3.2. Preparation and Characterization Data of Compounds
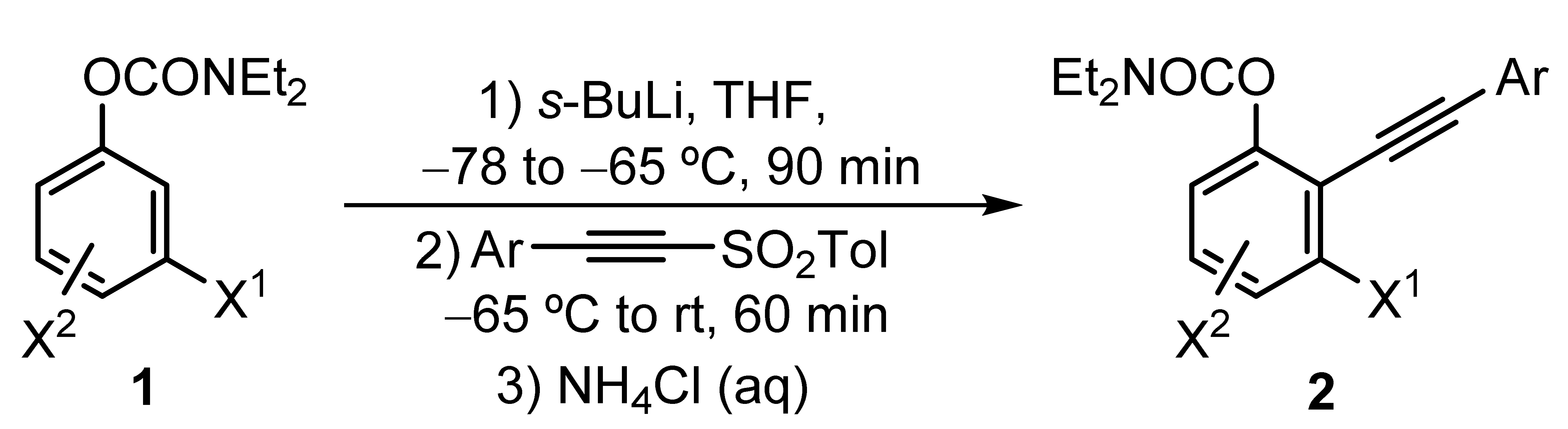
3.2.1. Synthesis of O-2-(Arylethynyl)phenyl N,N-Diethylcarbamates 2
General procedure: A solution of starting carbamate
1 (1 mmol) in THF (2 mL) at −78 °C under nitrogen atmosphere was treated with a solution of
s-BuLi (0.85 mL of a 1.4 M solution in cyclohexane, 1.2 mmol). The reaction mixture was allowed to reach −65 °C for 5 min and was then stirred at this temperature for 90 min. Then, the corresponding alkynyl sulfone [
29] (1.3 mmol) was added, and the resulting solution was stirred for 15 min at −65 °C. The solution was allowed to warm to room temperature and was then stirred for an additional 30 min. The reaction mixture was quenched with NH
4Cl (aq), diluted with EtOAc, and the layers were separated. The aqueous phase was extracted with EtOAc (3 × 10 mL), and the combined organic layers were dried over anhydrous Na
2SO
4. The solvents were removed under reduced pressure, and the residue was purified by silica gel (VWR chemicals, Radnor, PA, USA) column chromatography (hexane/EtOAc), affording the
O-2-(arylethynyl)phenyl
N,
N-diethylcarbamates
2.
O-3-Fluoro-2-(phenylethynyl)phenyl N,N-diethylcarbamate (2aa): The reaction of O-3-fluorophenyl N,N-diethylcarbamate (1a) (221 mg, 1 mmol), following the general procedure, yielded the product as a yellow oil (73% yield); Rf = 0.26 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.55–7.52 (m, 2H), 7.38–7.28 (m, 4H), 7.09 (dt, J = 8.3, 1.0 Hz, 1H), 7.00 (td, J = 8.3, 1.0 Hz, 1H), 3.54 (q, J = 7.0 Hz, 2H), 3.42 (q, J = 7.0 Hz, 2H), 1.32 (t, J = 7.0 Hz, 3H), 1.20 (t, J = 7.0 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 163.1 (d, J = 252.2 Hz, C), 153.32 (d, J = 4.1 Hz, C), 153.29 (C), 131.7 (2 × CH), 129.3 (d, J = 9.7 Hz, CH), 128.8 (CH), 128.4 (2 × CH), 123.0 (C), 118.5 (d, J = 3.3 Hz, CH), 112.3 (d, J = 21.0 Hz, CH), 107.4 (d, J = 18.0 Hz, C), 98.6 (d, J = 3.1 Hz, C), 78.5 (C), 42.5 (CH2), 42.2 (CH2), 14.3 (CH3), 13.4 (CH3). EI-LRMS m/z (%): 311 (M+, 4), 183 (7), 100 (100), 72 (56). ESI-HRMS was calculated for C19H19FNO2 [M + H]+ 312.139, and found 312.1402.
O-4-Chloro-3-fluoro-2-(phenylethynyl)phenyl N,N-diethylcarbamate (2ba): The reaction of O-4-chloro-3-fluorophenyl N,N-diethylcarbamate (1b) (245 mg, 1 mmol), following the general procedure, yielded the product as a yellow oil (75% yield); Rf = 0.29 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.54–7.51 (m, 2H), 7.39–7.28 (m, 4H), 7.05 (dd, J = 8.9, 1.7 Hz, 1H), 3.53 (q, J = 7.0 Hz, 2H), 3.42 (q, J = 7.1 Hz, 2H), 1.31 (t, J = 7.1 Hz, 3H), 1.20 (t, J = 7.0 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 158.4 (d, J = 254.1 Hz, C), 153.1 (C), 151.7 (d, J = 2.3 Hz, C), 131.7 (2 × CH), 129.6 (CH), 129.1 (CH), 128.5 (2 × CH), 122.6 (C), 119.0 (d, J = 4.0 Hz, CH), 117.9 (d, J = 17.7 Hz, C), 109.0 (d, J = 17.8 Hz, C), 99.7 (d, J = 3.7 Hz, C), 77.7 (C), 42.6 (CH2), 42.3 (CH2), 14.3 (CH3), 13.4 (CH3). EI-LRMS m/z (%): 347 (M+ + 2, 58), 345 (M+, 66), 181 (58), 100 (100), 72 (70). ESI-HRMS was calculated for C19H18ClFNO2 [M + H]+ 346.1005, and found 346.1013.
O-5-Chloro-3-fluoro-2-(phenylethynyl)phenyl N,N-diethyl carbamate (2ca): The reaction of O-3-chloro-5-fluorophenyl N,N-diethylcarbamate (1c) (245 mg, 1 mmol), following the general procedure, yielded the product as a yellow oil (80% yield); Rf = 0.31 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.51–7.48 (m, 2H), 7.37–7.32 (m, 3H), 7.14–7.13 (m, 1H), 7.03–7.00 (m, 1H), 3.49 (q, J = 7.1 Hz, 2H), 3.39 (q, J = 7.1 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H), 1.18 (t, J = 7.1 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 162.6 (d, J = 254.6 Hz, C), 153.4 (d, J = 5.6 Hz, C), 152.7 (C), 134.5 (d, J = 12.6 Hz, C), 131.6 (2 × CH), 129.0 (CH), 128.4 (2 × CH), 122.7 (C), 119.5 (d, J = 3.7 Hz, CH), 113.4 (d, J = 24.7 Hz, CH), 106.4 (d, J = 18.1 Hz, C), 99.3 (d, J = 3.5 Hz, C), 42.63 (CH2), 42.3 (CH2), 14.2 (CH3), 13.4 (CH3). EI-LRMS m/z (%): 347 (M+ + 2, 5), 345 (M+, 15), 181 (30), 100 (100), 72 (45). EI-HRMS was calculated for C19H17ClFNO2 [M]+ 345.0932, and found 345.0936.
O-3-Chloro-2-(phenylethynyl)phenyl N,N-diethylcarbamate (2da): The reaction of O-3-chlorophenyl N,N-diethylcarbamate (1d) (227 mg, 1 mmol), following the general procedure, yielded the product as a yellow oil (88% yield); Rf = 0.27 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.60–7.53 (m, 2H), 7.46–7.28 (m, 5H), 7.23–7.16 (m, 1H), 3.56 (q, J = 7.0 Hz, 2H), 3.43 (q, J = 7.0 Hz, 2H), 1.33 (t, J = 7.0 Hz, 3H), 1.21 (t, J = 7.0 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 153.1 (C), 152.9 (C), 136.4 (C), 131.3 (2 × CH), 128.8 (CH), 128.6 (CH), 128.2 (2 × CH), 125.8 (CH), 122.6 (C), 120.9 (CH), 117.9 (C), 98.6 (C), 81.8 (C), 42.2 (CH2), 41.9 (CH2), 14.0 (CH3), 13.1 (CH3). EI-LRMS m/z (%): 329 (M+ + 2, 4), 327 (M+, 12), 100 (100), 72 (60). ESI-HRMS was calculated for C19H19ClNO2 [M + H]+ 328.1099, and found 328.1105.
