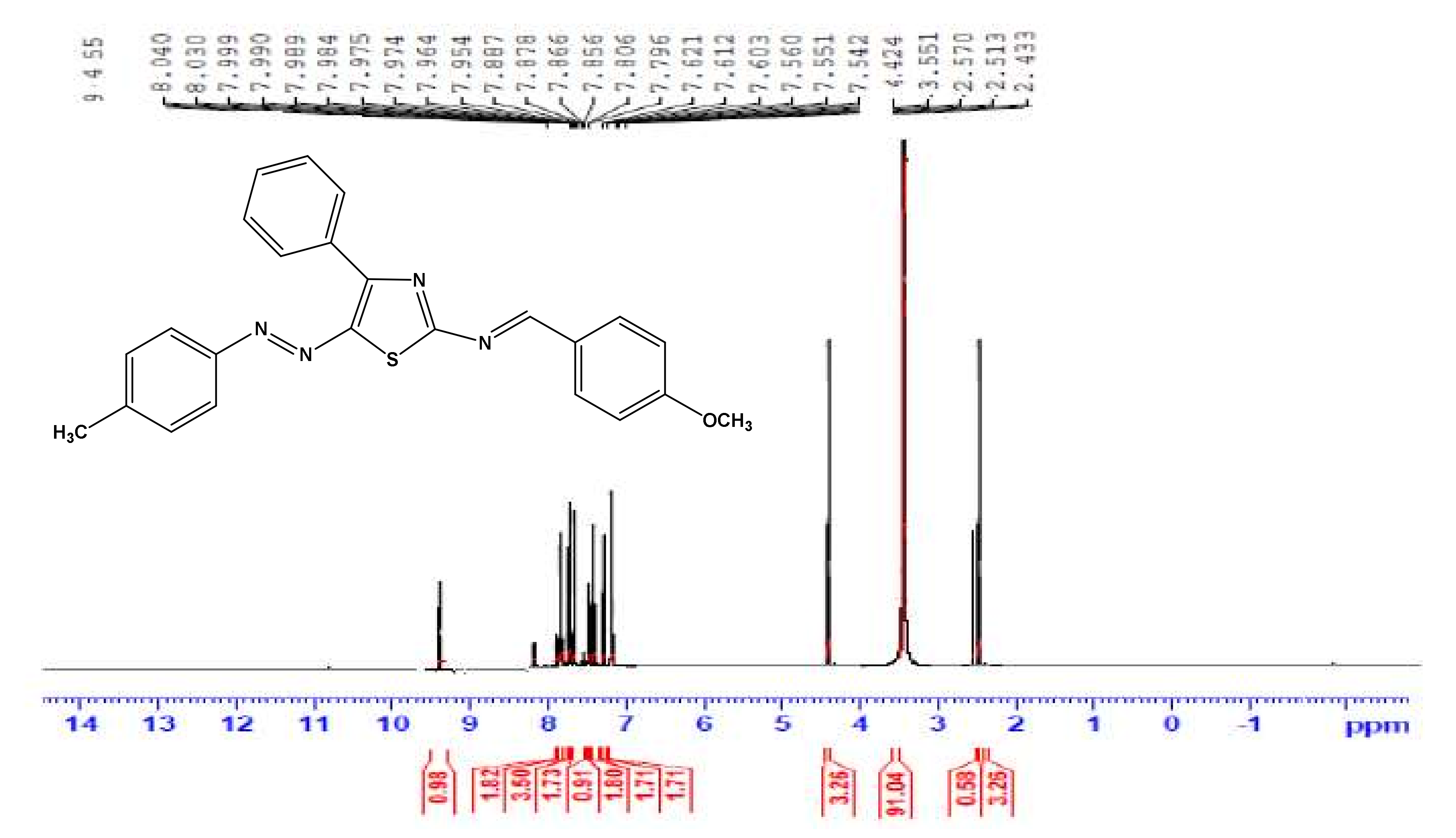
3.1. Instruments
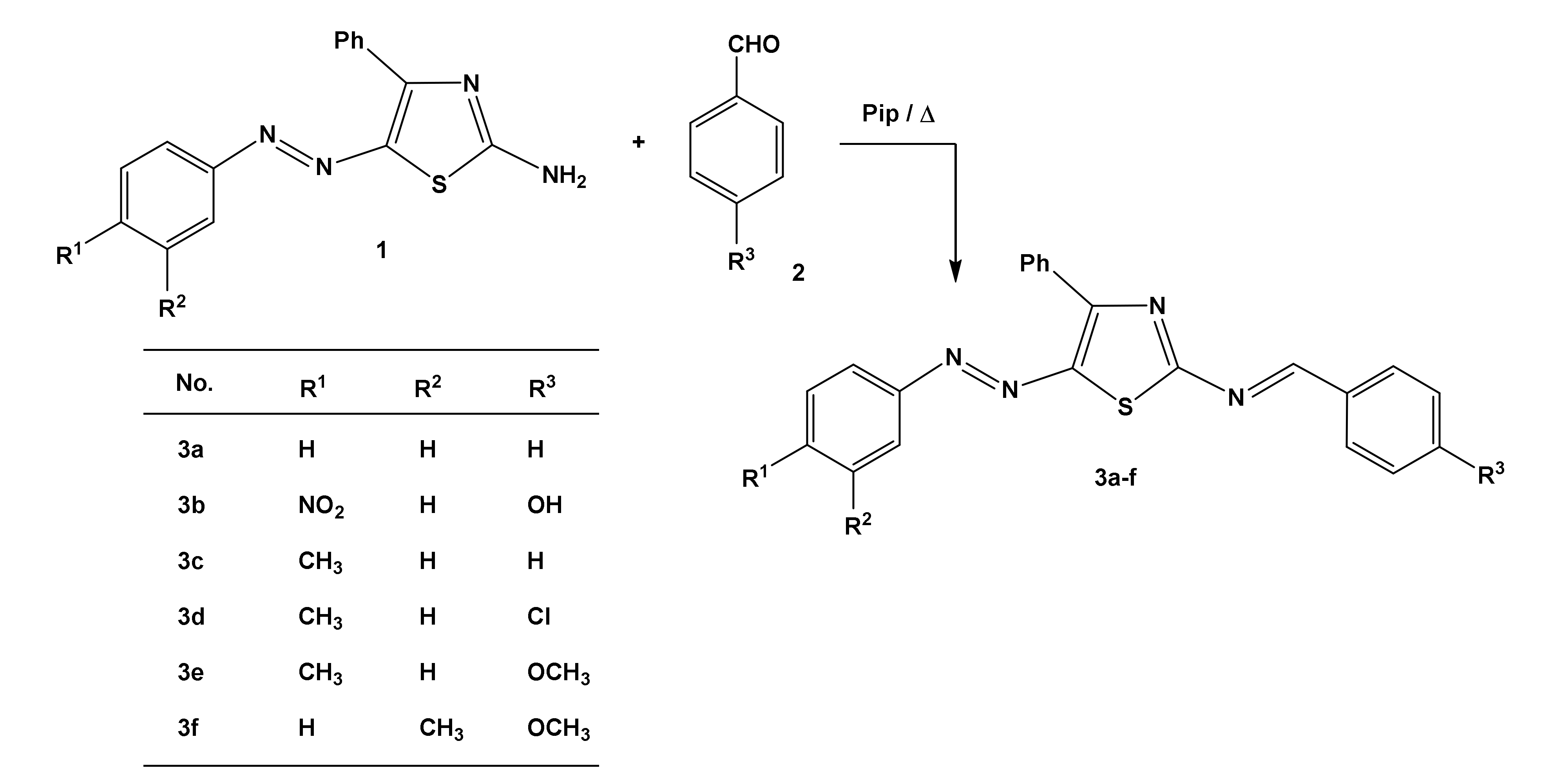
3.2. Synthesis of 2-Amino-4-phenyl-5-arylazothiazoles
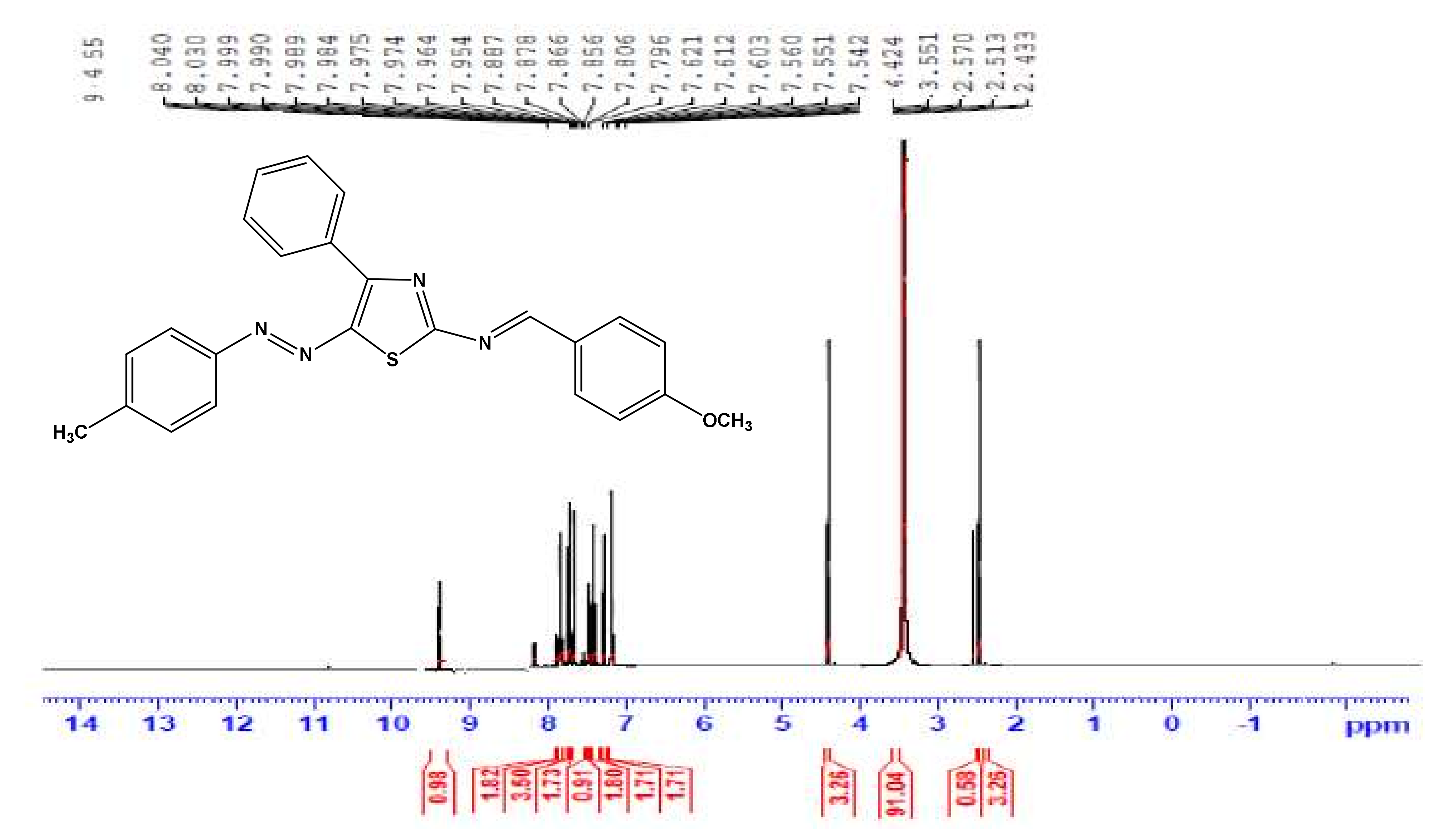
3.3. General Procedure for the Synthesis of Compounds 3a–f
3.4. Docking In Silico Studies
3.5. Statistical Analysis
3.6. In Silico Toxicity Prediction
3.7. SwissADME Drug-Likeness Properties Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Borbone, N.; Piccialli, G.; Roviello, G.N.; Oliviero, G. Nucleoside Analogs and Nucleoside Precursors as Drugs in the Fight against SARS-CoV-2 and Other Coronaviruses. Molecules 2021, 26, 986. [Google Scholar] [CrossRef]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Manickam, K.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharmacol. Rep. 2020, 72, 1479–1508. [Google Scholar] [CrossRef]
- Gao, Z.; Xu, Y.; Sun, C.; Wang, X.; Guo, Y.; Qiu, S.; Ma, K. A systematic review of asymptomatic infections with COVID-19. J. Microbiol. Immunol. Infect. 2021, 54, 12–16. [Google Scholar] [CrossRef]
- Yüce, M.; Filiztekin, E.; Özkaya, K.G. COVID-19 diagnosis—A review of current methods. Biosens. Bioelectron. 2021, 172, 112752. [Google Scholar] [CrossRef]
- Li, Y.; Tenchov, R.; Smoot, J.; Liu, C.; Watkins, S.; Zhou, Q. A Comprehensive Review of the Global Efforts on COVID-19 Vaccine Development. ACS Cent. Sci. 2021, 7, 512–533. [Google Scholar] [CrossRef]
- Noor, R. A Review on the Effectivity of the Current COVID-19 Drugs and Vaccines: Are They Really Working Against the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants? Curr. Clin. Microbiol. Rep. 2021, 8, 186–193. [Google Scholar] [CrossRef]
