Abstract
In the context of our SAR study concerning 6BrCaQ analogues as C-terminal Hsp90 inhibitors, we designed and synthesized a novel series of 3-(heteroaryl)quinolin-2(1H), of types 3, 4, and 5, as a novel class of analogues. A Pd-catalyzed Liebeskind–Srogl cross-coupling was developed as a convenient approach for easy access to complex purine architectures. This series of analogues showed a promising biological effect against MDA-MB231 and PC-3 cancer cell lines. This study led to the identification of the best compounds, 3b (IC50 = 28 µM) and 4e, which induce a significant decrease of CDK-1 client protein and stabilize the levels of Hsp90 and Hsp70 without triggering the HSR response.
1. Introduction
The 90-kDa heat shock protein (Hsp90) has emerged recently as a promising therapeutic target for the treatment of cancer [1,2,3,4,5] and other diseases [6,7]. As a chaperone protein, Hsp90 is evolved in the conformational maturation, folding, stabilization, activation, and degradation of over 400 client proteins in healthy cells as well as in cancerous cells which are directly associated with all hallmarks of cancer [8,9,10,11]. This Hsp90 chaperone cycle depends on the ATPase activity. ATP binding to the N-terminal domain (NTD) and hydrolysis by Hsp90 drive a conformational cycle necessary for chaperone function [12,13,14]. The binding of ATP to each monomer shifts Hsp90 to a “closed” formation that can bind, fold, and activate client proteins [15,16]. Thus, inhibition of Hsp90 function results in the simultaneous interruption of many signal transduction pathways which are pivotal to tumor progression and survival.
Several structurally distinct Hsp90 inhibitors that target the ATP binding pocket are currently being evaluated for anticancer activity in numerous Phase II and several Phase III clinical trials. However, they are ineffective over time due to the compensatory mechanism involving the induction of a heat shock response. The expression of chaperones Hsp27, Hsp70, Hsp40, and Hsp90 increases [17,18,19], leading to undesirable chemoprotective effects [20,21,22]. Clinical resistance has been attributed to this chemoprotective effect, and dosage increases to overcome resistance are not a viable option due to toxicity. These results continue to motivate the pursuit of alternative strategies for modulating heat shock protein complexes [23,24,25].
An alternative molecular mechanism of inhibition is through binding to the C-terminal domain of Hsp90. The CTD has been implicated biochemically as the site of a possible second cryptic ATP-binding site on the protein. Its contribution to the overall regulation of chaperone function is not clear, but some small molecules that interact with the C-terminal domain, such as the antibiotic novobiocin [26] (Nvb, Figure 1) and coumermycin A1 (Cm A1), induce client protein degradation without heat shock response induction [27,28,29,30,31], giving new promise to Hsp90 inhibition for cancer treatments.
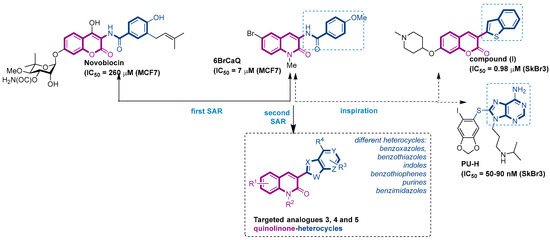
Figure 1.
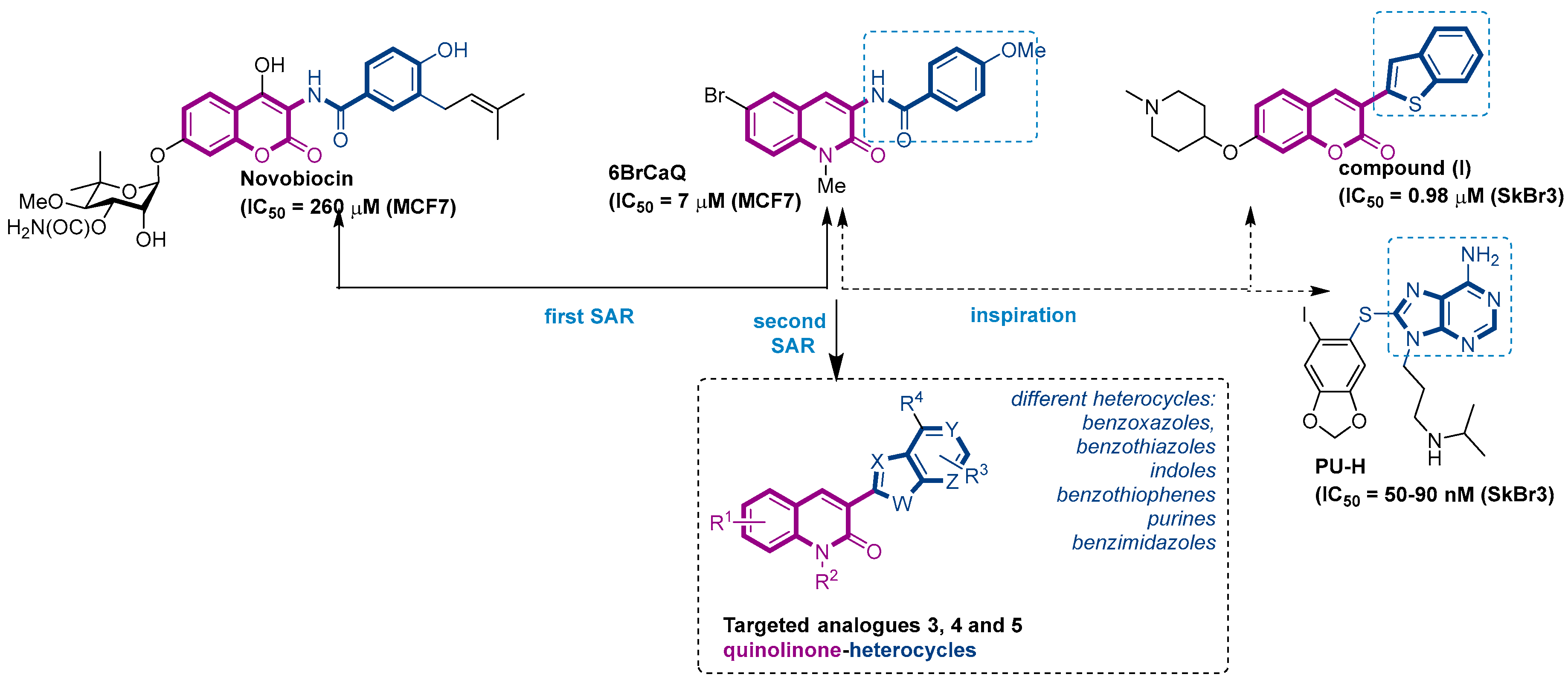
Structure of novobiocin and 6BrCaQ compounds and the approach for the design of targeted compounds 3, 4 and 5.
In this context, we previously reported a novel series of simplified 3 amido-quinolin-2-one analogues related to Nvb as a class of highly potent hsp90 inhibitors [32,33,34,35,36]. From the structure–activity relationship (SAR) studies, 6BrCaQ (Figure 1) [37,38] was identified as a very promising C-terminal Hsp90 inhibitor displaying an antiproliferative activity LC50 of 5–50 µM [39,40] against various cancer cell lines (MCF7, MDA MB231, Caco2, IGROV, ISHIKAWA, PC3, and HT29 cells). Further studies on its mode of action revealed that 6BrCaQ manifests downregulation of several Hsp90 client proteins (HER2, Raf-1 and cdk-4), induces a high apoptosis level in MCF-7 breast cancer cell line and PC3. In addition, encapsulated in liposomes, 6BrCaQ exerted an improved in vitro activity on breast cancer cells (MDA-MB-231) and displays an in vivo anti-tumor activity on an orthotopic breast cancer model in nude mice [40].
More recently, we demonstrated that conjugation of 6BrCaQ with the cationic head triphenylphosphonium (TPP) leads to the conjugate 6BrCaQ-C10-TPP for the targeting of the mitochondrial heat shock protein TRAP1. Hence, 6BrCaQ-C10-TPPdisplays an anti-proliferative activity with mean GI50 values at a nanomolar level in a diverse set of human cancer cells (GI50 = 0.008–0.30 µM) including MDA-MB-231, HT-29, HCT116, K562 and PC-3 cancer cell lines. This study showed that this compound 6BrCaQ-C10-TPP induces a significant mitochondrial membrane disruption and interferes with TRAP1 function in colon carcinoma cells without inducing the heat-shock response HSF1 [41].
On the other hand, Blagg and co-workers reported during their various SAR studies that the coumarin analogue (I) (Figure 1), which possesses a benzothiophen heterocycle at the C3 position, is able to induce the cell death with an IC50 of 0.98 µM against SkBr3 cell lines [42]. Inspired by this study and the promising activity displayed by 6BrCaQ, we proposed to design a news series of quinolinone based heterocycle analogues (Figure 1) in which the amide function of 6BrCaQ will be replaced by various heterocycles, including benzoxazoles, benzothiazoles, indoles, benzimidazoles, and purines, in the aim to better understand the SAR in this novel series. In this article, the synthesis and biological evaluation of analogues of type 3, 4, and 5 are described.