O-3,6-Dichloro-2-(phenylethynyl)phenyl N,N-diethylcarbamate (2ea): The reaction of O-3,6-dichlorophenyl N,N-diethylcarbamate (1e) (262 mg, 1 mmol), following the general procedure, yielded the product as a colorless solid (70% yield); mp = 119–121 °C; Rf = 0.32 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.55–7.52 (m, 2H), 7.39–7.29 (m, 5H), 3.56 (q, J = 7.1 Hz, 2H), 3.44 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.1 Hz, 3H), 1.21 (t, J = 7.1 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 152.1 (C), 149.6 (C), 135.1 (C), 131.9 (2 × CH), 129.6 (CH), 129.1 (CH), 128.5 (2 × CH), 127.0 (C), 126.9 (CH), 122.7 (C), 120.8 (C), 99.8 (C), 81.5 (C), 42.8 (CH2), 42.4 (CH2), 14.3 (CH3), 13.5 (CH3). EI-LRMS m/z (%): 363 (M+ + 2, 1), 361 (M+), 100 (100), 72 (42). ESI-HRMS was calculated for C19H18Cl2NO2 [M + H]+ 362.0709, and found 362.0716.
O-3-Fluoro-2-((4-methoxyphenyl)ethynyl)phenyl N,N-diethylcarbamate (2ab): The reaction of O-3-fluorophenyl N,N-diethylcarbamate (1a) (211 mg, 1 mmol), following the general procedure, yielded the product as a colorless oil (71% yield); Rf = 0.24 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.48–7.44 (m, 2H), 7.32–7.25 (m, 1H), 7.07 (dt, J = 8.3, 1.0 Hz, 1H), 6.99 (td, J = 8.3, 1.0 Hz, 1H), 6.91–6.86 (m, 2H), 3.84 (s, 3H), 3.53 (q, J = 6.8 Hz, 2H), 3.42 (q, J = 7.0 Hz, 2H), 1.31 (t, J = 6.8 Hz, 3H), 1.20 (t, J = 7.0 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 163.0 (d, J = 251.9 Hz, C), 160.1 (C), 153.4 (C), 153.2 (d, J = 4.2 Hz, C), 133.2 (2 × CH), 128.9 (d, J = 9.6 Hz, CH), 118.5 (d, J = 3.3 Hz, CH), 115.2 (C), 114.1 (2 × CH), 112.3 (d, J = 21.1 Hz, CH), 107.8 (d, J = 18.0 Hz, C), 98.7 (C), 77.2 (C), 55.4 (CH3), 42.5 (CH2), 42.2 (CH2), 14.3 (CH3), 13.5 (CH3). EI-LRMS m/z (%): 341 (M+, 8), 100 (100), 72 (84). ESI-HRMS was calculated for C20H21FNO3 [M + H]+ 342.1500, and found 342.1504.
O-2-((4-Chlorophenyl)ethynyl)-3-fluorophenyl N,N-diethylcarbamate (2ac): The reaction of O-3-fluorophenyl N,N-diethylcarbamate (1a) (211 mg, 1 mmol), following the general procedure, yielded the product as a colorless solid (68% yield); mp = 60–62 °C; Rf = 0.14 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.46–7.43 (m, 2H), 7.36–7.30 (m, 3H), 7.08 (d, J = 8.3 Hz, 1H), 7.00 (td, J = 8.3, 0.9 Hz, 1H), 3.52 (q, J = 7.0 Hz, 2H), 3.42 (q, J = 7.0 Hz, 2H), 1.30 (t, J = 7.0 Hz, 3H), 1.20 (t, J = 7.0 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 163.1 (d, J = 252.7 Hz, C), 153.36 (d, J = 4.1 Hz, C), 153.2 (C), 134.9 (C), 132.9 (2 × CH), 129.6 (d, J = 9.7 Hz, CH), 128.8 (2 × CH), 121.5 (C), 118.5 (d, J = 3.3 Hz, CH), 112.4 (d, J = 21.0 Hz, CH), 107.1 (d, J = 18.0 Hz, C), 97.3 (C), 79.6 (C), 42.6 (CH2), 42.2 (CH2), 14.3 (CH3), 13.4 (CH3). EI-LRMS m/z (%): 345 (M+, 6), 100 (100), 72 (62). ESI-HRMS was calculated for C19H18ClFNO2 [M + H]+ 346.1005, and found 346.1007.
O-2-((4-Chlorophenyl)ethynyl)-3-chlorophenyl N,N-diethylcarbamate (2dc): The reaction of O-3-chlorophenyl N,N-diethylcarbamate (1d) (227 mg, 1 mmol), following the general procedure, yielded the product as a colorless solid (79% yield); mp = 80–82 °C; Rf = 0.19 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.47–7.44 (m, 2H), 7.36–7.25 (m, 4H), 7.17 (dd, J = 7.3, 2.1 Hz, 1H), 3.52 (q, J = 7.1 Hz, 2H), 3.42 (q, J = 7.1 Hz, 2H), 1.30 (t, J = 7.1 Hz, 3H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 153.3 (C), 136.8 (C), 134.9 (C), 132.9 (2 × CH), 129.3 (CH), 129.0 (C), 128.8 (2 × CH), 126.3 (CH), 121.5 (C), 121.2 (CH), 117.9 (C), 97.6 (C), 83.0 (C), 42.6 (CH2), 42.2 (CH2), 14.3 (CH3), 13.5 (CH3). EI-LRMS m/z (%): 363 (M+ + 2, 4), 361 (M+, 2), 100 (100), 72 (50). ESI-HRMS was calculated for C19H18Cl2NO2 [M + H]+ 362.0709, and found 362.0715.
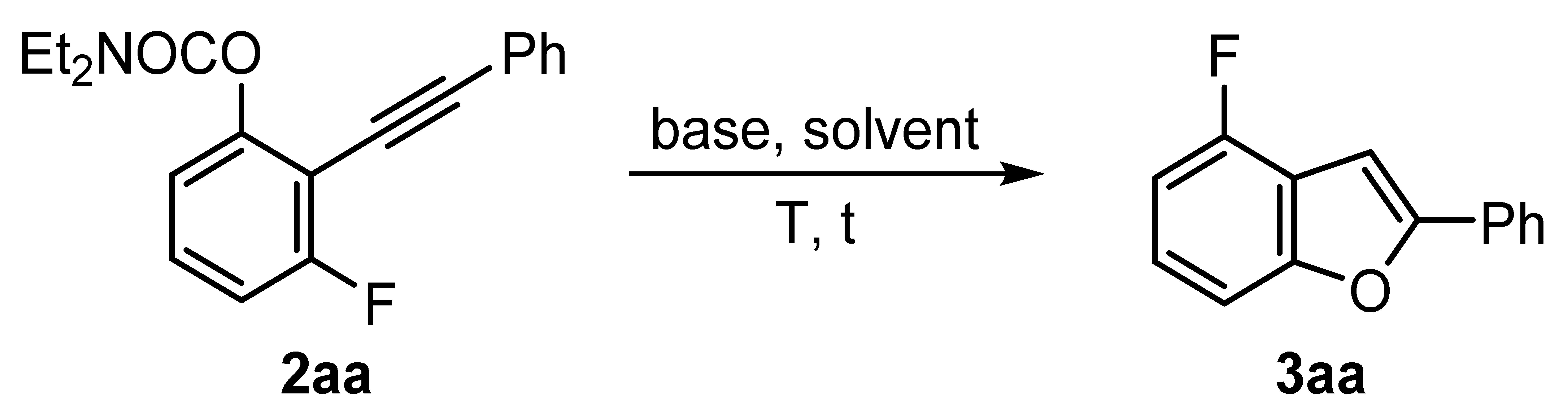
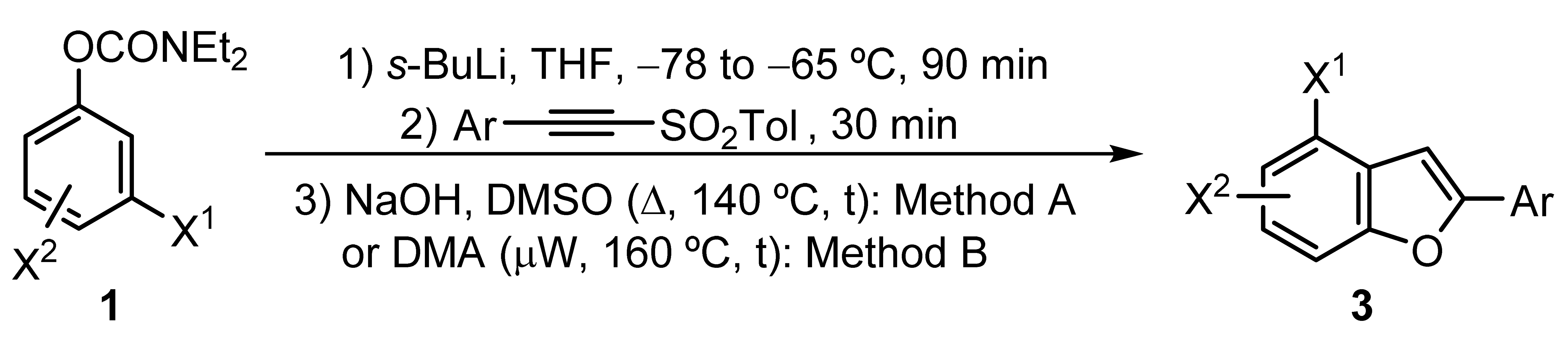
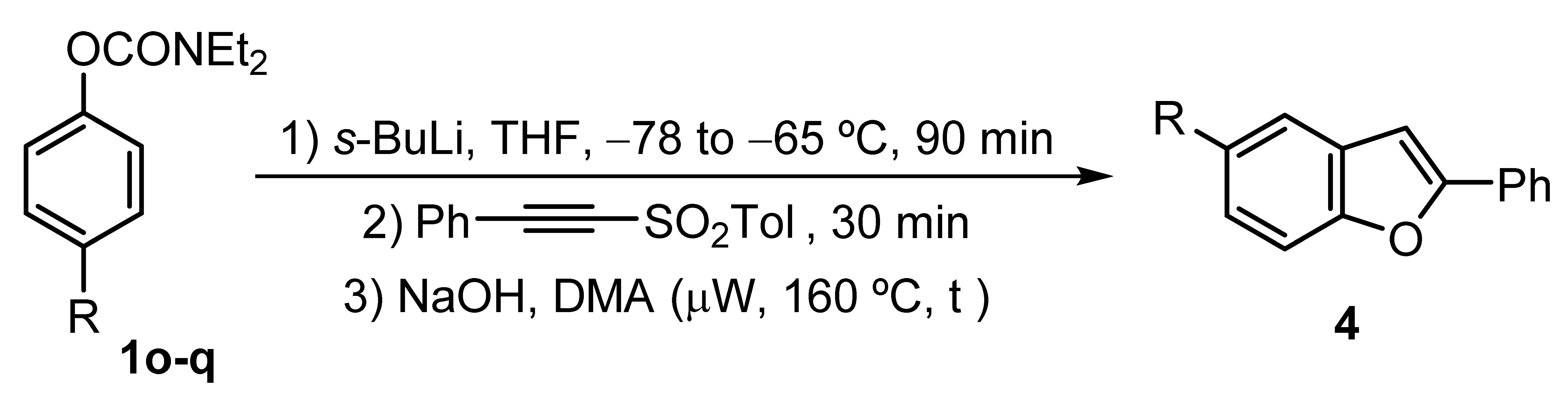
3.2.2. Synthesis of 2-Arylbenzo[b]furans 3 and 4
General procedure: A solution of starting carbamate
1 (1 mmol) in THF (2 mL) at –78 °C under nitrogen atmosphere was treated with a solution of
s-BuLi (0.85 mL of a 1.4 M solution in cyclohexane, 1.2 mmol). The reaction mixture was allowed to reach –65 °C for 5 min (–70 °C for 4-substituted carbamates