- Costanzo, M.; De Giglio, M.A.R.; Roviello, G. Anti-Coronavirus Vaccines: Past Investigations on SARS-CoV-1 and MERS-CoV, the Approved Vaccines from BioNTech/Pfizer, Moderna, Oxford/AstraZeneca and others under Development Against SARS-CoV-2 Infection. Curr. Med. Chem. 2021, in press. [Google Scholar] [CrossRef]
- Sultana, J.; Mazzaglia, G.; Luxi, N.; Cancellieri, A.; Capuano, A.; Ferrajolo, C.; de-Waure, C.; Ferlazzo, G.; Trifiro, G. Potential effects of vaccinations on the prevention of COVID-19: Rationale, clinical evidence, risks, and public health considerations. Exp. Rev. Vacc. 2020, 19, 919–936. [Google Scholar] [CrossRef]
- Germain, R.N.; Meier-Schellersheim, M.; Nita-Lazar, A.; Fraser, I.D.C. Systems biology in immunology: A computational modeling perspective. Annu. Rev. Immunol. 2011, 29, 527–585. [Google Scholar] [CrossRef] [Green Version]
- Roviello, G.; Roviello, V.; Autiero, I.; Saviano, M. Solid phase synthesis of TyrT, a thymine–tyrosine conjugate with poly(A) RNA-binding ability. RCS Adv. 2016, 6, 27607–27613. [Google Scholar] [CrossRef] [Green Version]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef]
- Ali, S.H.; Sayed, A.R. Review of the synthesis and biological activity of thiazoles. Synth. Commun. 2021, 51, 670–700. [Google Scholar] [CrossRef]
- Altamimi, M.A.; Hussain, A.; Alshehri, S.; Imam, S.S.; Alnami, A.; Bari, A. Novel hemocompatible imine compounds as alternatives for antimicrobial therapy in pharmaceutical application. Processes 2020, 8, 1476. [Google Scholar] [CrossRef]