2. Results and Discussion
2.1. Chemistry
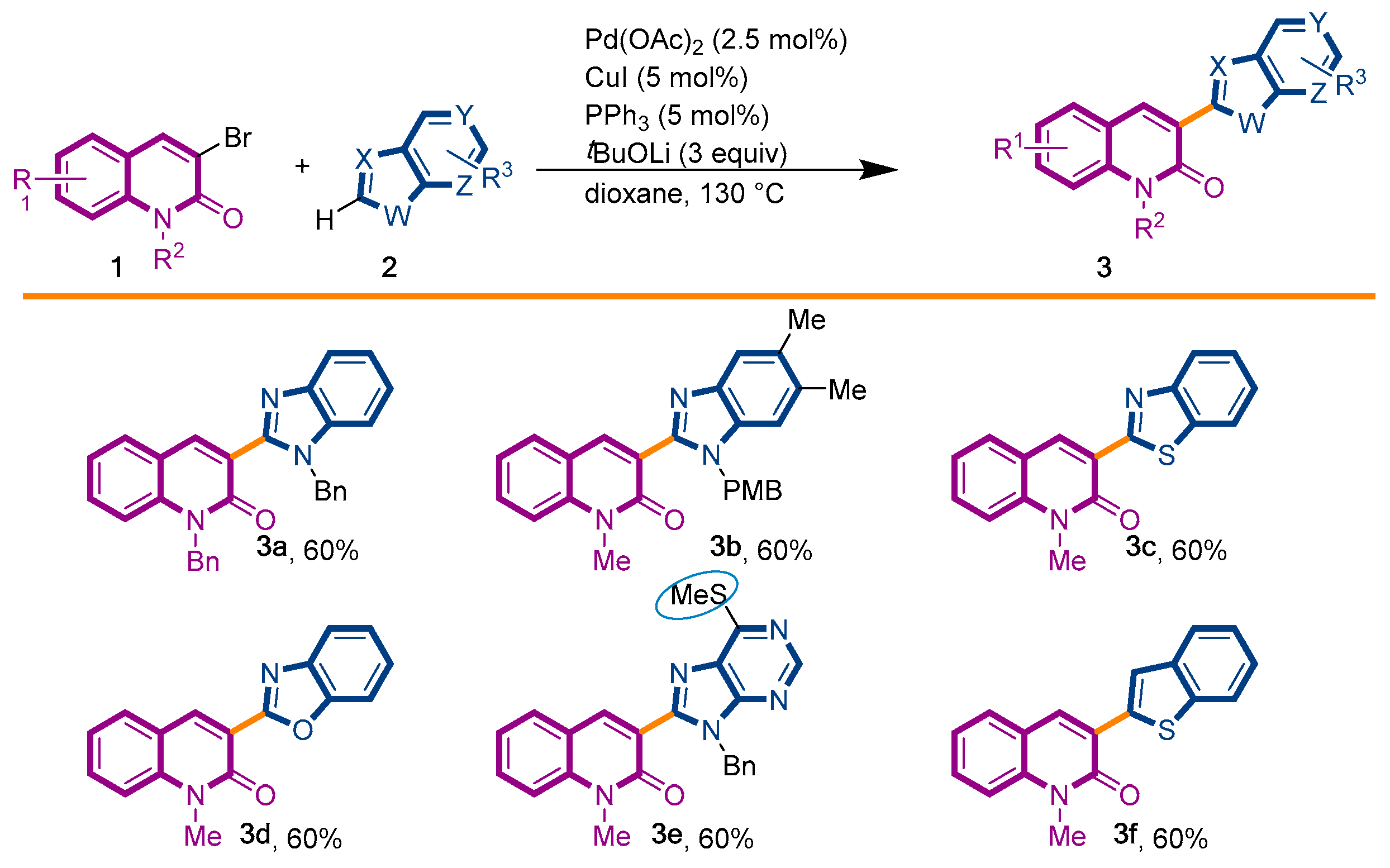
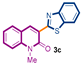
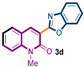
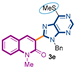
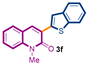
The first library of compounds targeted is simplified 3-(heteroaryl)quinolin-2(1H)-ones 3 (Scheme 1). These analogues were synthesized by the palladium-catalyzed C-H functionalization reaction of 3-bromoquinolin-2-(1H)-ones 1 with various azoles according to our previously reported conditions [43]. The reactions take place rapidly in 1,4-dioxane and proceed in good to excellent yields using bimetallic Pd(OAc)2/CuI as catalysts, PPh3 as the ligand, and LiOtBu as the base. Under this convergent protocol, compounds 3a–f were synthesized. These compounds were already reported in reference [43], fully characterized, and their physical properties can also be found in ref [43]. Various heterocycles could be introduced at the C3-position of the quinolin-2(1H)-one nucleus, including benzoxazole, benzothiazole, indole, benzimidazole, and the SMe-purine (Scheme 1).
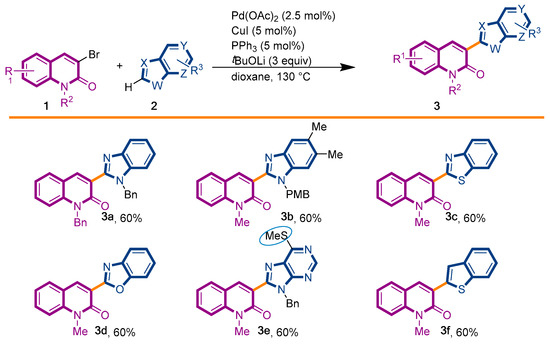
Scheme 1.
Synthetic strategy to target 3-(heteroaryl)quinolin-2(1H)-ones 3.
In the pursuit of our SAR-study, we considered the possibility of functionalization of the thiomethyl group attached to 6′-position of the purine motif in the derivative 3e. This motif is found in a series of hsp90 inhibitors, such as PU-H71 disclosed in Figure 1. If succeeded, this approach would provide a fast and easy access to a small library of more sophisticated purine-quinolinone analogues. We have rationalized that an additional aromatic group in molecule 1 would modify the intercalation ability due to changes in the planarity and in the extension of conjugation.
After a detailed survey about this topic, only few examples of this derivatization of thiopurines were found in the literature. One of the methodologies available to introduce diversity in this particular position is the scarcely explored Liebeskind–Srogl coupling [44]. This approach exploits the pseudo-halogen character of CH3S- group (thio-organyl in general) as partner in a Suzuki-like coupling reaction, involving an arylboronic acid under palladium catalysis in the presence of a copper salt [45,46].
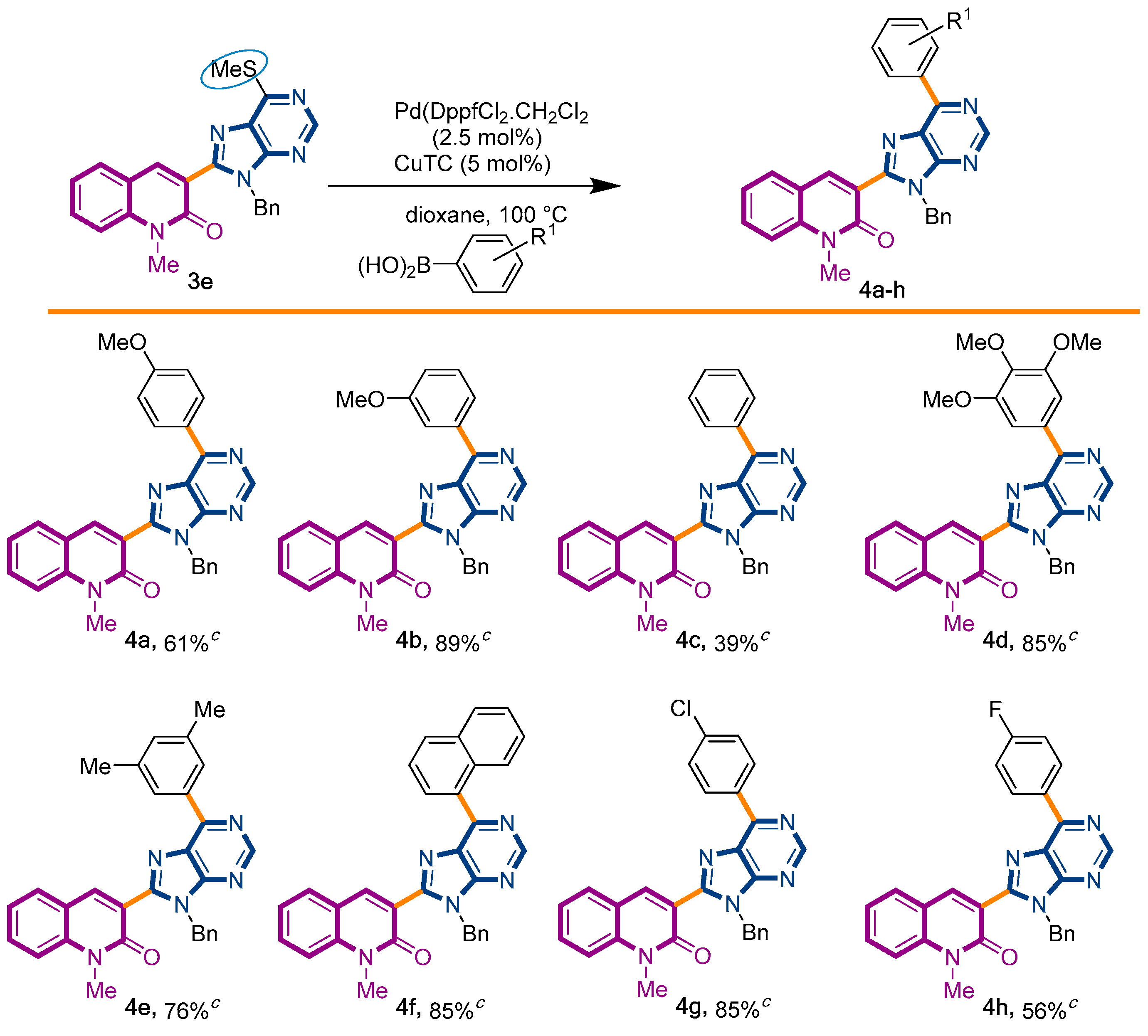
We decided to start our investigations with the adoption of two approaches. The first one involved the coupling of thiopurinoquinolone 3e with p-methoxyphenyl-boronic acid under PdDppfCl2·CH2Cl2 catalysis and conventional heating [47], while the second method chosen to promote the desired transformation employed Pd(OAc)2 and 1,10-phenanthroline under microwave irradiation [48]. To our delight, both approaches were capable to afford the expected product 4a, in 61% and 49% yield, respectively (Scheme 2). In order to explore the scope of this synthetic transformation, we decided to use the first method with a sort of arylboronic acid. This selection was based on the rationale of the electronic and steric effects exerted by the chosen substituents. The reactions proceeded smoothly, affording the expected products 4a–h with yields ranging from 39% to 89% (Scheme 2).