1o-q) and was stirred at this temperature for 90 min. Then, the corresponding alkynylsulfone (1.3 mmol) was added, and the resulting solution was allowed to warm to room temperature and was then stirred for 30 min. The solvents were removed under reduced pressure and the crude solution was treated with NaOH (2 mmol, 80 mg) in DMA (2 mL) or DMSO (2 mL). The resulting solution was warmed by conventional heating (Method A: DMSO, 140 °C) or under microwave irradiation (Method B: DMA, 160 °C), and stirred at this temperature until the completion of the reaction (see
Table 3). The mixture was diluted with EtOAc and NH
4Cl (aq), and the layers were separated. The aqueous phase was extracted with EtOAc (3 × 10 mL), and the combined organic layers were dried over anhydrous Na
2SO
4. The solvents were removed under reduced pressure, and the residue was purified by silica gel column chromatography (hexane/EtOAc), affording the 2-arylbenzo[
b]furans
3 and
4.
4-Fluoro-2-phenylbenzo[b]furan (
3aa) [
17]: The reaction of
O-3-fluorophenyl
N,N-diethylcarbamate (
1a) (211 mg, 1 mmol), following the general procedure with Method A, yielded the product as a white solid (51% yield);
Rf = 0.16 (hexane/EtOAc, 10:1).
1H NMR (300 MHz, CDCl
3) δ (ppm): 7.96–7.87 (m, 2H), 7.56–7.35 (m, 4H), 7.33–7.25 (m, 1H), 7.15 (d,
J = 0.8 Hz, 1H), 7.03–6.96 (m, 1H).
13C NMR (75.4 MHz, CDCl
3) δ (ppm): 156.8 (d,
J = 9.8 Hz, C), 156.1 (C), 156.0 (d,
J = 249.5 Hz, C), 130.0 (C), 129.0 (CH), 128.9 (2 × CH), 125.1 (2 × CH), 124.7 (d,
J = 7.6 Hz, CH), 118.5 (d,
J = 22.0 Hz, C), 108.6 (d,
J = 18.9 Hz, CH), 107.5 (d,
J = 4.1 Hz, CH), 97.4 (d,
J = 1.7 Hz, CH). EI-LRMS
m/z (%): 212 (M
+, 60), 193 (100), 116 (42).
4-Fluoro-2-(4-methoxyphenyl)benzo[b]furan (3ab): The reaction of O-3-fluorophenyl N,N-diethylcarbamate (1a) (211 mg, 1 mmol), following the general procedure with Method B, yielded the product as a colorless solid (62% yield); mp = 99–101 °C; Rf = 0.33 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.84–7.81 (m, 2H), 7.33 (dd, J = 8.2, 0.7 Hz, 1H), 7.24–7.19 (m, 1H), 7.03–6.9 (m, 4H), 3.89 (s, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 160.4 (C), 156.3 (C), 155.9 (d, J = 248.9 Hz, C), 126.7 (2 × CH), 124.2 (d, J = 7.4 Hz, CH), 122.9 (C), 118.8 (d, J = 21.8 Hz, C), 114.5 (2 × CH), 108.5 (d, J = 19.0 Hz, CH), 107.4 (d, J = 3.6 Hz, CH), 95.8 (CH), 55.5 (CH3), one aromatic C does not appear due to overlapping. EI-LRMS m/z (%): 242 (M+, 100), 227 (84), 199 (80), 170 (28). ESI-HRMS was calculated for C15H12FO2 [M + H]+ 243.0816, and found 243.0819.
2-(4-Chlorophenyl)-4-fluorobenzo[b]furan (3ac): The reaction of O-3-fluorophenyl N,N-diethylcarbamate (1a) (211 mg, 1 mmol), following the general procedure with Method B, yielded the product as a colorless solid (72% yield); mp = 105–107 °C; Rf = 0.32 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.83–7.78 (m, 2H), 7.47–7.43 (m, 2H), 7.33–7.24 (m, 2H), 7.10 (d, J = 0.9 Hz, 1H), 6.96 (ddd, J = 9.4, 8.0, 0.9 Hz, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 156.8 (d, J = 9.7 Hz, C), 156.0 (d, J = 250.0 Hz, C), 155.0 (C), 134.9 (C), 129.3 (2 × CH), 128.6 (C), 126.4 (2 × CH), 125.1 (d, J = 7.6 Hz, CH), 118.5 (d, J = 21.9 Hz, C), 108.7 (d, J = 18.9 Hz, CH), 107.5 (d, J = 4.1 Hz, CH), 97.9 (d, J = 1.2 Hz, CH). EI-LRMS m/z (%): 246 (M+, 100), 127 (12), 183 (46). APCI-HRMS was calculated for C14H9ClFO [M + H]+ 247.0320, and found 247.0324.
5-Chloro-4-fluoro-2-phenylbenzo[b]furan (3ba): The reaction of O-4-chloro-3-fluorophenyl N,N-diethylcarbamate (1b) (245 mg, 1 mmol), following the general procedure with Method B, yielded the product as a colorless solid (52% yield); mp = 92–94 °C; Rf = 0.13 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.87–7.84 (m, 2H), 7.49–7.42 (m, 3H), 7.28–7.27 (m, 2H), 7.08 (d, J = 0.4 Hz, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.3 (C), 154.9 (d, J = 8.9 Hz, C), 151.0 (d, J = 252.1 Hz, C), 129.6 (C), 129. (CH), 129.0 (2 × CH), 125.4 (CH), 125.2 (2 × CH), 119.7 (d, J = 21.0 Hz, C), 114.3 (d, J = 15.9 Hz, C), 108.0 (d, J = 4.4 Hz, CH), 97.3 (d, J = 1.6 Hz, CH). EI-LRMS m/z (%): 248 (M+ + 2, 35), 246 (M+, 100), 183 (32), 81 (19). APCI-HRMS was calculated for C14H9ClFO [M + H]+ 247.0320, found 247.0326.