- Bashiri, M.; Jarrahpour, A.; Rastegari, B.; Iraji, A.; Irajie, C.; Amirghofran, Z.; Malek-Hosseini, S.; Motamedifar, M.; Haddadi, M.; Zomorodian, K.; et al. Synthesis and evaluation of biological activities of tripodal imines and β-lactams attached to the 1,3,5-triazine nucleus. Monatsh. Chem. 2020, 151, 821–835. [Google Scholar] [CrossRef]
- Da Silva, E.T.; Araújo, A.S.; Moraes, A.M.; de Souza, L.A.; Lourenço, M.C.S.; de Souza, M.V.N.; Wardell, J.L.; Wardell, S.M.S.V. Synthesis and Biological Activities of Camphor Hydrazone and Imine Derivatives. Sci. Pharm. 2016, 84, 467. [Google Scholar] [CrossRef] [Green Version]
- Sakthinathan, S.P.; Suresh, R.; Kamalakkannan, D.; Mala, V.; Sathiyamoorthi, K.; Vanangamudi, G.; Thirunarayanan, G. Microwave assisted synthesis, spectral correlation and antimicrobial Evaluation of some aryl imines. J. Chil. Chem. Soc. 2018, 63, 3918–3923. [Google Scholar]
- Khan, M.S.; Siddiqui, S.P.; Tarannum, N. A systematic review on the synthesis and biological activity of hydrazide derivatives. Hygeia. J. Drugs Med. 2017, 9, 61–79. [Google Scholar] [CrossRef]
- Jeelani, A.; Muthu, S.; Narayana, B. Molecular structure determination, Bioactivity score, Spectroscopic and Quantum computational studies on (E)-N′-(4-Chlorobenzylidene)-2-(napthalen-2-yloxy) acetohydrazide. J. Mol. Struct. 2021, 1241, 130558. [Google Scholar] [CrossRef]
- Alsaygh, A.; Al-Humaidi, J.; Al-Najjar, I. Synthesis of Some New Pyridine-2-yl-Benzylidene-Imines. Int. J. Org. Chem. 2014, 4, 116–121. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, R.; Patrinos, H.A. Learning loss during COVID-19: An early systematic review. Prospects 2021, 1–9. [Google Scholar] [CrossRef]