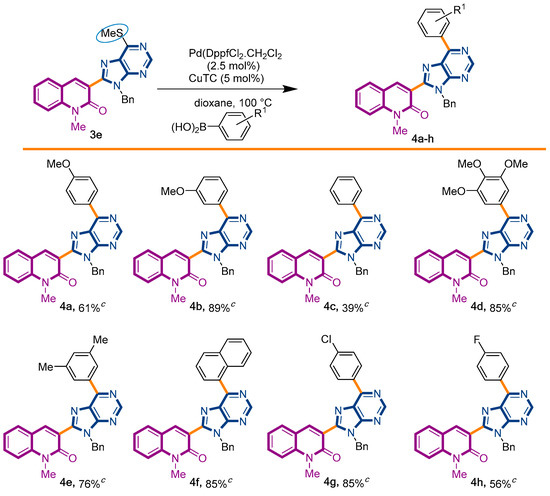
Scheme 2.
Synthetic strategy to target 3-(purino)-quinolin-2(1H)-ones 4a–h.
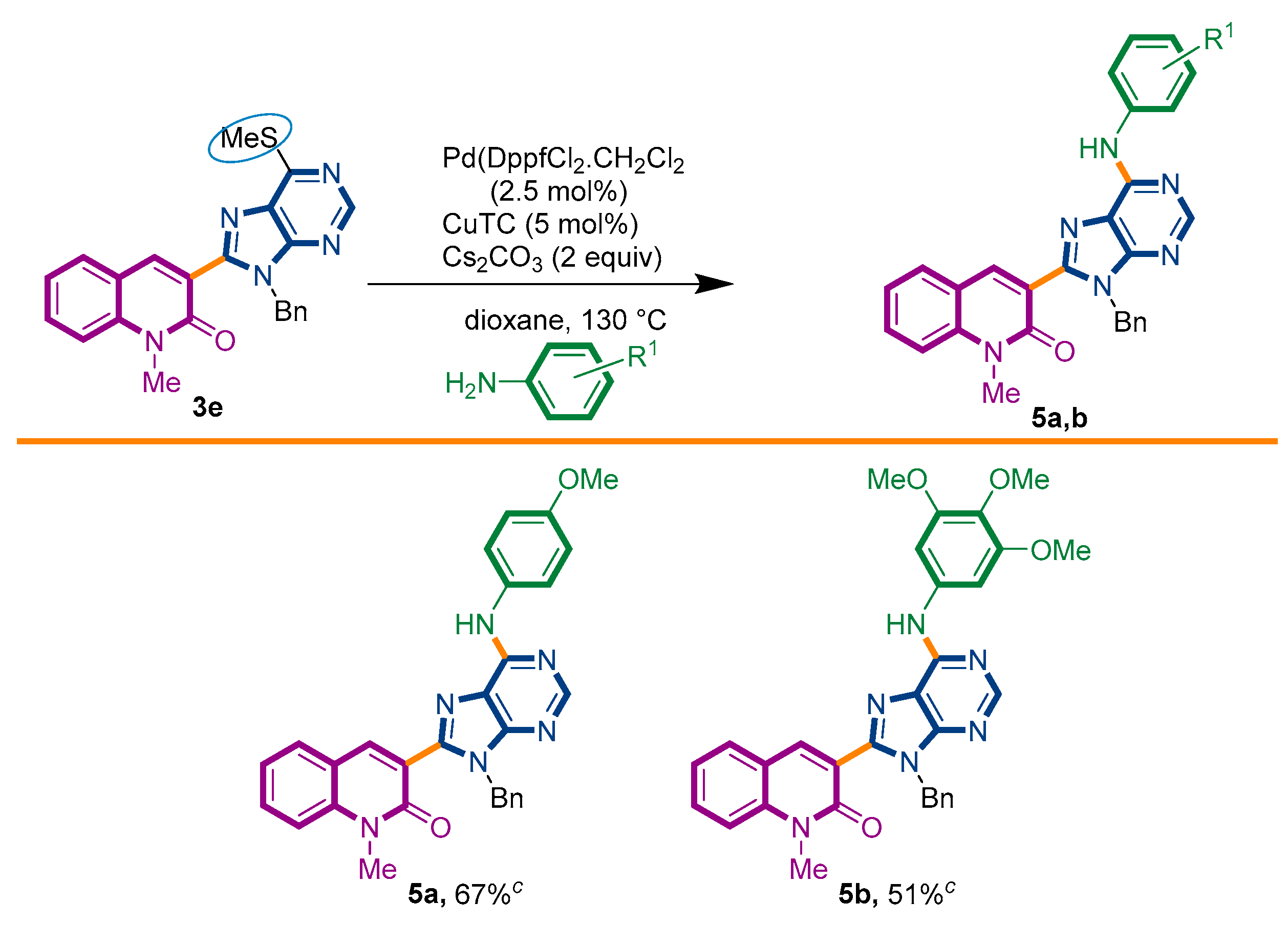
Taking advantage of this synthetic procedure, we decided to examine the scope of Liebeskind–Srogl reaction with anilines (Scheme 3). The introduction of a nitrogen atom at the 6′-position of the purine ring would imply its overall transformation into a nucleic acid analogue, i.e., an [(N-phenyl)adenine] motif. The possibility to have an adenine ring attached to a quinolone nucleus would increase its biological resemblance [49,50].
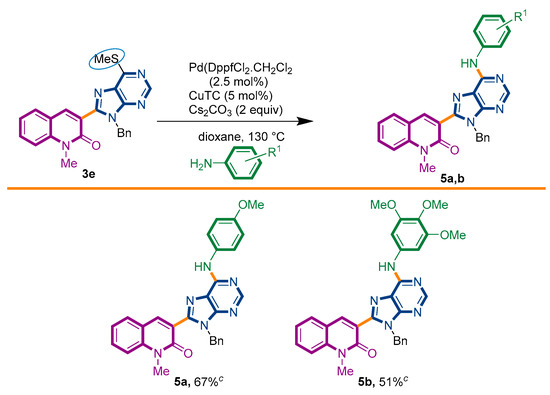
Scheme 3.
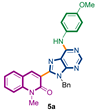
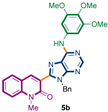
Synthetic strategy to target 3-adenines-quinolin-2(1H)-ones 5a,b.
It is interesting to denote that this reaction can also be reached by an alternative two-step procedure involving the initial thioether-sulfone oxidation, followed by a nucleophilic heteroaromatic substitution with the appropriate amine. This protocol has been previously employed by Piguel and coworkers during the synthesis of 6,8,9-purine-derivatives [47]. To the best of our knowledge, the introduction of an amine to the purine ring in 6′-position through a Liebeskind–Srogl reaction has never been reported in the literature.
We started our investigations by adapting the arylsulfide amination protocol described by the group of Murakami [51]. Unfortunately, only the degradation of starting material 3e was detected. Several conditions were assayed, including the palladium source, ligand, base, and microwave heating [52,53,54]. To our surprise, with a slight modification of the previously used PdDppf·Cl2/CuTC protocol, the reaction proceeded until completeness, giving the desired product 5a in 67% yield (Scheme 3). It is important to observe that the presence of CuTC and the Cs2CO3 base were mandatory to accomplish the expected transformation.
Under this condition, we succeed to generate the coupling product from 3,4,5-trimethoxyaniline (5b, 51%). Unfortunately, however, the reaction of 3e with aniline, benzyamine, butylamine, and pyrrolidine could not be driven to completeness and the expected product could not be separated from the starting material by current chromatographic purification conditions (CC and preparative TLC).