6-Chloro-4-fluoro-2-phenylbenzo[b]furan (3ca): The reaction of O-5-chloro-3-fluorophenyl N,N-diethylcarbamate (1c) (245 mg, 1 mmol), following the general procedure with Method B, yielded the product as a colorless solid (47% yield); mp = 78–80 °C; Rf = 0.14 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.85–7.82 (m, 2H), 7.50–7.35 (m, 4H), 7.03 (d, J = 0.9 Hz, 1H), 6.99 (dd, J = 9.2, 1.6 Hz, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 156.7 (d, J = 1.0 Hz, C), 156.3 (d, J = 11.3 Hz, C), 155.1 (d, J = 252.7 Hz, C), 130.0 (d, J = 10.1 Hz, C), 129.5 (C), 129.3 (CH), 129.0 (2 × CH), 125.1 (2 × CH), 117.4 (d, J = 21.8 Hz, C), 110.2 (d, J = 22.7 Hz, CH), 108.3 (d, J = 4.5 Hz, CH), 97.2 (d, J = 2.0 Hz, CH). EI-LRMS m/z (%): 248 (M+ + 2, 35), 246 (M+, 100), 183 (36), 81 (17). APCI-HRMS was calculated for C14H9ClFO [M + H]+ 247.0320, and found 247.0325.
4-Chloro-2-phenylbenzo[b]furan (
3da) [
17]: The reaction of
O-3-chlorophenyl
N,N-diethylcarbamate (
1d) (227 mg, 1 mmol), following the general procedure with Method A, yielded the product as a white solid (67% yield);
Rf = 0.18 (hexane/EtOAc, 10:1).
1H NMR (300 MHz, CDCl
3) δ (ppm): 7.92–7.89 (m, 2H), 7.52–7.41 (m, 4H), 7.27–7.23 (m, 2H), 7.14 (d,
J = 0.9 Hz, 1H).
13C NMR (75.4 MHz, CDCl
3) δ (ppm): 156.7 (C), 155.2 (C), 130.0 (C), 129.1 (CH), 129.0 (2 × CH), 128.9 (C, 125.9 (C), 125.2 (2 × CH), 124.9 (CH), 123.0 (CH), 109.8 (CH), 99.9 (CH). EI-LRMS
m/z (%): 230 (M
+ + 2, 12), 228 (M
+, 36), 193 (100), 116 (51).
4,7-Dichloro-2-phenylbenzo[b]furan (3ea): The reaction of O-3,5-dichlorophenyl N,N-diethylcarbamate (1e) (262 mg, 1 mmol), following the general procedure with Method A, yielded the product as a colorless solid (52% yield); mp = 109–111 °C; Rf = 0.33 (hexane). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.95–7.91 (m, 2H), 7.53–7.43 (m, 3H), 7.23 (d, J = 8.4 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.14 (s, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.5 (C), 150.5 (C), 130.0 (C), 129.5 (CH), 129.3 (C), 129.0 (2 × CH), 125.3 (2 × CH), 124.8 (CH), 124.3 (C), 123.7 (CH), 115.4 (C), 100.4 (CH). EI-LRMS m/z (%): 264 (M+ + 2, 64), 262 (M+, 100), 199 (22), 163 (24). APCI-HRMS was calculated for C14H9Cl2O [M + H]+ 263.0025, and found 263.0028.
4-Chloro-5-fluoro-2-phenylbenzo[b]furan (3fa): The reaction of O-3,4-difluorophenyl N,N-diethylcarbamate (1f) (245 mg, 1 mmol), following the general procedure with Method A, yielded the product as a light yellow solid (57% yield); mp = 103–105 °C; Rf = 0.13 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.89 (d, J = 7.8 Hz, 2H), 7.53–7.35 (m, 4H), 7.17–7.05 (m, 4H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 158.3 (C), 154.9 (d, J = 241.1 Hz, C), 150.6 (C), 129.9 (d, J = 2.5 Hz, C), 129.6 (C), 129.4 (CH), 129.0 (2 × CH), 125.2 (2 × CH), 112.4 (d, J = 25.5 Hz, CH), 111.6 (d, J = 21.3 Hz, C), 110.0 (d, J = 8.4 Hz, CH), 100.1 (d, J = 4.4 Hz, CH). EI-LRMS m/z (%): 248 (M+ + 2, 9), 246 (M+, 27), 193 (100), 116 (51). APCI-HRMS was calculated for C14H9ClFO [M + H]+ 247.0320, and found 247.0327.
4,5-Difluoro-2-phenylbenzo[b]furan (3ga): The reaction of O-3,4-difluorophenyl N,N-diethylcarbamate (1g) (229 mg, 1 mmol), following the general procedure with Method B, yielded the product as a colorless solid (53% yield); mp = 86–88 °C; Rf = 0.15 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.91–7.79 (m, 2H), 7.51–7.36 (m, 3H), 7.27– 7.19 (m, 1H), 7.15–7.02 (m, 2H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.6 (C), 151.7 (d, J = 7.9 Hz, C), 146.5 (dd, J = 238.8, 11.1 Hz, C), 142.8 (dd, J = 251.4, 15.3 Hz, C), 129.6 (C), 129.4 (CH), 129.0 (2 × CH), 125.2 (2 × CH), 120.0 (dd, J = 17.6, 2.4 Hz), 113.0 (d, J = 21.8 Hz, CH), 106.8 (dd, J = 7.4, 4.5 Hz, CH), 97.8 (dd, J = 4.6, 1.6 Hz, CH). EI-LRMS m/z (%): 230 (M+, 31), 193 (100), 116 (51). APCI-HRMS was calculated for C14H9F2O [M + H]+ 231.0616, and found 231.0621.
4,5-Dichloro-2-phenylbenzo[b]furan (3ha): The reaction of O-3,4-dichlorophenyl N,N-diethylcarbamate (1h) (262 mg, 1 mmol), following the general procedure with Method A, yielded the product as a colorless solid (70% yield); mp = 114–116 °C; Rf = 0.25 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.90–7.79 (m, 2H), 7.52–7.32 (m, 5H), 7.09–7.05 (m, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.7 (C), 153.0 (C), 130.2 (C), 129.5 (C), 129.4 (CH), 128.9 (2 × CH), 126.7 (C), 125.4 (CH), 125.2 (2 × CH), 123.7 (C), 110.5 (CH), 100.2 (CH). EI-LRMS m/z (%): 265 (M + + 4, 3), 263 (M+ + 2, 9), 261 ( 27), 211 (41), 193 (100). APCI-HRMS was calculated for C14H9Cl2O [M + H]+ 263.0025, and found 263.0032.
4,5-Dichloro-2-(4-methoxyphenyl)benzo[b]furan (3hb): The reaction of O-3,4-dichlorophenyl N,N-diethylcarbamate (1h) (262 mg, 1 mmol), following the general procedure with Method B, yielded the product as a colorless solid (65% yield); mp = 127–129 °C; Rf = 0.35 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.81–7.78 (m, 2H), 7.36–7.28 (m, 2H), 7.01–6.98 (m, 2H), 6.94 (d, J = 0.6 Hz, 1H), 3.89 (s, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 160.7 (C), 158.1 (C), 152.9 (C), 130.6 (C), 126.8 (2 × CH), 126.7 (C), 125.0 (CH), 123.4 (C), 122.4 (C), 114.5 (2 × CH), 110.4 (CH), 98.6 (CH), 55.5 (CH3). EI-LRMS m/z (%): 294 (M+ + 2, 80), 292 (M+, 100), 278 (75), 249 (38). ESI-HRMS was calculated for C15H11Cl2O2 [M + H]+ 293.0131, and found 293.0131.
4,6-Dichloro-2-phenylbenzo[b]furan (3ia): The reaction of O-3,5-dichlorophenyl N,N-diethylcarbamate (1i) (262 mg, 1 mmol), following the general procedure with Method A yielded the product as a colorless solid (46% yield); mp = 92–94 °C; Rf = 0.11 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.88–7.85 (m, 2H), 7.51–7.42 (m, 4H), 7.27 (d, J = 1.6 Hz, 1H), 7.05 (d, J = 0.9 Hz, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.4 (C), 154.8 (C), 130.1 (C), 129.6 (C), 129.4 (CH), 129.1 (2 × CH), 127.7 (C), 126.2 (C), 125.2 (2 × CH), 123.6 (CH), 110.6 (CH), 99.7 (CH) EI-LRMS m/z (%): 265 (M++4, 6), 263 (M+ + 2, 18), 261 (M+, 54), 211 (42), 193 (100). APCI-HRMS was calculated for C14H9Cl2O [M + H]+ 263.0025, and found 263.0027.
4,6-Dichloro-2-(4-chlorophenyl)benzo[b]furan (3ic): The reaction of O-3,5-dichlorophenyl N,N-diethylcarbamate (1i) (262 mg, 1 mmol), following the general procedure with Method B yielded the product as a colorless solid (68% yield); mp = 143–145 °C; Rf = 0.45 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.77–7.74 (m, 2H), 7.46–7.42 (m, 3H), 7.26 (d, J = 1.6 Hz, 1H), 7.03 (d, J = 0.8 Hz, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 156.2 (C), 154.8 (C), 135.3 (C), 130.4 (C), 129.3 (2 × CH), 128.0 (C), 127.6 (C), 126.4 (2 × CH), 126.3 (C), 123.7 (CH), 110.6 (CH), 100.1 (CH) EI-LRMS m/z (%): 298 (M+ + 2, 100), 296 (M+, 98), 233 (51), 163 (62). ESI-HRMS could not be recorded.