- Marin, B.G.; Aghagoli, G.; Lavine, K.; Yang, L.; Siff, E.J.; Chiang, S.S.; Salazar-Mather, T.P.; Dumenco, L.; Savaria, M.C.; Aung, S.N.; et al. Predictors of COVID-19 severity: A literature review. Rev. Med. Virol. 2021, 31, 1–10. [Google Scholar] [CrossRef]
- Zhang, X.; Tan, Y.; Ling, Y.; Lu, G.; Liu, F.; Yi, Z.; Jia, X.; Wu, M.; Shi, B.; Xu, S.; et al. Viral and host factors related to the clinical outcome of COVID-19. Nature 2020, 583, 437–440. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Vallianou, N.G.; Tsilingiris, D.; Christodoulatos, G.S.; Karampela, I.; Dalamaga, M. Anti-viral treatment for SARS-CoV-2 infection: A race against time amidst the ongoing pandemic. Metab. Open 2021, 10, 100096. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Thai, N.Q.; Truong, D.T.; Li, M.S. Remdesivir Strongly Binds to Both RNA-Dependent RNA Polymerase and Main Protease of SARS-CoV-2: Evidence from Molecular Simulations. J. Phys. Chem. B 2020, 124, 11337–11348. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Karplus, M. Protein Dynamics in Solution and in a Crystalline Environment: A Molecular Dynamics Study. Biochemistry 1982, 21, 2259–2274. [Google Scholar] [CrossRef]
- Musumeci, D.; Ullah, S.; Ikram, A.; Roviello, G.N. Novel insights on nucleopeptide binding: A spectroscopic and In Silico investigation on the interaction of a thymine-bearing tetrapeptide with a homoadenine DNA. J. Mol. Liq. 2021, 117975. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Wishart, D.S.; Sykes, B.D.; Richards, F.M.; Pastone, A.; Hayes, D.M. Hyper Chem Version 8.pdf PYRX, USA 2001. Available online: http://www.swissadme.ch/ (accessed on 22 December 2021).