2.2. Biological Evaluation of Quinolones Analogues
Antiproliferative Activity
Upon completion of their syntheses, the in vitro activity of quinolone derivatives 3a–f, 4a–h, and 5a,b was evaluated by their growth-inhibitory potency in three cancer cell lines. At first, the viability of the synthesized compounds was examined with the MDA-MB-231 MCF-7 breast cancer cell line at concentrations of 10 µM, 15 µM, and 25 µM. Prostate cancer PC-3 cells and human fetal lung fibroblast MRC-5 cell lines were also subjected to this series of compounds at a unique concentration of 15 µM. The quantification of cell survival in these cell lines was established using MTS assays after 72 h exposure (Table 1), and GI50 values were estimated at the concentration required to produce 50% inhibition (Table 2).

Table 1.
Cell viability effect of 3a–f, 4a–h and 5a,b derivatives against MDA-MB-231, PC-3 and MRC-5 cell lines measured through cell metabolic activity (MTS-based assay).
Table 2.
GI50 (µM) values for anti-proliferative effects of selected compounds 3a–e [a].
As shown in Table 1, all these series of analogues induced a significant decrease of the cell viability in MDA-MB-231 cells in a concentration depend manner. At 10 µM concentration the viability percentage of MDA-MB-231 cells decreased until less than 47% under 3a and 5a exposure (Table 1). In addition, increasing the concentration at 25 µM, analogues 3a, 3b, 4g, and 4h importantly affect the growth of MDA-MB-231 cells (~30% survival), clearly demonstrating the bioactivity potential of these compounds.
Then, the cytotoxicity activity was examined with two other cancer cell lines: PC-3 cells and human fetal lung fibroblast MRC-5. As shown in Table 1, almost all the reported compounds do not present any effect against MRC-5 cell lines (>82% survival) at 15 µM concentration, except compounds 4d, 4e, and 4g, which induce a slight effect on the growth of MRC-5 cells (71% to 79% survival). In contrast, PC-3 cells seem to be more sensitive to these derivatives than MRC-5 cells, as we can see in Table 1. Upon exposure of these cell lines at 15 µM concentration, compounds 3b, 3h, 3e, and 4e were able to decrease the cell viability in PC-3 cells until 56%.
Then, the growth inhibitory activities against PC-3 prostate cancer cell line were measured for the selected 3-heteroaryl-quinolin-2(1H)-one derivatives 3a–e. All the compounds shown in Table 2 display an estimated GI50 ranging between 28 and 48 µM. Of the selected derivatives, 3b showed a significant ability to inhibit cell growth and was the most cytotoxic (GI50 = 28 µM) against the PC-3 prostate cancer cell lines.
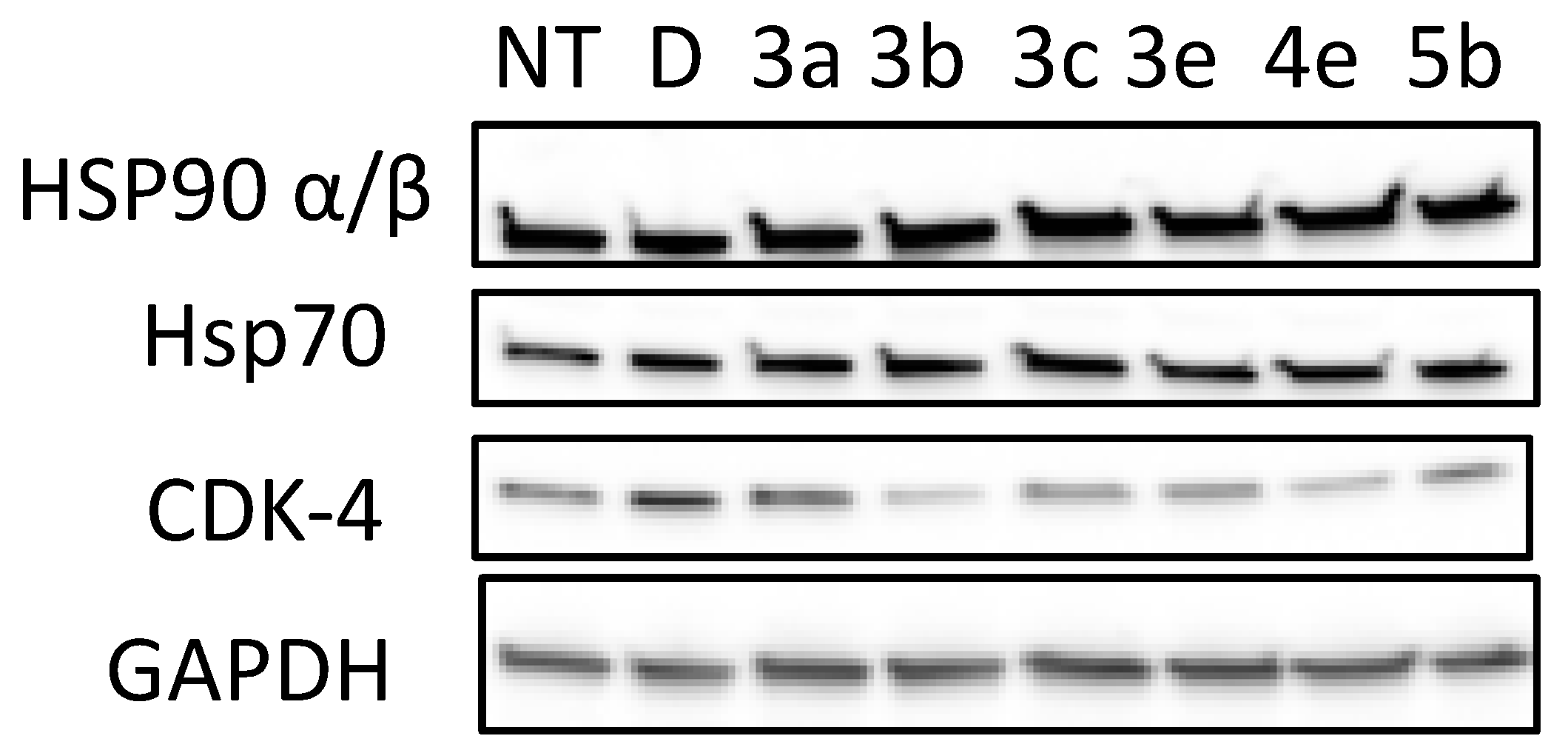
To provide additional evidence of the growth inhibitory activity manifested by the derivatives, the most active compounds 3a–e, 4e, and 5b were evaluated for their ability to induce the degradation of Hsp90-dependent client protein Cdk4, the most widely studied molecular signature indicative of Hsp90 blockade.
As depicted in Figure 2, the cyclin-dependant kinase CdK4 was degraded following treatment with 3a–e, 4e, and 5b. The GAPDH protein was not affected by the tested compounds, indicating the selective degradation of hsp90-dependent clients. CDK-4 level was more decreased by compounds 3b and 4e at a concentration of 15 μM. One can note that the anti-proliferative activity of 3b (IC50 = 28 µM, Table 2) and 4e correlate well with the concentration needed to induce Hsp90/CDK-4 client protein degradation.
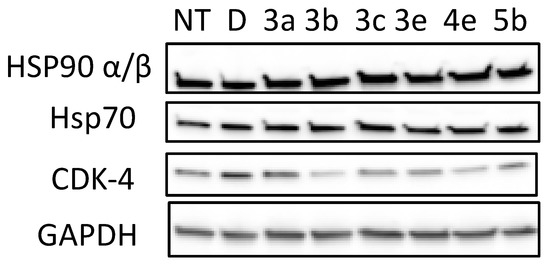
Figure 2.
Effects of quinolone analogues 3a–e, 4e and 5b on HSP90 machinery protein levels and on CDK-4 stability. PC-3 cells were grown and exposed to Hsp90 inhibitors (3a–e, 4e and 5b, 15 µM) as described in Experimental section for 72 h and cell lysates were analyzed by Western blotting with regard to the levels of CDK-4, Hsp90α/β and Hsp70. NT corresponds to untreated cells; D, DMSO-treated cells were used as controls, GADPH level is used for control in protein loading on gels.
Hsp90 N-terminal inhibitors induce a Heat-shock-response by releasing a transcription factor (HSF1) of the genes of Hsp27, Hsp70, and Hsp90. This increase in transcription leads to opposition, to apoptosis, and thus resistance to treatment.
It is important to check that the levels of these proteins are not increasing with our compounds. We showed by Western blot that 3a–e, 4e, and 5b stabilize the levels of Hsp90 and Hsp70 without triggering the HSR. This result was already observed with 6-BrCaQ in PC-3 cell lines, as we reported previously: liposomal 6-BrCaQ stabilized levels of Hsp70 and decreased the level of Hsp90 [40].
3. Conclusions
In summary, we have designed and synthesized a new series of 3-heteroaryl-quinolin-2(1H)-one derivatives as potential Hsp90 inhibitors. During this study, we developed a Pd-catalyzed Liebeskind–Srogl cross-coupling reaction between an SMe-containing quinolinyl-purine derivative and various aryl boronic acids. We reported also, for the first time, that anilines may be used as nucleophilic partners during this coupling. From these SAR studies, 3a–e, 4e, and 5b were found to display the strongest cell viability effect against MDA-MB 231 and PC-3 cancer cell lines. In addition, compounds 3b and 4e were found to be able to induce a significant decrease of CDK-1 client protein and stabilize the levels of Hsp90 and Hsp70 without triggering the HSR response.