5-Bromo-4-fluoro-2-phenylbenzo[b]furan (3ja): The reaction of O-4-bromo-3-fluorophenyl N,N-diethylcarbamate (1j) (290 mg, 1 mmol), following the general procedure with Method B yielded the product as a yellow solid (53% yield); mp = 119–121 °C; Rf = 0.33 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.88–7.85 (m, 2H), 7.51–7.39 (m, 4H), 7.24 (d, J = 8.7 Hz, 1H), 7.09 (s, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.2 (C), 155.6 (d, J = 9.0 Hz, C), 152.0 (d, J = 250.4 Hz, C), 129.6 (C), 129.4 (CH), 129.1 (2 × CH), 128.0 (CH), 125.3 (2 × CH), 119.8 (d, J = 22.2 Hz, C), 108.6 (d, J = 4.2 Hz, CH), 101.7 (d, J = 19.2 Hz, C), 97.3 (d, J = 1.9 Hz, CH). EI-LRMS m/z (%): 292 (M+ + 2, 100), 290 (M+, 93), 183 (17). APCI-HRMS was calculated for C14H9BrFO [M + H]+ 290.9815, and found 290.9824.
5-Bromo-4-chloro-2-phenylbenzo[b]furan (3ka): The reaction of O-4-bromo-3-chlorophenyl N,N-diethylcarbamate (1k) (306 mg, 1 mmol), following the general procedure with Method A yielded the product as a colorless solid (60% yield); mp = 118–120 °C; Rf = 0.38 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.80 (dd, J = 8.1, 1.3 Hz, 2H), 7.49–7.38 (m, 4H), 7.24 (dd, J = 8.7, 0.8 Hz, 1H), 7.00 (s, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.5 (C), 153.5 (C), 130.4 (C), 129.4 (C), 129.3 (CH), 128.9 (2 × CH), 128.4 (CH), 125.7 (C), 125.2 (2 × CH), 116.1 (C), 110.9 (CH), 100.3 (CH). EI-LRMS m/z (%): 309 (M+ + 4, 11), 307 (M+ + 2, 11), 305 (M+, 33), 193 (100). APCI-HRMS was calculated for C14H9BrClO [M + H]+ 306.9520, and found 306.9524.
4,7-Difluoro-2-phenylbenzo[b]furan (3la): The reaction of O-2,5-difluorophenyl N,N-diethylcarbamate (1l) (229 mg, 1 mmol), following the general procedure with Method A yielded the product as a colorless solid (44% yield); mp = 97–99 °C; Rf = 0.41 (hexane). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.92–7.89 (m, 2H), 7.52–7.43 (M, 3H), 7.13 (d, J = 2.6 Hz, 1H), 6.97 (ddd, J = 9.3, 3.9 Hz, 1H), 6.86 (ddd, J = 8.8, 3.1 Hz, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.1 (CH), 151.54 (d, J = 245.7 Hz, C), 151.50 (d, J = 245.7 Hz, C), 146.2 (d, J = 3.9 Hz, C), 143.0 (d, J = 3.9 Hz, C), 129.4 (CH), 129.3 (C), 129.0 (2 × CH), 125.3 (2 × CH), 121.2 (dd, J = 24.2, 3.1 Hz, C), 110.4 (dd, J = 19.1, 8.1 Hz, CH), 108.4 (dd, J = 21.9, 6.4 Hz, CH), 97.8 (t, J = 1.9 Hz, CH). EI-LRMS m/z (%): 230 (M+, 100), 201 (42), 181 (8). ESI-HRMS was calculated for C14H9F2O [M + H]+ 231.0616, and found 231.0615.
7-Bromo-4-fluoro-2-phenylbenz[b]ofuran (3ma): The reaction of O-2-bromo-5-fluorophenyl N,N-diethylcarbamate (1m) (290 mg, 1 mmol), following the general procedure with Method A yielded the product as a colorless solid (41% yield); mp = 84–86 °C; Rf = 0.31 (hexane). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.94–7.91 (m, 2H), 7.53–7.36 (m, 4H), 7.17 (s, 1H), 6.91–6.85 (m, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.0 (C), 155.3 (d, J = 243.9 Hz, C), 129.5 (CH), 129.5 (C), 129.1 (2 × CH), 127.3 (d, J = 7.4 Hz, CH), 125.4 (2 × CH), 125.2 (C), 119.7 (d, J = 23.0 Hz, C), 110.2 (d, J = 20.6 Hz, CH), 98.8 (d, J = 4.4 Hz, C), 98.2 (d, J = 1.6 Hz, CH). EI-LRMS m/z (%): 291 (M+ + 2, 98), 289 (M+, 100), 211 (40). ESI-HRMS could not be recorded.
4-Bromo-2-phenylbenzo[b]furan (
3na) [
17]: The reaction of
O-3-bromophenyl
N,N-diethylcarbamate (
1n) (272 mg, 1 mmol), following the general procedure with Method A yielded the product as a colorless solid (56% yield);
Rf = 0.18 (hexane/EtOAc, 10:1)).
1H NMR (300 MHz, CDCl
3) δ (ppm): 7.92–7.88 (m, 2H), 7.59–7.46 (m, 3H), 7.45–7.38 m, 2H), 7.17 (t,
J = 8.1 Hz, 1H), 7.08 (s, 1H).
13C NMR (75.4 MHz, CDCl
3) δ (ppm): 156.6 (C), 154.6 (C), 130.9 (CH), 129.9 (C), 129.1 (2 × CH), 128.9 (CH), 126.1 (2 × CH), 125.2 (C), 113.9 (C), 110.4 (CH), 102.4 (CH), 101.5 (CH). EI-LRMS
m/z (%): 274 (M
+ + 2, 96), 272 (M
+, 100).
5-Fluoro-2-phenylbenzo[b]furan (
4oa) [
34]: The reaction of
O-4-fluorophenyl
N,N-diethylcarbamate (
1o) (211 mg, 1 mmol), following the general procedure with Method B yielded the product as a colorless solid (59% yield);
Rf = 0.42 (hexane/EtOAc, 5:1).
1H NMR (300 MHz, CDCl
3) δ (ppm): 7.91–7.87 (m, 2H), 7.53–7.39 (m, 4H), 7.27 (dd,
J = 8.6, 2.6 Hz, 1H), 7.05 (dt,
J = 8.6, 2.6 Hz, 1H), 7.00 (d,
J = 0.8 Hz, 1H).
13C NMR (75.4 MHz, CDCl
3) δ (ppm): 159.5 (d,
J = 237.9 Hz, C), 157.8 (C), 151.2 (C), 130.2 (C), 130.1 (d,
J = 11.5 Hz, C), 129.0 (CH), 128.9 (2 × CH), 125.1 (2 × CH), 112.0 (d,
J = 17.7 Hz, CH), 111.8 (CH), 106.4 (d,
J = 25.1 Hz, CH), 101.5 (d,
J = 4.0 Hz, CH). EI-LRMS
m/z (%): 212 (M
+, 100), 183 (66), 106 (16).
5-Chloro-2-phenylbenzo[b]furan (
4pa) [
35]: The reaction of
O-4-chlorophenyl
N,N-diethylcarbamate (
1p) (227 mg, 1 mmol), following the general procedure with Method B yielded the product as a colorless solid (55% yield);
Rf = 0.15 (hexane/EtOAc, 10:1).
1H NMR (300 MHz, CDCl
3) δ (ppm): 7.87–7.83 (m, 2H), 7.54 (d,
J = 2.1 Hz, 1H), 7.49–7.38 (m, 4H), 7.24 (dd,
J = 8.7, 2.1 Hz, 1H), 6.96 (s, 1H).
13C NMR (75.4 MHz, CDCl
3) δ (ppm): 157.5 (C), 153.3 (C), 130.7 (C), 130.1 (C), 129.1 (CH), 129.0 (2 × CH), 128.6 (C), 125.2 (2 × CH), 124.5 (CH), 120.5 (CH), 112.2 (CH), 100.9 (CH). EI-LRMS
m/z (%): 230 (M
+ + 2, 12), 228 (M
+, 36), 193 (100), 116 (51).
5-Methoxy-2-phenylbenzo[b]furan (
4qa) [
36]: The reaction of
O-4-methoxyphenyl
N,N-diethylcarbamate (
1q) (223 mg, 1 mmol), following the general procedure with Method B yielded the product as a colorless oil (56% yield);
Rf = 0.15 (hexane/EtOAc, 10:1).
1H NMR (300 MHz, CDCl
3) δ (ppm): 7.90–7.87 (m, 2H), 7.51–7.36 (m, 4H), 7.08 (d,
J = 2.6 Hz, 1H), 6.99 (d,
J = 0.9 Hz, 1H), 6.93 (dd,
J = 8.9, 2.6 Hz, 1H), 3.89 (s, 3H).
13C NMR (75.4 MHz, CDCl
3) δ (ppm): 156.8 (C), 156.2 (C), 150.1 (C), 130.7 (C), 129.9 (C), 128.9 (2 × CH), 128.6 (CH), 125.0 (2 × CH), 113.1 (CH), 111.7 (CH), 103.4 (CH), 101.6 (CH), 56.0 (CH
3). EI-LRMS
m/z (%): 224 (M
+, 100), 153 (44), 76 (41).