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in-silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. PyMOL Reference Guide; Delano Scientific: San Carlos, CA, USA, 2004; pp. 1–68. [Google Scholar]
- Rubin, D.; Chan-Tack, K.; Farley, J.; Sherwat, A. FDA Approval of Remdesivir—A Step in the Right Direction. N. Engl. J. Med. 2020, 383, 2598–2600. [Google Scholar] [CrossRef]
- Shah, B.; Modi, P.; Sagar, S.R. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020, 252, 117652. [Google Scholar] [CrossRef]
- Alsafi, M.A.; Hughes, D.L.; Said, M.A. First COVID-19 molecular docking with a chalcone-based compound: Synthesis, single-crystal structure and Hirshfeld surface analysis study. Acta Crystallogr. Sect. C Struct. Chem. 2020, 76, 1043–1050. [Google Scholar] [CrossRef]
- Imran, M.; Alshrari, A.S.; Asdaq, S.M.B.; Abida. Trends in the development of remdesivir based inventions against COVID-19 and other disorders: A patent review. J. Infect. Public Health 2021, 14, 1075–1086. [Google Scholar] [CrossRef]
- Pardo, J.; Shukla, A.M.; Chamarthi, G.; Gupte, A. The journey of remdesivir: From Ebola to COVID-19. Drugs Context 2020, 9, 4–14. [Google Scholar] [CrossRef]
- Yoon, J.J.; Toots, M.; Lee, S.; Lee, M.E.; Ludeke, B.; Luczo, J.M.; Ganti, K.; Cox, R.M.; Sticher, Z.M.; Edpuganti, V.; et al. Orally Efficacious Broad-Spectrum Ribonucleoside Analog Inhibitor of Influenza and Respiratory Syncytial Viruses. Antimicrob. Agents Chemother. 2018, 62, e00766-18. [Google Scholar] [CrossRef] [Green Version]
- Fischer, W.; Eron, J.J.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.J.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. Molnupiravir, an Oral Antiviral Treatment for COVID-19. Preprint. medRxiv 2021. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Parlak, C.; Alver, Ö.; Ouma, C.N.M.; Rhyman, L.; Ramasami, P. Interaction between favipiravir and hydroxychloroquine and their combined drug assessment: In silico investigations. Chem. Pap. 2021, in press. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Ertl, P.; Lewis, R. The graphical representation of ADME-related molecule properties for medicinal chemists. Drug Discov. Today 2011, 16, 65–72. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Yen, M.S. Synthesis and solvatochromism of heterocyclic bichromophoric dyes derived from 2-aminothiazole. Pigment. Resin Tech. 2012, 41, 216–222. [Google Scholar] [CrossRef]
- Derkowska-Zielinska, B.; Skowronski, L.; Biitseva, A.; Grabowski, A.; Naparty, M.K.; Smokal, V.; Kysil, A.; Krupka, O. Optical characterization of heterocyclic azo dyes containing polymers thin films. Appl. Surf. Sci. 2017, 421, 361–366. [Google Scholar] [CrossRef]
- Prajapati, A.K.; Modi, V.P. Synthesis and biological activity of n-{5-(4-methylphenyl) diazenyl-4-phenyl-1, 3-thiazol-2-yl}benzamide derivatives. Quim. Nova 2011, 34, 771–774. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimisation, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. Ligplot: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
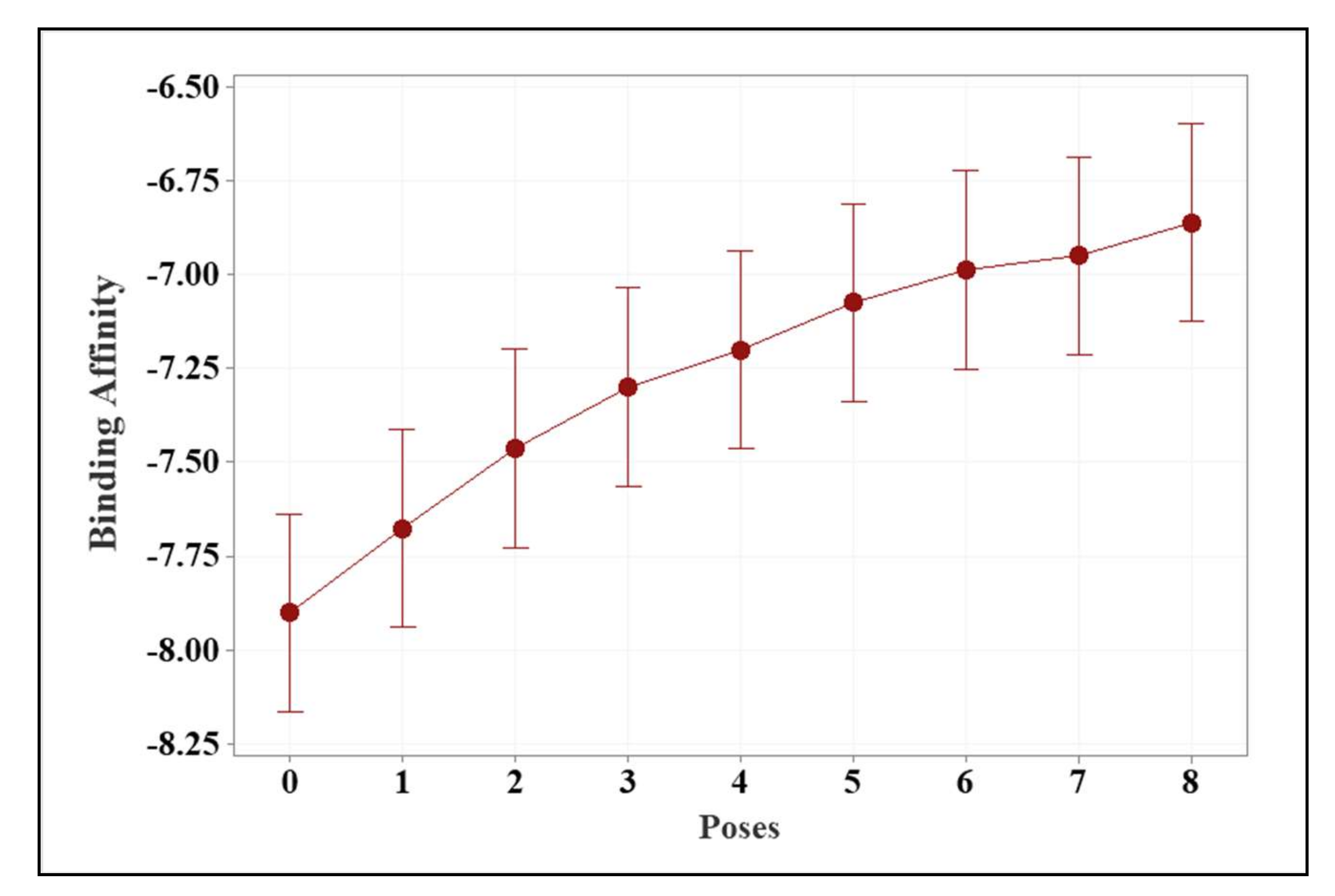
| Source of Variance | DF | F-Value | p-Value |
|---|---|---|---|
| Ligands and approved drugs | 7 | 7.42 | 0.00 |
| Number of poses | 8 | 7.12 | 0.00 |
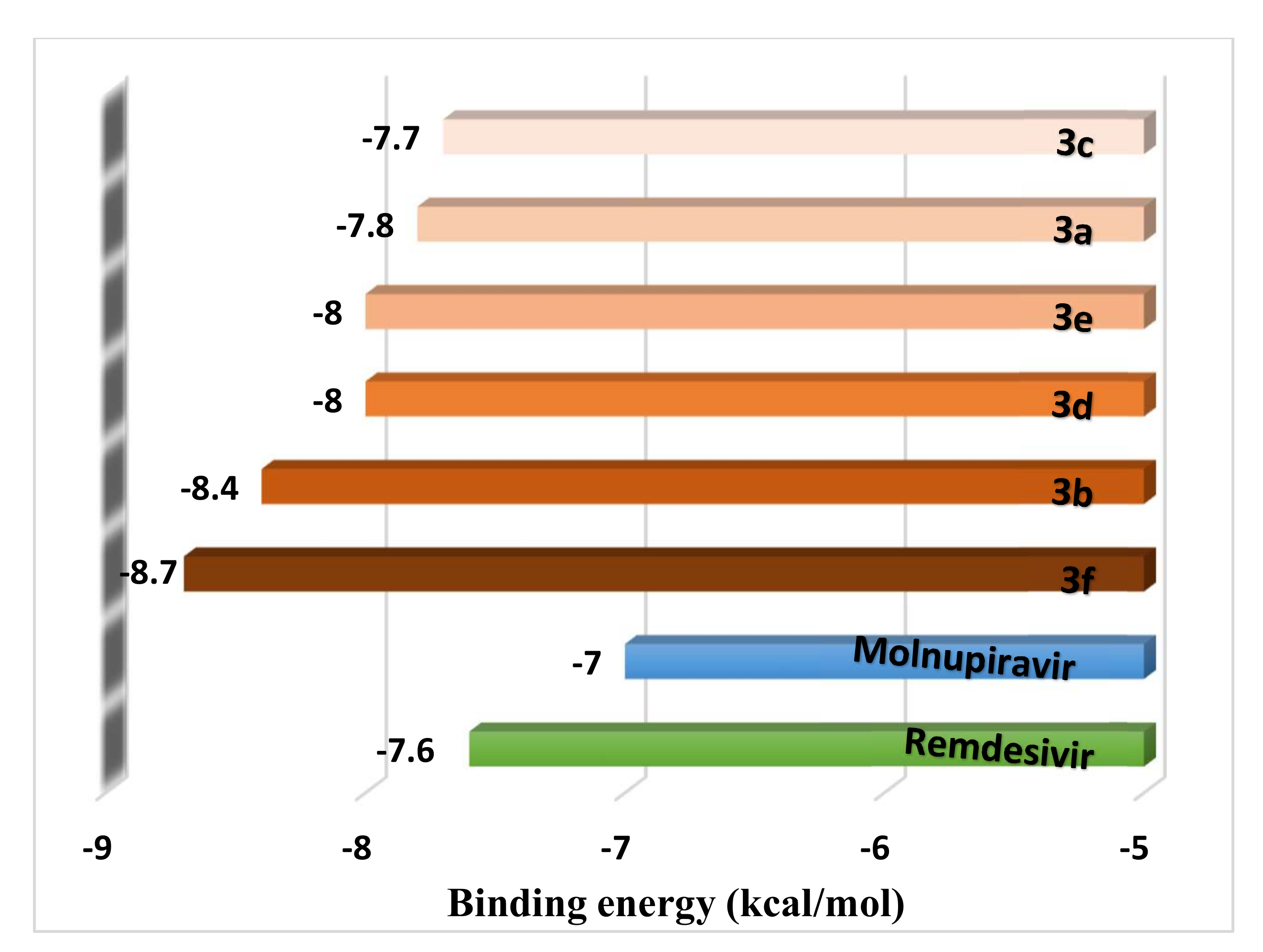
| Ligand and Approved Medicines | Average Binding Energy (kcal/mol) | |
|---|---|---|
| Ligands | 3f | −7.81 ± 0.48 a |
| 3d | −7.52 ± 0.21 a,b | |
| 3a | −7.41 ± 0.29 a,b,c | |
| 3b | −7.36 ± 0.50 a,b,c | |
| 3e | −7.25 ± 0.46 a,b,c,d | |
| 3c | −7.13 ± 0.46 b,c,d | |
| Approved medicine | Remdesivir | −6.93 ± 0.33 c,d |
| Molnupiravir | −6.71 ± 0.23 d | |
| Ligand | Oral Toxicity Prediction Results | |||
|---|---|---|---|---|
| Predicted LD50 (mg/kg) | Predicted Toxicity Class | Average Similarity (%) | Prediction Accuracy (%) | |
| 3a | 300 | 3 | 44.61 | 54.26 |
| 3b | 300 | 3 | 42.28 | 54.26 |
| 3c | 300 | 3 | 44.32 | 54.26 |
| 3d | 300 | 3 | 45.74 | 54.26 |