4. Materials and Methods
4.1. General Experimental Methods
The compounds were all identified by the usual physical methods, namely 1H-NMR, 13C-NMR, IR, and HRMS (ESI) (See supplementary materials). The 1H and 13C NMR spectroscopic data were recorded on a Bruker Avance 300 FT-NMR (300.13 MHz for 1H NMR and 75.48 MHz for 13C NMR). 1H and 13C NMR spectra were measured in CDCl3 unless otherwise stated. 1H chemical shifts are reported in ppm using tetramethylsilane (TMS) as internal standard. The following abbreviations are used: m (multiplet), s (singlet), bs (broad singlet), d (doublet), t (triplet), dd (doublet of doublet), td (triplet of doublet), q (quadruplet), qui (quintuplet), sex (sextuplet). 13C NMR chemical shifts are reported in ppm from the central peak of deuterochloroform (77.1). IR spectra were measured on a Bruker Vector 22 spectrophotometer [neat, ATR] and are reported in wave numbers (cm−1). High resolution mass spectra (HRMS) were recorded by direct infusion in a mass spectrometer LCT Premier/XE (Waters).
General Methods: All glassware was oven-dried at 140 °C and all reactions were conducted under dry argon atmosphere. The solvents cyclohexane, ethyl acetate and MeOH for chromatography were purchased to Aldrich and were used as received. The reactions were monitored by TLC run in cyclohexane/EtOAc mixtures. The plates were visualized by UV light (254 and 365 nm) and immersed into a solution of phosphomolybdic acid in ethanol, and carefully heated to improve selectivity. Preparative TLC was performed on 2.0-mm thick Merck pre-coated silica gel PLC plates. Merck silica gel 60 (230–400 mesh) was used for column chromatography, employing cyclohexane/EtOAc polarity gradient techniques under positive pressure. Melting points were recorded on a Büchi B-450 apparatus and are uncorrected.
4.2. General Procedure for the Liebeskind–Srogl Coupling of 3-(6-Methylthiopurine)-2-quinolone (3e) and Boronic Acids
In a reaction tube under argon atmosphere, quinolone 3e (1 equiv.), boronic acid (2.0 equiv.), PdDppfCl2·CH2Cl2 (0.02 equiv.), and CuTC (2 equiv.) were mixed in dry dioxane (10 mL/mmol). The tube was sealed and placed into a pre-heated oil bath at 80–90 °C until reaction completeness (4–8 h) was ascertained by TLC. The volatiles were removed in the rotavapor, and the crude material was purified by column chromatography or preparative TLC.
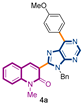
3-[9-benzyl-6-(4-methoxyphenyl)-9H-purin-8-yl]-1-methylquinolin-2(1H)-one (4a): 27 mg, 0.056 mmol, Yield 61%. Colorless oil. Rf = 0.67 (C6H12/AcOEt 50%). IR (ATR-diamond, ν): 2963, 1649, 1642, 1598, 1578, 1512, 1453, 1413, 1322, 1292, 1251, 1174, 1103, 1028, 1013, 873, 846, 804, 750, 734, 699 cm−1. 1H NMR δ: 3.84 (s, 3H, NCH3), 3.90 (s, 3H, ArOCH3), 5.74 (s, 2H, NCH2Ph), 7.00–7.12 (m, 7H, Bn, ArOCH3), 7.28 (dt, J = 7.9, 0.7 Hz, 1H), 7.42 (d, J = 8.7 Hz, 1H), 7.57 (dd, J = 7.9, 1.5 Hz, 1H), 7.66 (dd, J = 8.3, 0.7 Hz, 1H), 8.05 (s, 1H, H-4), 8.87 (d, J = 9.0 Hz, 2H, ArOCH3), 9.01 (s, 1H, H-4′). 13C NMR δ: 30.1, 47.1, 55.4, 113.5, 114.1, 114.3, 119.7, 122.8, 123.2, 127.4, 128.3, 128.5, 129.9, 130.3, 131.7, 132.4, 136.4, 140.6, 143.2, 151.9, 152.3, 153.8, 154.1, 160.1, 162.0. HRMS m/z calcd for C29H25N6O2: 489.2039 [M + H]+, found: 489.2030.
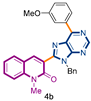
3-[9-benzyl-6-(3-methoxyphenyl)-9H-purin-8-yl]-1-methylquinolin-2(1H)-one (4b): 47 mg, 0.099 mmol, Yield 89%. Colorless oil. Rf = 0.29 (C6H12/AcOEt 50%). IR (ATR-diamond, ν): 3250, 1646, 1461, 1323, 1218, 1161, 1071, 954, 791 cm−1. 1H NMR δ (d6-acetone): 3.87 (s, 3H, NCH3), 3.91 (s, 3H, OCH3), 5.76 (s, 2H, NCH2Ph), 7.07–7.15 (m, 6H, Bn, ArOCH3), 7.35 (dt, J = 7.9, 0.9 Hz, 1H), 7.49 (d, J = 7.9 Hz, 1H), 7.66 (d, J = 8.3 Hz, 1H), 7.77 (dt, J = 7.9, 1.5 Hz, 1H), 7.81 (d, J = 7.9 Hz, 1H), 8.29 (s, 1H), 8.64–8.67 (m, 2H, ArOCH3), 9.01 (s, 1H, H-4′). 13C NMR δ (d6-acetone): 30.2, 47.6, 55.7, 115.7, 117.5, 120.5, 123.2, 123.5, 128.3, 128.4, 129.3, 130.3, 130.8, 133.3, 143.9, 153.7, 160.6, 160.8. HRMS m/z calcd for C29H24N5O2: 474.1930 [M + H]+, found: 474.1937.
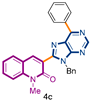
3-(9-benzyl-6-phenyl-9H-purin-8-yl)-1-methylquinolin-2(1H)-one (4c): 10 mg, 0.024 mmol, Yield 39%. Colorless oil. Rf = 0.52 (C6H12/AcOEt 50%). IR (ATR-diamond, ν): 3060, 2931, 1641, 1586, 1466, 1331, 1298, 1166, 954, 768, 668 cm−1. 1H NMR δ: 3.84 (s, 3H, NCH3), 5.74 (s, 2H, NCH2Ph), 7.00–7.13 (m, 5H, Bn), 7.29 (t, J = 8.1 Hz, 1H), 7.43 (d, J = 8.7 Hz, 1H), 7.48–7.62 (m, 8H), 7.67 (dd, J = 8.7, 1.5 Hz, 2H), 8.05 (s, 1H, H-4), 8.25 (d, J = 6.8 Hz, 1H, H-4″), 8.81 (dd, J = 8.3, 1.9 Hz, 2H, H-2″,6″), 9.08 (s, 1H, H-4′). 13C NMR δ: 30.1, 47.1, 114.3, 119.7, 122.9, 123.1, 127.4, 127.7, 127.8, 128.0, 128.5, 128.7, 129.9, 130.7, 130.9, 132.4, 132.7, 133.2, 133.5, 135.9, 136.4, 140.6, 143.3, 152.2, 152.5, 154.3, 154.4, 160.1. HRMS m/z calcd for C28H21N5ONa: 466.1644 [M + Na]+, found: 466.1646.
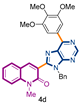
3-[9-benzyl-6-(3,4,5-trimethoxyphenyl)-9H-purin-8-yl]-1-methylquinolin-2(1H)-one (4d): 24 mg, 0.053 mmol, Yield 85%. Colorless oil. Rf = 0.28 (C6H12/AcOEt 50%). IR (ATR-diamond, ν): 2939, 2836, 1641, 1505, 1444, 1346, 1217, 11,241,072, 1002, 952, 858, 723, 697 cm−1. 1H NMR δ: 3.84 (s, 3H, NCH3), 3.93 (s, 3H, OCH3), 3.938 (s, 6H, OCH3), 5.72 (s, 2H, NCH2Ph), 6.96–7.02 (m, 2H, Bn), 7.07–7.11 (m, 3H, Bn), 7.28 (t, J = 7.4 Hz, 1H), 7.44 (d, J = 8.5 Hz, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.68 (dt, J = 7.4, 1.5 Hz, 1H) 8.03 (s, 1H), 8.25 (s, 2H, ArOCH3), 9.03 (s, 1H, H-4′). 13C NMR δ: 30.1, 47.1, 56.3, 61.0, 107.3, 114.4, 119.7, 122.9, 123.4, 127.4, 127.7, 128.5, 129.9, 130.8, 131.3, 132.4, 136.4, 140.6, 143.0, 152.0, 153.3, 153.7, 154.3, 160.1. HRMS m/z calcd for C31H28N5O4: 534.2141 [M + H]+, found: 534.2148.