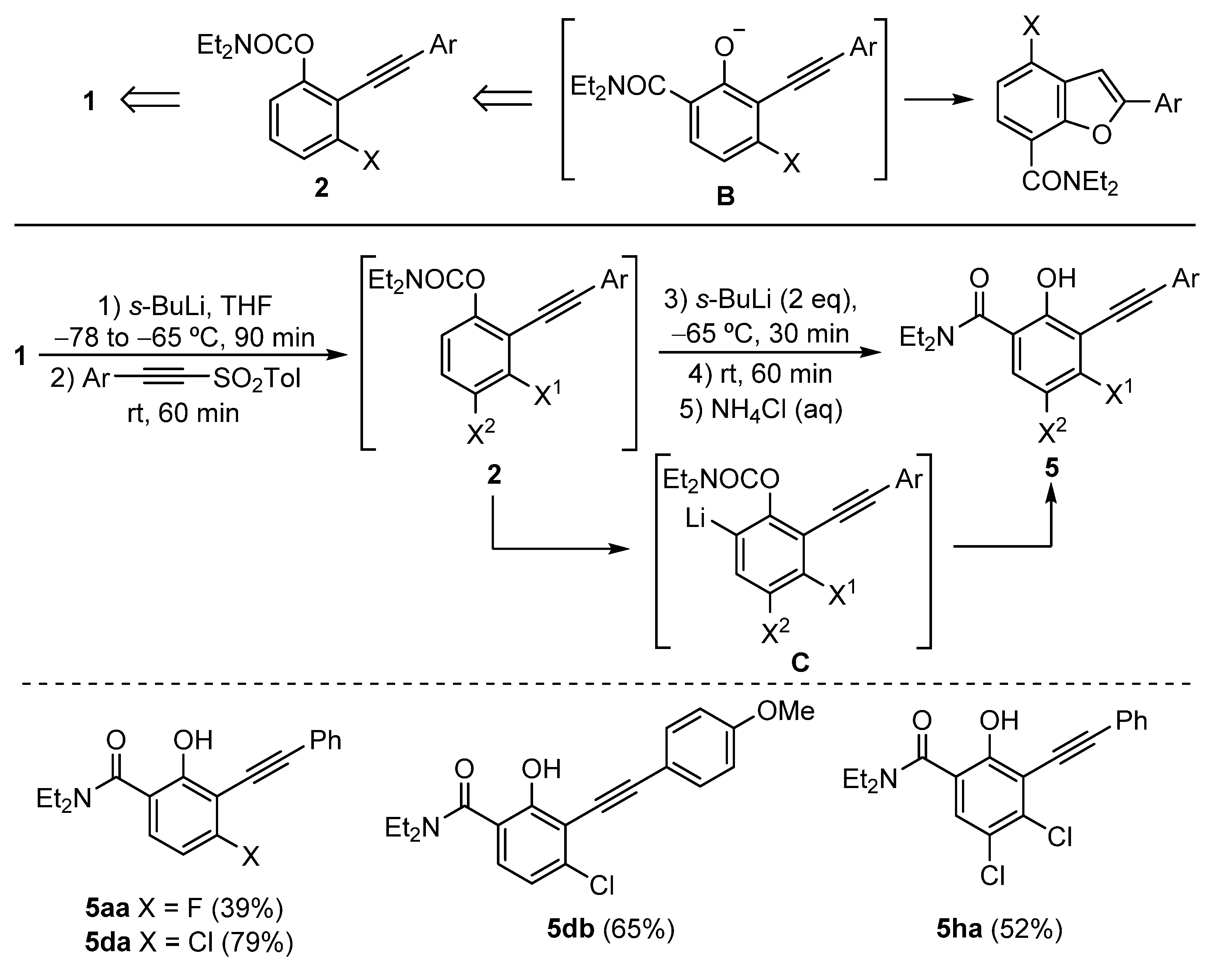
3.2.3. Synthesis of o-Alkynyl Salicylamides 5
General Procedure: A solution of starting carbamate 1 (1 mmol) in THF (2 mL) at −78 °C under nitrogen was treated with a solution of s-BuLi (0.85 mL of a 1.4 M solution in cyclohexane, 1.2 mmol). The reaction mixture was allowed to reach −65 °C for 5 min and then stirred at this temperature for 90 min. Then, the corresponding alkynyl sulfone (1.3 mmol) was added at −65 °C, and the resulting solution was allowed to reach room temperature and then stirred for 60 min. Then, it was treated with s-BuLi (1.42 mL of a 1.4 M solution in cyclohexane, 2 mmol) at −65 °C and stirred at that temperature for 30 min. The mixture was allowed to warm slowly to room temperature and then stirred for an additional 60 min. The reaction mixture was quenched with NH4Cl (aq) and diluted with EtOAc. The layers were separated, the aqueous phase was extracted with EtOAc (3 × 10 mL), and the combined organic layers were dried over anhydrous Na2SO4. The solvents were removed under reduced pressure, and the residue was purified by column chromatography (hexane/EtOAc), affording the N,N-diethyl-2-hydroxy-3-(phenylethynyl) benzamides 5.
N,N-Diethyl-4-fluoro-2-hydroxy-3-(phenylethynyl) benzamide (5aa): The reaction of O-3-fluorophenyl N,N-diethylcarbamate (1a) (211 mg, 1 mmol), following the general procedure, yielded the product as a yellow oil (39% yield); Rf = 0.10 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 10.28 (s, 1H), 7.63–7.60 (m, 2H), 7.39–7.37 (m, 3H), 7.26 (dd, J = 8.9, 6.3 Hz, 1H), 6.67 (t, J = 8.9 Hz, 1H), 3.52 (q, J = 7.1 Hz, 4H), 1.29 (t, J = 7.1 Hz, 6H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 170.3 ©, 164.4 (d, J = 255.6 Hz, C), 160.8 (d, J = 5.9 Hz, C), 131.94 (2 × CH), 128.8 (CH), 128.5 (2 × CH), 128.2 (CH), 125.2 (C), 122.9 (C), 115.0 (d, J = 3.1 Hz, C), 106.1 (d, J = 22.0 Hz, CH), 99.7 (C), 77.9 (C), 42.3 (2 × CH2), 13.5 (2 × CH3). EI-LRMS m/z (%): 311 (M+ + 2, 25), 310 (M+, 22), 239 (100), 183 (51). ESI-HRMS was calculated for C19H19FNO2 [M + H]+ 312.1394, and found 312.1400.
4-Chloro-N,N-diethyl-2-hydroxy-3-(phenylethynyl) benzamide (5da): The reaction of O-3-chlorophenyl N,N-diethylcarbamate (1d) (227 mg, 1 mmol), following the general procedure, yielded the product as a dark yellow oil (79% yield); Rf = 0.12 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 9.77 (s, 1H), 7.64–7.61 (m, 2H), 7.40–7.36 (m, 3H), 7.15 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 3.49 (q, J = 7.1 Hz, 4H), 3.25 (t, J = 7.1 Hz, 6H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 169.7 (C), 158.9 (C), 138.7 (C), 131.9 (2 × CH), 128.9 (CH), 128.4 (2 × CH), 127.5 (CH), 122.8 (C), 119.8 (CH), 118.2 (C), 113.1 (C), 100.1 (C), 81.5 (C), 42.1 (2 × CH2), 13.5 (2 × CH3). EI-LRMS m/z (%): 329 (M+ + 2, 1), 327 (M+, 3), 292 (32), 58 (100). ESI-HRMS was calculated for C19H19ClNO2 [M + H]+ 328.1099, and found 328.1105.
4-Chloro-N,N-diethyl-2-hydroxy-3-((4-methoxyphenyl)ethynyl)benzamide (5db): The reaction of O-3-chlorophenyl N,N-diethylcarbamate (1d) (227 mg, 1 mmol), following the general procedure, yielded the product as a yellow solid (65% yield); mp = 121–123 °C; Rf = 0.25 (hexane/EtOAc, 2:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 9.44 (s, 1H), 7.54 (d, J = 9.0 Hz, 2H), 7.13 (d, J = 8.4 Hz, 1H), 6.97 (d, J = 8.4 Hz, 1H), 6.89 (d, J = 9.0 Hz, 2H), 3.83 (s, 3H), 3.47 (q, J = 7.1 Hz, 4H), 1.24 (t, J = 7.1 Hz, 6H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 169.6 (C), 160.2 (C), 158.2 (C), 138.3 (C), 133.4 (2 × CH), 127.2 (CH), 119.9 (CH), 118.4 (C), 114.8 (C), 114.1 (2 × CH), 113.3 (C), 100.4 (C), 80.1 (c), 55.4 (CH3), 42.0 (2 × CH2), 13.4 (2 × CH3). EI-LRMS m/z (%): 359 (M+ + 2, 40), 357 (M+, 53), 285 (100), 151 (50), 42 (49). ESI-HRMS was calculated for C20H21ClNO3 [M + H]+ 358.1204, and found 358.1213.