| 3e | 1000 | 4 | 43.73 | 54.26 |
| 3f | 1000 | 4 | 42.98 | 54.26 |
| Molnupiravir | 1000 | 4 | 40.93 | 54.26 |
| Remdesivir | 826 | 4 | 79.41 | 69.26 |
| Ligands | Hepatotoxicity | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Activity | Probability | Activity | Probability | Activity | Probability | Activity | Probability | Activity | Probability | |
| 3a | Yes | 0.58 | Yes | 0.59 | No | 0.99 | Yes | 0.57 | No | 0.80 |
| 3b | Yes | 0.55 | Yes | 0.77 | No | 0.99 | Yes | 0.75 | No | 0.76 |
| 3c | Yes | 0.55 | Yes | 0.60 | No | 0.99 | Yes | 0.58 | No | 0.82 |
| 3d | Yes | 0.57 | No | 0.59 | No | 0.99 | No | 0.55 | No | 0.82 |
| 3e | Yes | 0.58 | Yes | 0.57 | No | 0.99 | Yes | 0.63 | No | 0.73 |
| 3f | Yes | 0.58 | Yes | 0.57 | No | 0.99 | Yes | 0.63 | No | 0.73 |
| Molnupiravir | Yes | 0.56 | No | 0.50 | No | 0.96 | No | 0.50 | No | 0.74 |
| Remdesivir | No | 0.56 | No | 0.55 | No | 0.90 | No | 0.62 | No | 0.55 |
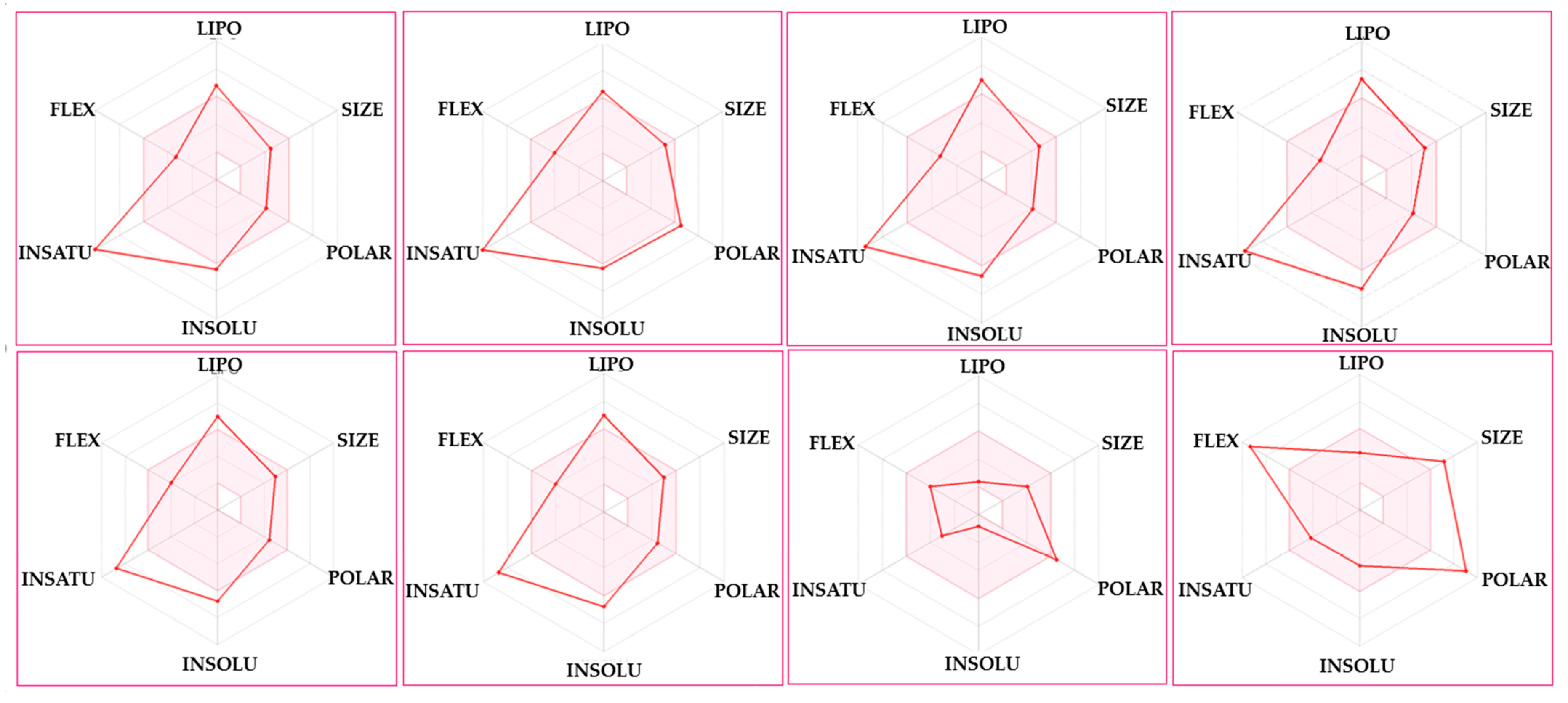
| Physicochemical | 3a | 3b | 3c | 3d | 3e | 3f | Molnupiravir | Remdesivir |
|---|---|---|---|---|---|---|---|---|
| Molecular weight (g/mol) | 368.45 | 429.45 | 382.48 | 416.93 | 412.51 | 412.51 | 329.31 | 602.58 |
| Heavy atom | 27 | 31 | 28 | 29 | 30 | 30 | 23 | 42 |
| Arom. heavy atom | 23 | 23 | 23 | 23 | 23 | 23 | 6 | 15 |
| Fraction Csp3 | 0.00 | 0.00 | 0.04 | 0.04 | 0.08 | 0.08 | 0.62 | 0.48 |
| Rotatable bond | 5 | 6 | 5 | 5 | 6 | 6 | 6 | 14 |
| H-bond acceptor | 4 | 7 | 4 | 4 | 5 | 5 | 8 | 12 |
| H-bond donor | 0 | 1 | 0 | 0 | 0 | 0 | 4 | 4 |
| Molar refractivity | 111.86 | 122.71 | 116.83 | 121.84 | 123.32 | 123.32 | 76.02 | 150.43 |
| Polar surface area (Å2) | 78.21 | 144.26 | 78.21 | 78.21 | 87.44 | 87.44 | 143.14 | 213.36 |
| Lipophilicity | ||||||||
| MLOGP | 4.05 | 2.57 | 4.27 | 4.74 | 3.92 | 3.92 | −1.15 | 0.18 |
| WLOGP | 6.98 | 6.59 | 7.28 | 7.94 | 7.29 | 7.29 | −1.65 | 2.21 |
| XLOGP3 | 6.36 | 5.83 | 6.72 | 7.35 | 6.69 | 6.69 | −1.34 | 1.91 |