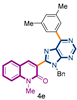
3-[9-benzyl-6-(3,5-dimethylphenyl)-9H-purin-8-yl]-1-methylquinolin-2(1H)-one (4e): 19 mg, 0.054 mmol, Yield 76%. Colorless oil. Rf = 0.61 (C6H12/AcOEt 50%). IR (ATR-diamond, ν): 3037, 2910, 1646, 1582, 1447, 1322, 1216, 1117, 1073, 955, 863, 725 cm−1. 1H NMR δ: 3.83 (s, 3H, NCH3), 5.73 (s, 2H, NCH2Ph), 6.98–7.01 (m, 2H, Bn), 7.07–7.12 (m, 3H, Bn), 7.14 (s, 1H, ArCH3), 7.28 (t, J = 7.4 Hz, 1H), 7.43 (d, J = 8.5 Hz, 1H), 7.59 (d, J = 7.7 Hz, 1H), 7.67 (t, J = 7.4 Hz, 1H) 8.07 (s, 1H), 8.42 (s, 2H, ArCH3), 9.06 (s, 1H, H-4′). 13C NMR δ: 21.5, 30.1, 47.1, 114.3, 119.7, 122.8, 123.2, 127.4, 127.6, 127.7, 128.5, 130.0, 130.9, 132.4, 132.7, 135.7, 136.4, 138.2, 140.6, 143.3, 152.1, 152.4, 154.2, 154.8, 160.1. HRMS m/z calcd for C30H26N5O: 472.2137 [M + H]+, found: 472.2139.
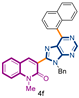
3-[9-benzyl-6-(naphthalen-2-yl)-9H-purin-8-yl]-1-methylquinolin-2(1H)-one (4f): 21 mg, 0.059 mmol, Yield 85%. Colorless oil. Rf = 0.61 (C6H12/AcOEt 50%). IR (ATR-diamond, ν): 3067, 1642, 1570, 1446, 1320, 1276, 1168, 920, 847, 754, 696 cm−1. 1H NMR δ: 3.85 (s, 3H, NCH3), 5.76 (s, 2H, NCH2Ph), 7.02–7.12 (m, 5H, Bn), 7.29 (t, J = 8.1 Hz, 1H), 7.43 (d, J = 8.5 Hz, 1H), 7.48–7.56 (m, 2H), 7.60 (d, J = 7.3 Hz, 1H), 7.67 (d, J = 7.7 Hz, 1H), 7.88 (d, J = 8.7 Hz, 1H), 7.99 (d, J = 8.7 Hz, 1H), 8.05 (d, J = 7.3 Hz, 1H), 8.11 (s, 1H, H-4), 8.97 (d, J = 8.3 Hz, 1H, Naphthyl), 9.12 (s, 1H, H-4′), 9.46 (s, 1H, Naphthyl). 13C NMR δ: 30.1, 47.2, 114.3, 119.7, 122.9, 123.2, 126.2, 126.3, 127.3, 127.5, 127.70, 127.74, 128.2, 128.5, 123.0, 130.8, 131.2, 132.4, 133.3, 133.4, 133.6, 134.6, 136.4, 140.6, 143.4, 152.3, 152.5, 154.1, 154.4, 160.1. HRMS m/z calcd for C32H24N5O: 494.1981 [M + H]+, found: 494.1976.
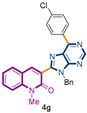
3-[9-benzyl-6-(4-chlorophenyl)-9H-purin-8-yl]-1-methylquinolin-2(1H)-one (4g): 34 mg, 0.083 mmol, Yield 85%. Colorless solid, m.p.: 202–203 °C (CH2Cl2/MeOH). Rf = 0.65 (C6H12/AcOEt 50%). IR (ATR-diamond, ν): 3044, 2928, 1641, 1582, 1492, 1381, 1298, 1177, 1089, 953, 803, 736, 720 cm−1. 1H NMR δ: 3.85 (s, 3H, NCH3), 5.73 (s, 2H, NCH2Ph), 6.99–7.03 (m, 2H, Bn), 7.07–7.12 (m, 3H, Bn), 7.30 (dt, J = 7.9, 0.7 Hz, 1H), 7.44 (d, J = 8.5 Hz, 1H), 7.51 (d, J = 8.5 Hz, 2H, ArCl), 7.69 (dd, J = 8.7, 1.7 Hz, 1H), 8.04 (s, 1H), 8.85 (d, J = 8.5 Hz, 2H, ArCl), 9.06 (s, 1H, H-4′). 13C NMR δ: 30.1, 47.2, 114.3, 119.7, 122.9, 123.0, 127.5, 127.8, 128.9, 130.0, 130.8, 131.2, 132.5, 134.4, 136.3, 137.0, 140.6, 143.3, 152.4, 152.9, 154.4, 160.0. HRMS m/z calcd for C28H21N5OCl: 478.1435 [M + H]+, found: 478.1440.
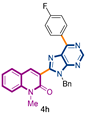
3-[9-benzyl-6-(4-fluorophenyl)-9H-purin-8-yl]-1-methylquinolin-2(1H)-one (4h): 22 mg, 0.047 mmol, Yield 56%. Colorless oil. Rf = 0.57 (C6H12/AcOEt 50%). IR (ATR-diamond, ν): 3058, 2958, 1642, 1569, 1463, 1320, 1297, 1159, 1070, 952, 847, 724, 698 cm−1. 1H NMR δ: 3.63 (s, 3H, NCH3), 5.65 (s, 2H, NCH2Ph), 6.91–6.94 (m, 2H, Bn), 6.99–7.05 (m, 3H, Bn), 7.14 (t, J = 8.9 Hz, 2H, ArF), 7.22 (t, J = 7.9 Hz, 1H), 7.36 (d, J = 8.5 Hz, 1H), 7.51 (dd, J = 7.9, 1.3 Hz, 1H), 7.61 (dt, J = 8.5, 1.3 Hz, 1H), 7.97 (s, 1H, H-4), 8.84 (dd, J = 8.9, 5.6 Hz, 1H, ArF), 8.97 (s, 1H, H-4′). 13C NMR δ: 30.1, 47.1, 67.1 114.3, 115.7 (d, 2JC-F = 21.6 Hz), 119.7, 122.9, 123.1, 127.5, 127.8, 128.5, 130.0, 132.1 (d, 3JC-F = 8.3 Hz), 136.3, 140.6, 143.3, 152.3, 152.5, 153.0, 154.3, 160.1, 164.5 (d, 1JC-F = 249.8 Hz). HRMS m/z calcd for C28H20N5OFNa: 484.1550 [M + Na]+, found: 484.1552.
4.3. Procedure for the Liebeskind–Srogl Coupling of 3-(6-Methylthiopurine)-2-quinolone (3e) and Anilines
In a reaction tube equipped with a stirring bar under argon atmosphere, quinolone 3e (1 equiv.), the appropriate aniline (2.0 equiv.), PdDppfCl2·CH2Cl2 (0.1 equiv.), Ph3P (0.2 equiv.), Cs2CO3 (2.0 equiv.), and CuTC (2 equiv.) were admixed in dry dioxane (20 mL/mmol). The tube was sealed and poured into a pre-heated oil bath at 130 °C until consumption of quinolone 1, monitored by TLC (15–18 h). The volatiles were removed in the rotavapor, and the crude material was suspended with AcOEt, filtered through a short pad of cotton, concentrated under vacuum, and the remaining solid was purified by preparative TLC (2 × C6H12/AcOEt 50%).
3-{9-benzyl-6-[(4-methoxyphenyl)amino]-9H-purin-8-yl}-1-methylquinolin-2(1H)-one (5a): 20 mg, 0.039 mmol, Yield 67%. Yellowish oil. Rf = 0.36 (2 × C6H12/AcOEt 50%). IR (ATR-diamond, ν): 3283, 3038, 2928, 2847, 1641, 1588, 1573, 1461, 1380, 1241, 1180, 1087, 953, 829, 755, 697 cm−1. 1H NMR δ: 3.74 (s, 3H, NCH3), 3.75 (s, 3H, OCH3), 5.54 (s, 2H, NCH2Ph), 6.92 (d, J = 8.9 Hz, 2H, Ar), 6.99–7.02 (m, 2H, Bn), 7.09–7.13 (m, 3H, Bn), 7.26 (t, J = 8.2 Hz, 1H, H-6), 7.41 (d, J = 8.5 Hz, 1H, H-8), 7.48 (d, J = 7.7 Hz, 1H, H-5), 7.62–7.68 (m, 3H, H-7, ArOCH3), 7.71 (sb, 1H, NH) 7.86 (s, 1H, H-4), 8.55 (s, 1H, H-4′). 13C NMR δ: 30.0, 47.1, 55.5, 114.3, 119.6, 119.9, 122.5, 122.8, 123.0, 127.4, 127.9, 128.5, 129.8, 131.7, 132.2, 136.6, 140.5, 142.4, 147.9, 151.2, 152.2, 153.2, 156.1, 160.1. HRMS m/z calcd for C29H25N6O2: 489.2039 [M + H]+, found: 489.2030.