4,5-Dichloro-N,N-diethyl-2-hydroxy-3-(phenylethynyl)benzamide (5ha): The reaction of O-3,4-dichlorophenyl N,N-diethylcarbamate (1h) (262 mg, 1 mmol), following the general procedure, yielded the product as a yellow solid (52% yield); mp = 155–157 °C; Rf = 0.18 (hexane/EtOAc, 2:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 9.34 (s, 1H), 7.63 (dd, J = 6.6, 3.1 Hz, 2H), 7.41–7.39 (m, 3H), 7.31 (s, 1H), 3.49 (q, J = 7.1 Hz, 4H), 1.27 (t, J = 7.1 Hz, 6H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 168.3 (C), 156.1 (C), 136.2 (C), 131.9 (2 × CH), 129.1 (CH), 128.5 (2 × CH), 127.9 (CH), 123.4 (C), 122.5 (C), 120.5 (C), 114.8 (C), 101.0 (C), 81.4 (C), 42.0 (2 × CH2), 13.4 (2 × CH3). EI-LRMS m/z (%): 363 (M+ + 2, 40), 361 (M+, 48), 289 (100), 163 (34). ESI-HRMS was calculated for C19H18Cl2NO2 [M + H]+ 362.0709, and found 362.0718.
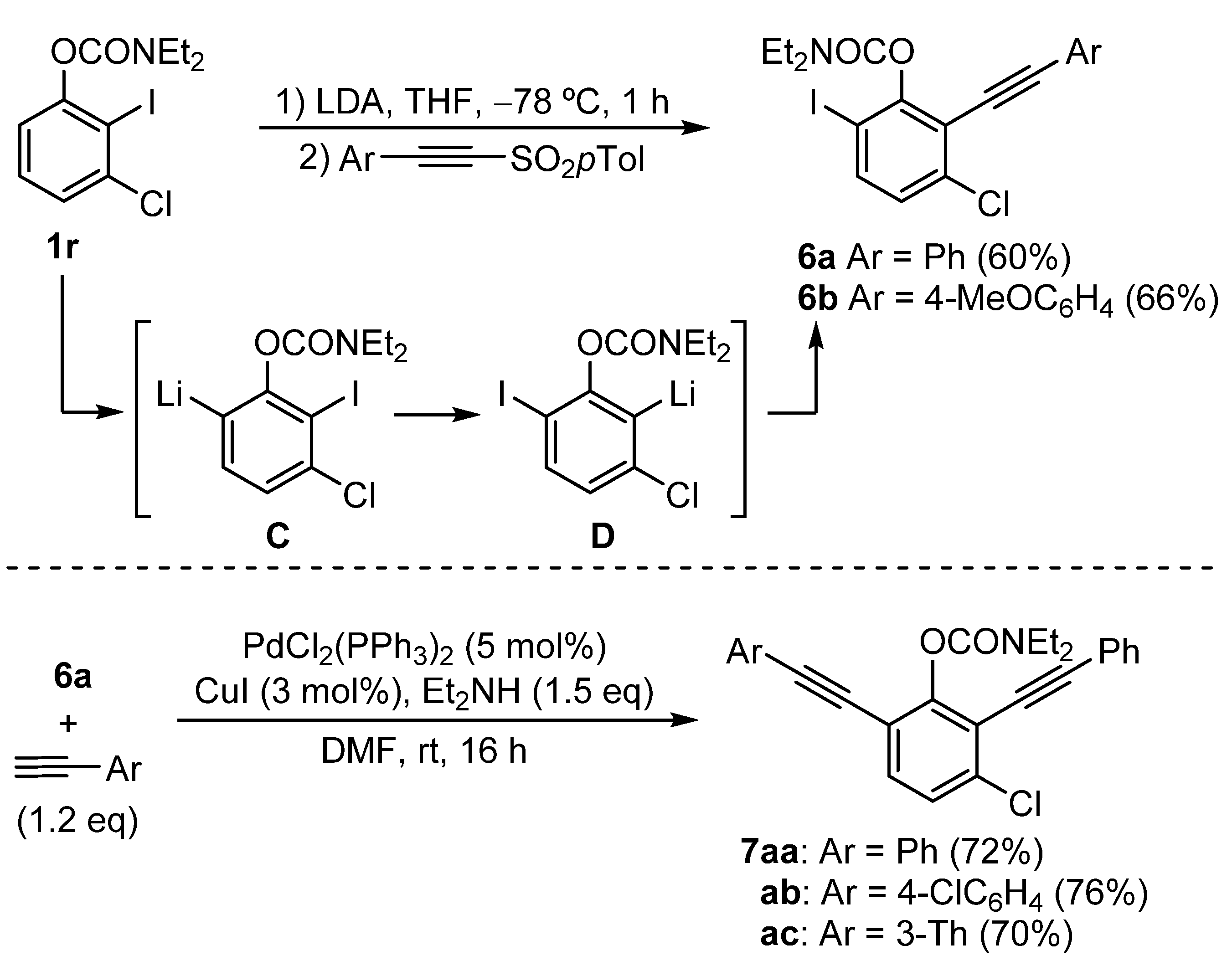
3.2.4. Synthesis of O-3-Chloro-6-iodo-2-(arylethynyl)phenyl N,N-Diethylcarbamates 6
General Procedure: A solution of O-3-chloro-2-iodophenyl N,N-diethylcarbamate (1r) (317 mg, 0.9 mmol) in THF (2 mL) at −78 °C under nitrogen was treated with a solution of LDA (1.08 mmol). The reaction mixture was stirred at −78 °C for 1 h. Then, the corresponding alkynyl sulfone (1.17 mmol) was added, and the resulting solution was stirred for 2 h at −78 °C. Finally, the resulting solution was warmed to room temperature and stirred overnight. The reaction mixture was quenched with NH4Cl (aq), and the aqueous phase was extracted with Et2O (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, and the solvents were removed under reduced pressure. The residue was purified by column chromatography (hexane/EtOAc), affording O-3-chloro-6-iodo-2-(arylethynyl)phenyl N,N-diethylcarbamates 6.
O-3-Chloro-6-iodo-2-(phenylethynyl)phenyl N,N-diethylcarbamate (6a): The use of 2-phenyl p-toluenesulfonylacetylene (300 mg), following the general procedure, yielded the product as a colorless solid (60% yield); mp = 87–89 °C; Rf = 0.27 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.69 (dd, J = 8.6, 0.9 Hz, 1H), 7.54–7.51 (m, 2H), 7.39–7.36 (m, 3H), 7.08 (dd, J = 8.6, 0.9 Hz, 1H), 3.60–3.55 (m, 2H), 3.46–3.42 (m, 2H), 1.39 (t, J = 7.1 Hz, 3H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 153.7 (C), 152.0 (C), 138.3 (2 × CH), 137.1 (C), 131.8 (2 × CH), 129.1 (CH), 128.5 (2 × CH), 127.7 (CH), 122.7 (C), 119.9 (C), 99.6 (C), 89.5 (C), 81.7 (C), 42.8 (CH2), 42.4 (CH2), 14.5 (CH3), 13.5 (CH3). EI-LRMS m/z (%): 455 (M+ + 2, 4), 453 (M+, 10), 163 (42), 100 (100). ESI-HRMS was calculated for C19H18ClINO2 [M + H]+ 454.0065, and found 454.0074.
O-3-Chloro-6-iodo-2-((4-methoxyphenyl)ethynyl)phenyl N,N-diethylcarbamate (6b): The use of 2-(4-methoxyphenyl) p-toluenesulfonylacetylene (334 mg), following the general procedure, yielded the product as a colorless solid (66% yield); mp = 88–90 °C; Rf = 0.46 (hexane/EtOAc, 3:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.66 (d, J = 8.6 Hz, 1H), 7.45 (d, J = 8.9 Hz, 2H), 7.06 (d, J = 8.6 Hz, 1H), 6.89 (d, J = 8.9 Hz, 2H), 3.84 (s, 3H), 3.61–3.52 (m, 2H), 3.49–3.39 (m, 2H), 1.38 (t, J = 7.1 Hz, 3H), 1.20 (t, J = 7.1 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 160.2 (C), 153.5 (C), 152.0 (C), 137.9 (CH), 136.7 (C), 133.3 (2 × CH), 127.6 (CH), 120.2 (C), 114.7 (C), 114.1 (2 × CH), 99.9 (C), 89.5 (C), 80.5 (C), 55.4 (CH3), 42.7 (CH2), 42.4 (CH2), 14.5 (CH3), 13.5 (CH3). EI-LRMS m/z (%): 485 (M+ + 2, 14) 483 (M+, 29), 100 (100), 72 (36). ESI-HRMS was calculated for C20H20ClINO3 [M + H]+ 484.0171, and found 484.0180.
3.2.5. Synthesis of O-3-Chloro-2,6-bis(alkynyl)phenyl N,N-Diethylcarbamates 7
General Procedure: To a solution of starting carbamate 6a (176 mg, 0.5 mmol) in DMF (3 mL) under nitrogen, Et2NH (55 mg, 0.75 mmol), the corresponding terminal alkyne (0.6 mmol), PdCl2(PPh3)2 (10.5 mg, 0.025 mmol), and CuI (2.9 mg, 0.015 mmol) were added. The resulting mixture was stirred at room temperature overnight. After completion of the reaction, the solvents were removed under reduced pressure, and the residue was purified by silica gel column chromatography (hexane/EtOAc = 10:1), affording the O-3-chloro-2,6-bis(alkynyl)phenyl N,N-diethylcarbamates 7.