| Drug-likeness evaluation | ||||||||
| Lipinski | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 2 |
| Pharmacokinetics | 3a | 3b | 3c | 3d | 3e | 3f | Molnupiravir | Remdesivir | |
|---|---|---|---|---|---|---|---|---|---|
| Gastrointestinal (GI) absorption | Low | Low | Low | Low | Low | Low | Low | Low | |
| Blood–brain barrier (BBB) permeant | No | No | No | No | No | No | No | No | |
| P-glycoprotein (P-gp) substrate | No | No | No | Yes | No | No | No | Yes | |
| Inhibitor | CYP1A2 | Yes | No | No | No | No | No | No | No |
| CYP2C19 | Yes | No | Yes | No | No | No | No | No | |
| CYP2C9 | Yes | Yes | Yes | No | Yes | Yes | Yes | No | |
| CYP2D6 | No | No | No | No | No | No | No | No | |
| CYP3A4 | No | No | No | No | No | No | No | Yes | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu-Melha, S.; Edrees, M.M.; Said, M.A.; Riyadh, S.M.; Al-Kaff, N.S.; Gomha, S.M. Potential COVID-19 Drug Candidates Based on Diazinyl-Thiazol-Imine Moieties: Synthesis and Greener Pastures Biological Study. Molecules 2022, 27, 488. https://doi.org/10.3390/molecules27020488
Abu-Melha S, Edrees MM, Said MA, Riyadh SM, Al-Kaff NS, Gomha SM. Potential COVID-19 Drug Candidates Based on Diazinyl-Thiazol-Imine Moieties: Synthesis and Greener Pastures Biological Study. Molecules. 2022; 27(2):488. https://doi.org/10.3390/molecules27020488
Chicago/Turabian StyleAbu-Melha, Sraa, Mastoura Mohamed Edrees, Musa A. Said, Sayed M. Riyadh, Nadia S. Al-Kaff, and Sobhi M. Gomha. 2022. "Potential COVID-19 Drug Candidates Based on Diazinyl-Thiazol-Imine Moieties: Synthesis and Greener Pastures Biological Study" Molecules 27, no. 2: 488. https://doi.org/10.3390/molecules27020488
APA StyleAbu-Melha, S., Edrees, M. M., Said, M. A., Riyadh, S. M., Al-Kaff, N. S., & Gomha, S. M. (2022). Potential COVID-19 Drug Candidates Based on Diazinyl-Thiazol-Imine Moieties: Synthesis and Greener Pastures Biological Study. Molecules, 27(2), 488. https://doi.org/10.3390/molecules27020488