3-{9-benzyl-6-[(3,4,5-trimethoxyphenyl)amino]-9H-purin-8-yl}-1-methylquinolin-2(1H)-one (5b): 14 mg, 0.025 mmol, Yield 51%. Yellowish oil. Rf = 0.18 (3 × C6H12/AcOEt 50%). IR (ATR-diamond, ν): 3296, 3039, 2933, 2834, 1642, 1588, 1463, 1323, 1126, 1088, 954, 697 cm−1. 1H NMR δ: 3.82 (s, 3H, NCH3), 3.83 (s, 3H, OCH3), 3.87 (s, 6H, OCH3), 5.63 (s, 2H, N-CH2Ph), 6.99–7.02 (m, 2H, Bn), 7.09–7.14 (m, 3H, Bn), 7.16 [s, 2H, Ar(OCH3)3], 7.27 (t, J = 7.4 Hz, 1H, H-6), 7.42 (d, J = 8.5 Hz, 1H, H-8), 7.49 (d, J = 7.2 Hz, 1H, H-5), 7.66 (t, J = 7.9 Hz, 1H, H-7), 7.87 (sb, 1H, NH), 8.31 (s, 1H, H-4), 8.59 (s, 1H, H-10). 13C NMR δ: 30.1, 47.1, 56.1, 61.0, 98.0, 114.3, 119.6, 119.8, 122.8, 122.8, 127.4, 127.7, 128.5, 129.8, 132.3, 135.1, 136.5, 140.5, 142.5, 148.2, 151.9, 153.1, 153.3, 160.2. HRMS m/z calcd for C31H29N6O4: 549.2250 [M + H]+, found: 549.2250.
4.4. Materials and Methods for Cell Culture and Western Blot Analysis
MDA-MB-231 cells were grown in L15 supplemented with 15% serum, 2 mM glutamine, and 22 mM sodium bicarbonate in the presence of Penicillin/Streptomycin antibiotic mixture. PC-3 cells were cultured in RPMI 1640 supplemented with 10% serum and 2 mM glutamine in the presence of Penicillin/Streptomycin antibiotic mixture. MRC-5 cells were cultured in EMEM supplemented with 10% serum and 2 mM glutamine in the presence of Penicillin/Streptomycin antibiotic mixture (Reagents were purchased from Sigma Aldrich, Saint-Quentin-Fallavier, France).
Cells were treated for 72 h with 10, 15, and 25 µM of the selected substances. Control cells were treated with the equivalent in DMSO.
Cell survival was assessed using the CellTiter Aqueous One Proliferation assay (Promega, Charbonnières-les-Bains, France) whereby 2500 or 5000 cells per well were seeded in 96-well plates in 100 µL. At the end of the treatment, 20 µL of reagent was added, and absorbance readings were taken at 492 nm on a 96-well plate reader after 3 h of contact (on average).
For protein expression analysis, cells were seeded at a rate of 0.75 × 106 cells on a 25 cm2 surface. At the end of the treatment the cells were washed and lysed with RIPA buffer (Sigma Aldrich, Saint-Quentin-Fallavier, France city, country) to which protease inhibitors (Sigma Aldrich, Saint-Quentin-Fallavier, France SIGMA) were added. After 30 min of lysis on ice, the samples were centrifuged (3500 rpm, 10 min) and stored at −20 °C. Total protein concentration was obtained using the Bio-rad Protein Assay reagent (Bio-Rad Laboratories, Marnes-la-Coquette, France city, country). Hence, 30 µg of protein (denatured in the presence of Laemmli buffer, sample buffer from Bio-Rad) was plated on a 4–15% pre-cast acrylamide gel (Bio-Rad, Marnes-la-Coquette, France) and subjected to SDS-PAGE (150 V, 1 h) and PVDF membrane transfer (100 V, 45 min). Immunorevelation was performed as follows: 1 h saturation of the membrane with TBS-Tween (0.1%)–5% skim milk followed by overnight incubation in primary antibody solution (antibodies were from Santa Cruz Biotechnologies, Clinisciences, Nanterre, France or from Sigma Aldrich, Saint-Quentin-Fallavier, France)(See Table 3). After 1 h of washing in 0.1% TBS-Tween, the membranes were contacted with the second horseradish peroxidase-coupled antibody. Detection of the chemiluminescence signal (SuperSignal Pierce reagent, Fisher Scientific, Illkirch, France) was performed using the ChemiBis gel Imager (DNR Bio-Imaging Systems, Israel).
Table 3.
Informations concerning antibodies used during this study.
Supplementary Materials
The following are available online, 1H and 13C NMR spectra for compounds 4a–h and 5a, 5b.
Author Contributions
S.M. and M.A. conceived and designed the experiments; E.L.L. developed and performed the synthetic approach for the synthesis of the 6BrCaQ analogues; A.B. and F.S. performed the biological evaluations under the supervision of J.V.-G., E.L.L., A.B., F.S., M.A., J.V.-G., and S.M. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data supporting the reported results will be available from the corresponding author (E. L. Larghi, or S. Messaoudi).
Acknowledgments
Authors acknowledge the support of this project by CNRS, University Paris Paris Sacaly. E.L.L. also thanks CONICET for the fellowship and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, PICT 2018-01933).
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Messaoudi, S.; Peyrat, J.-F.; Brion, J.-D.; Alami, M. Recent advances in Hsp90 inhibitors as antitumor agents. Anticancer Agents Med. Chem. 2008, 8, 761–782. [Google Scholar] [CrossRef]
- Sgobba, M.; Rastelli, G. Structure-based and in silico design of Hsp90 inhibitors. ChemMedChem. 2009, 4, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Janin, Y.L. ATPase inhibitors of heat-shock protein 90, second season. Drug Discov. Today 2010, 15, 342–353. [Google Scholar] [CrossRef]
- Messaoudi, S.; Peyrat, J.-F.; Brion, J.-D.; Alami, M. Heat-shock protein 90 inhibitors as antitumor agents: A survey of the literature from 2005 to 2010. Expert Opin. Ther. Pat. 2011, 21, 1501–1542. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, S.; Peyrat, J.-F.; Brion, J.-D.; Alami, M. Recent advances in hsp90 inhibitors as antitumor agents. In Advances in Anti-Cancer Agents in Medicinal Chemistry, 1st ed.; Prudhomme, M., Ed.; Bentham Sciences Publishers: Oak Park, IL, USA, 2013; Volume 1, pp. 107–183. [Google Scholar]
- Neckers, L. Heat shock protein 90: The cancer chaperone. J. Biosci. 2007, 32, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Amolins, M.W.; Blagg, B.S.J. Natural product inhibitors of Hsp90: Potential leads for drug discovery. Mini Rev. Med. Chem. 2009, 9, 140–152. [Google Scholar] [CrossRef] [Green Version]
- Maloney, A.; Workman, P. HSP90 as a new therapeutic target for cancer therapy: The story unfolds. Expert Opin. Biol. Ther. 2002, 2, 3–24. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.-L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer. 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Zhang, H.; Burrows, F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J. Mol. Med. 2004, 82, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Blagg, B.S.J.; Kerr, T.D. Hsp90 inhibitors: Small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med. Res. Rev. 2006, 26, 310–338. [Google Scholar] [CrossRef]
- Chène, P. ATPases as drug targets: Learning from their structure. Nat. Rev. Drug Discov. 2002, 1, 665–673. [Google Scholar] [CrossRef]
- Taldone, T.; Sun, W.; Chiosis, G. Discovery and development of heat shock protein 90 inhibitors. Bioorg. Med. Chem. 2009, 17, 2225–2235. [Google Scholar] [CrossRef] [Green Version]
- Taldone, T.; Gozman, A.; Maharaj, R.; Chiosis, G. Targeting Hsp90: Small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 2008, 8, 370–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalingam, D.; Swords, R.; Carew, J.S.; Nawrocki, S.T.; Bhalla, K.; Giles, F.J. Targeting Hsp90 for cancer therapy. Br. J. Cancer 2009, 100, 1523–1529. [Google Scholar] [CrossRef] [Green Version]
- Panaretou, B.; Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998, 17, 4829–4836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, P.A.; Hostein, I.; Banerji, U.; Stefano, F.D.; Maloney, A.; Walton, M.; Judson, I.; Workman, P. Gene expression profiling of human colon cancer cells following inhibition of signal transduction by 17-allylamino-17-demethoxygeldanamycin, an inhibitor of the Hsp90 molecular chaperone. Oncogene 2000, 19, 4125–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlichman, C. Tanespimycin: The opportunities and challenges of targeting heat shock protein 90. Expert Opin. Investig. Drugs 2009, 18, 861–868. [Google Scholar] [CrossRef]
- McCollum, A.K.; Teneyck, C.J.; Sauer, B.M.; Toft, D.O.; Erlichman, C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res. 2006, 66, 10967–10975. [Google Scholar] [CrossRef] [Green Version]
- Demidenko, Z.N.; Vivo, C.; Halicka, H.D.; Li, C.J.; Bhalla, K.; Broude, E.V.; Blagosklonny, M.V. Pharmacological induction of Hsp70 protects apoptosis-prone cells from doxorubicin: Comparison with caspase-inhibitor- and cycle-arrest- mediated cytoprotection. Cell. Death Differ. 2006, 13, 1434–1441. [Google Scholar] [CrossRef] [Green Version]