O-3-Chloro-2,6-bis(phenylethynyl)phenyl N,N-diethyl carbamate (7aa): The use of phenylacetylene (61 mg), following the general procedure, yielded the product as a colorless solid (72% yield); mp = 94–96 °C; Rf = 0.17 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.59–7.49 (m, 4H), 7.46 (d, J = 8.4 Hz, 1H), 7.40–7.28 (m, 7H), 3.60 (q, J = 7.1 Hz, 2H), 3.45 (q, J = 7.1 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H), 1.18 (t, J = 7.1 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 154.0 (C), 152.7 (C), 136.6 (C), 132.1 (CH), 131.8 (2 × CH), 131.7 (2 × CH), 129.0 (CHs), 128.8 (CH), 128.5 (4 × CH), 126.4 (CH), 123.0 (C), 122.9 (C), 119.6 (C), 117.5 (C), 99.3 (C), 94.8 (C), 83.7 (C), 81.6 (C), 42.7 (CH2), 42.4 (CH2), 14.4 (CH3), 13.5 (CH3). EI-LRMS m/z (%): 429 (M+ + 2, 2), 427 (M+, 6), 100 (100), 72 (47). ESI-HRMS was calculated for C27H23ClNO2 [M + H]+ 428.1412, and found 428.1418.
O-3-Chloro-6-((4-chlorophenyl)ethynyl)-2-(phenylethynyl)phenyl N,N-diethylcarbamate (7ab): The use of 1-chloro-4-ethynylbenzene (82 mg), following the general procedure, yielded the product as a dark yellow oil (76% yield); Rf = 0.20 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.57–7.54 (m, 2H), 7.46–7.30 (m, 9H), 3.59 (q, J = 7.0 Hz, 2H), 3.45 (q, J = 7.0 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H), 1.18 (t, J = 7.1 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 154.0 (C), 152.6 (C), 136.8 (C), 134.8 (C), 132.8 (2 × CH), 132.0 (CH), 131.8 (2 × CH), 129.0 (CH), 128.8 (2 × CH), 128.4 (2 × CH), 126.4 (CH), 122.8 (C), 121.4 (C), 119.6 (C), 117.2 (C), 99.4 (C), 93.6 (C), 84.7 (C), 81.5 (C), 42.7 (CH2), 42.3 (CH2), 14.4 (CH3), 13.5 (CH3). EI-LRMS m/z (%): 463 (M+ + 2, 8), 461 (M+, 12), 100 (100), 72 (25). ESI-HRMS was calculated for C27H22Cl2NO2 [M + H]+ 462.1022, and found 462.1031.
O-3-Chloro-2-(phenylethynyl)-6-(thiophen-3-ylethynyl)phenyl N,N-diethylcarbamate (7ac): The use of 3-ethynylthiophene (65 mg), following the general procedure, yielded the product as a dark yellow oil (70% yield); Rf = 0.19 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.57–7.51 (m, 3H), 7.45–7.30 (m, 6H), 7.17 (d, J = 5.0 Hz, 1H), 3.58 (q, J = 7.0 Hz, 2H), 3.44 (q, J = 7.0 Hz, 2H), 1.38 (t, J = 7.0 Hz, 3H), 1.18 (t, J = 7.0 Hz, 3H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 153.9 (C), 152.6 (C), 136.5 (C), 132.0 (CH), 131.8 (2 × CH), 129.8 (CH), 129.2 (CH), 129.0 (CH), 128.4 (2 × CH), 126.4 (CH), 125.6 (CH), 122.9 (C), 122.0 (C), 119.6 (C), 117.4 (C), 99.3 (C), 90.0 (C), 83.2 (C), 81.6 (C), 42.7 (CH2), 42.3 (CH2), 14.4 (CH3), 13.5 (CH3). EI-LRMS m/z (%): 435 (M+ + 2, 10), 433 (M+, 28), 100 (100), 72 (33). ESI-HRMS was calculated for C25H21ClNO2S [M + H]+ 434.0976, and found 434.0982.
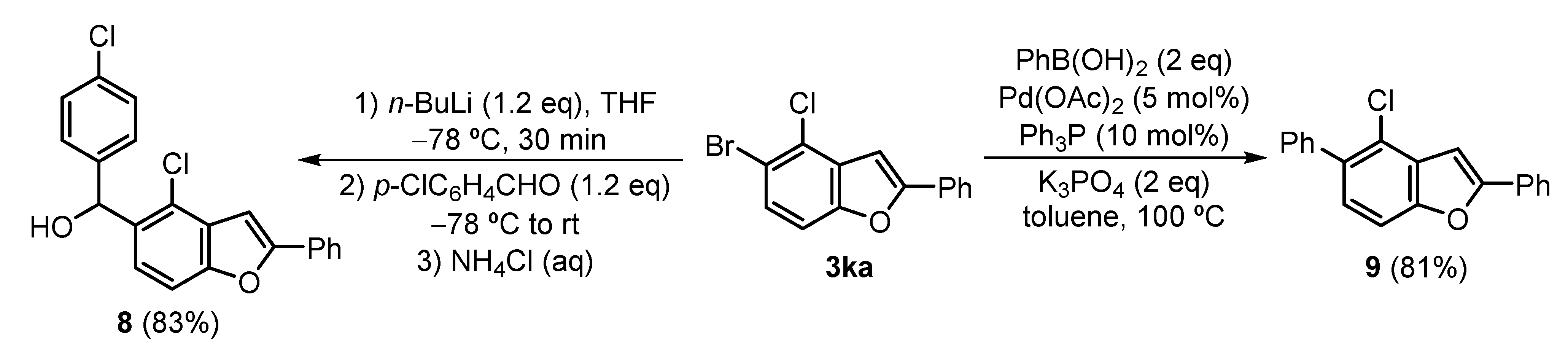
3.2.6. Synthesis of (4-Chloro-2-Phenylbenzofuran-5-yl)(4-Chlorophenyl) Methanol 8
A solution of starting carbamate 3ka (123 mg, 0.4 mmol) in THF (2 mL) at −78 °C under nitrogen was treated with a solution of n-BuLi (0.30 mL of a 1.6 M solution in hexane, 0.48 mmol). The reaction mixture was stirred at this temperature for 30 min. Then, 4-chlorobenzaldehyde (67 mg, 0.48 mmol) was added, and the resulting solution was warmed to room temperature. The mixture was quenched with aqueous NH4Cl, diluted with EtOAc, and the layers were separated. The aqueous phase was extracted with EtOAc (3 × 10 mL), and the combined organic layers were dried over anhydrous Na2SO4. The solvents were removed under reduced pressure, and the residue was purified by column chromatography (hexane/EtOAc = 5:1), affording functionalized benzo[b]furan derivative 8.
(4-Chloro-2-phenylbenzofuran-5-yl)(4-chlorophenyl) methanol (8): Colorless oil (83% yield); Rf = 0.26 (hexane/EtOAc, 5:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.89–7.63 (m, 2H), 7.48–7.34 (m, 9H), 7.10 (s, 1H), 6.34 (s, 1H), 2.58 (s, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.2 (C), 154.3 (C), 141.4 (C), 135.3 (C), 133.5 (C), 129.9 (C), 129.3 (C), 129.2 (CH), 129.0 (2 × CH), 128.7 (2 × CH), 128.1 (2 × CH), 127.4 (C), 125.2 (2 × CH), 123.8 (CH), 110.3 (CH), 100.2 (CH), 71.8 (CH). EI-LRMS m/z (%): 372 (M++4, 6), 370 (M+ + 2, 18), 368 (M+, 54), 351 (100), 281 (41). ESI-HRMS was calculated for C21H14Cl2NaO2 [M + Na]+ 391.0263, and found 391.0267.
3.2.7. Synthesis of 4-Chloro-2,5-Diphenylbenzofuran 9
A solution of starting carbamate 3ka (123 mg, 0.4 mmol) in toluene (2 mL) was mixed with acid phenylboronic acid (97 mg, 0.8 mmol), Pd(OAc)2 (5 mg, 5 mol%), PPh3 (11 mg, 10 mol%), and K3PO4 (169 mg, 0.8 mmol). The resulting mixture was stirred at 100 °C for 12 h. After completion of the reaction, the solvents were removed under reduced pressure, and the residue was purified by silica gel column chromatography (hexane/EtOAc = 10:1), affording benzo[b]furan derivative 9.
4-Chloro-2,5-diphenylbenzofuran (9): White solid (81% yield); mp = 116–118 °C; Rf = 0.18 (hexane/EtOAc, 10:1). 1H NMR (300 MHz, CDCl3) δ (ppm): 7.95–7.92 (m, 2H), 7.53–7.49 (m, 9H), 7.30 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 0.9 Hz, 1H). 13C NMR (75.4 MHz, CDCl3) δ (ppm): 157.2 (C), 154.2 (C), 139.6 (C), 135.4 (C), 130.1 (C), 130.0 (2 × CH), 129.6 (C), 129.2 (CH), 129.0 (2 × CH), 128.2 (2 × CH), 127.5 (CH), 127.3 (CH), 125.3 (2 × CH), 123.9 (C), 109.8 (CH), 100.6 (CH). EI-LRMS m/z (%): 306 (M+ + 2, 34), 304 (M+, 100), 239 (35), 119 (20). ESI-HRMS was calculated for C20H14ClO [M + H]+ 305.0728, and found 305.0739.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}