- Gabai, V.L.; Budagova, K.R.; Sherman, M.Y. Increased expression of the major heat shock protein Hsp72 in human prostate carcinoma cells is dispensable for their viability but confers resistance to a variety of anticancer agents. Oncogene 2005, 24, 3328–3338. [Google Scholar] [CrossRef] [Green Version]
- Pocaly, M.; Lagarde, V.; Etienne, G.; Ribeil, J.A.; Claverol, S.; Bonneu, M.; Moreau-Gaudry, F.; Guyonnet-Duperat, V.; Hermine, O.; Melo, J.V.; et al. Overexpression of the heat-shock protein 70 is associated to imatinib resistance in chronic myeloid leukemia. Leukemia 2007, 21, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.E. Targeting heat shock proteins 70/90 and proteasome for cancer therapy. Curr. Med. Chem. 2011, 18, 4250–4264. [Google Scholar] [CrossRef]
- Wang, H.; Tan, M.S.; Lu, R.C.; Yu, J.T.; Tan, L. Heat shock proteins at the crossroads between cancer and Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 239164. [Google Scholar] [CrossRef]
- Murphy, M.E. The Hsp70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, A.; Blagg, B.S.J. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem. 2008, 15, 2702–2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskew, J.D.; Sadikot, T.; Morales, P.; Duren, A.; Dunwiddie, I.; Swink, M.; Zhang, X.; Hembruff, S.; Donnelly, A.; Rajewski, R.A.; et al. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 2011, 11, 468. [Google Scholar] [CrossRef] [Green Version]
- Buckton, K.; Wahyudi, H.; McAlpine, S.R. The first report of direct inhibitors that target the C-terminal MEEVD region on heat shock protein 90 L. Chem. Commun. 2016, 52, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Burlison, J.A.; Avila, C.; Vielhauer, G.; Lubbers, D.J.; Holzbeierlein, J.; Blagg, B.S.J. Development of novobiocin analogues that manifest anti-proliferative activity against several cancer cell lines. J. Org. Chem. 2008, 73, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, H.K.; Koay, Y.C.; Irani, S.; Das, R.; Nassar, Z.D.; Australian Prostate Cancer BioResource; Selth, L.A.; Centenera, M.M.; McAlpine, S.R.; Butler, L.M. A novel class of Hsp90 C-Terminal modulators have pre-clinical efficacy in prostate tumor cells without induction of a heat shock response. Prostate 2016, 76, 1546–1559. [Google Scholar] [CrossRef] [PubMed]
- Moses, M.A.; Henry, E.C.; Ricke, W.A.; Gasiewicz, T.A. The heat shock protein 90 inhibitor, (-)-epigallocatechin gallate, has anticancer activity in a novel human prostate cancer progression model. Cancer Prev. Res. (Phila.) 2015, 8, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Le Bras, G.; Radanyi, C.; Peyrat, J.-F.; Brion, J.-D.; Alami, M.; Marsaud, V.; Stella, B.; Renoir, J.-M. New novobiocin analogues as antiproliferative agents in breast cancer cells and potential inhibitors of heat shock protein 90. J. Med. Chem. 2007, 50, 6189–6200. [Google Scholar] [CrossRef] [PubMed]
- Radanyi, C.; Le Bras, G.; Marsaud, V.; Peyrat, J.-F.; Messaoudi, S.; Catelli, M.G.; Brion, J.-D.; Alami, M.; Renoir, J.-M. Antiproliferative and apoptotic activities of tosylcyclonovobiocic acids as potent heat shock protein 90 inhibitors in human cancer cells. Cancer Lett. 2008, 274, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Radanyi, C.; Le Bras, G.; Bouclier, C.; Messaoudi, S.; Peyrat, J.-F.; Brion, J.-D.; Alami, M.; Renoir, J.-M. Tosylcyclonovobiocic acids promote cleavage of the hsp90-associated cochaperone p23. Biochem. Biophys. Res. Commun. 2009, 379, 514–518. [Google Scholar] [CrossRef]
- Audisio, D.; Methy-Gonnot, D.; Radanyi, C.; Renoir, J.-M.; Denis, S.; Sauvage, F.; Vergnaud-Gauduchon, J.; Brion, J.-D.; Messaoudi, S.; Alami, M. synthesis and antiproliferative activity of novobiocin analogues as potential Hsp90 inhibitors. Eur. J. Med. Chem. 2014, 83, 498–507. [Google Scholar] [CrossRef]
- Radanyi, C.; Le Bras, G.; Messaoudi, S.; Bouclier, C.; Peyrat, J.-F.; Brion, J.-D.; Marsaud, V.; Renoir, J.-M.; Alami, M. Synthesis and biological activity of simplified denoviose-coumarins related to novobiocin as potent inhibitors of heat-shock protein 90 (hsp90). Bioorg. Med. Chem. Lett. 2008, 18, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Audisio, D.; Messaoudi, S.; Cegielkowski, L.; Peyrat, J.-F.; Brion, J.-D.; Methy-Gonnot, D.; Radanyi, C.; Renoir, J.-M.; Alami, M. Discovery and biological activity of 6brcaq as an inhibitor of the Hsp90 protein folding machinery. ChemMedChem 2011, 6, 804–815. [Google Scholar] [CrossRef]
- Messaoudi, S.; Audisio, D.; Brion, J.-D.; Alami, M. Rapid access to 3-(N-substituted)-aminoquinolin-2(1H)-ones using palladium-catalyzed C–N bond coupling reaction. Tetrahedron 2007, 63, 10202–10210. [Google Scholar] [CrossRef]
- Sauvage, F.; Fattal, E.; Al-Shaer, W.; Denis, S.; Brotin, E.; Denoyelle, C.; Blanc-Fournier, C.; Toussaint, B.; Messaoudi, S.; Alami, M.; et al. Antitumor activity of nanoliposomes encapsulating the novobiocin analog 6brcaq in a triple-negative breast cancer model in mice. Cancer Lett. 2018, 432, 103–111. [Google Scholar] [CrossRef]
- Sauvage, F.; Franzè, S.; Bruneau, A.; Alami, M.; Denis, S.; Nicolas, V.; Lesieur, S.; Legrand, F.-X.; Barratt, G.; Messaoudi, S.; et al. Formulation and in vitro efficacy of liposomes containing the Hsp90 inhibitor 6BrCaQ in prostate cancer cells. Int. J. Pharm. 2016, 499, 101–109. [Google Scholar] [CrossRef]
- Mathieu, C.; Chamayou, Q.; Luong, T.T.H.; Naud, D.; Mahuteau, F.; Alami, M.; Fattal, E.; Messaoudi, S.; Vergnaud-Gauduchon, J. Synthesis and antiproliferative activity of 6BrCaQ-TPP conjugates for targeting the mitochondrial heat shock protein TRAP1. Eur. J. Med. Chem. 2022, 229, 114052–114065. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, B.; Peterson, L.B.; Blagg, B.S.J. 3-Arylcoumarin derivatives manifest anti-proliferative activity through hsp90 inhibition. ACS Med. Chem. Lett. 2012, 3, 327–331. [Google Scholar] [CrossRef]
- Bruneau, A.; Brion, J.-B.; Messaoudi, S.; Alami, M. A General Pd/Cu-catalyzed C-H heteroarylation of 3-bromoquinolin-2(1H)-ones. Org. Biomol. Chem. 2014, 12, 8533–8541. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, W.; Yu, Z. Transition-metal mediated carbon–sulfur bond activation and transformations. Chem. Soc. Rev. 2013, 42, 599–621. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Shi, Z.-J. Recent advances in transition-metal-catalyzed C–S activation: From thioester to (hetero)aryl thioether. ACS Catal. 2014, 4, 280–288. [Google Scholar] [CrossRef]
- Prokopcová, H.; Kappe, C.O. The Liebeskind–Srogl C-C cross-coupling reaction. Angew. Chem. Int. Ed. 2009, 48, 2276–2286. [Google Scholar] [CrossRef]
- Vabre, R. Fonctionnalisation Directe de Liaisons C-H et Couplages Croisés pour la Formation de Liaisons C-C et C-N: Synthèse de Purines 6,8,9-trisubstituées. Ph.D. Thesis, Université Paris Sud—Paris XI, Paris, France, 2013. [Google Scholar]
- Vabre, R.; Chevot, F.; Legraverend, M.; Piguel, S. Microwave-assisted Pd/Cu-catalyzed C-8 direct alkenylation of purines and related azoles: An alternative access to 6, 8, 9-trisubstituted purines. J. Org. Chem. 2011, 76, 9542–9547. [Google Scholar] [CrossRef]
- Benner, S.A. Understanding nucleic acids using synthetic chemistry. Acc. Chem. Res. 2004, 37, 784–797. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef]
- Sugahara, T.; Murakami, K.; Yorimitsu, H.; Osuka, A. Palladium-catalyzed amination of aryl sulfides with anilines. Angew. Chem. Int. Ed. 2014, 53, 9329–9333. [Google Scholar] [CrossRef] [Green Version]
- Pellegatti, L.; Vedrenne, E.; Leger, J.-M.; Jarry, C.; Routier, S. First efficient palladium-catalyzed aminations of pyrimidines, 1, 2, 4-triazines and tetrazines by original methyl sulfur release. Synlett 2009, 13, 2137–2142. [Google Scholar] [CrossRef]
- Bruneau, L.A.; Roche, M.; Alami, M.; Messaoudi, S. 2-Aminobiphenyl Palladacycles: The “Most Powerful” Precatalysts in C–C and C–Heteroatom Cross-Couplings. ACS Catal. 2015, 5, 1386–1396. [Google Scholar] [CrossRef]
- Liu, J.; Robins, M.J. Fluoro, alkylsulfanyl, and alkylsulfonyl leaving groups in Suzuki cross-coupling reactions of purine 2′-deoxynucleosides and nucleosides. Org. Lett. 2005, 7, 1149–1151. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).