

A Novel Two-Dimensional ZnSiP2 Monolayer as an Anode Material for K-Ion Batteries and NO2 Gas Sensing


Abstract
1. Introduction
2. Results and Discussion
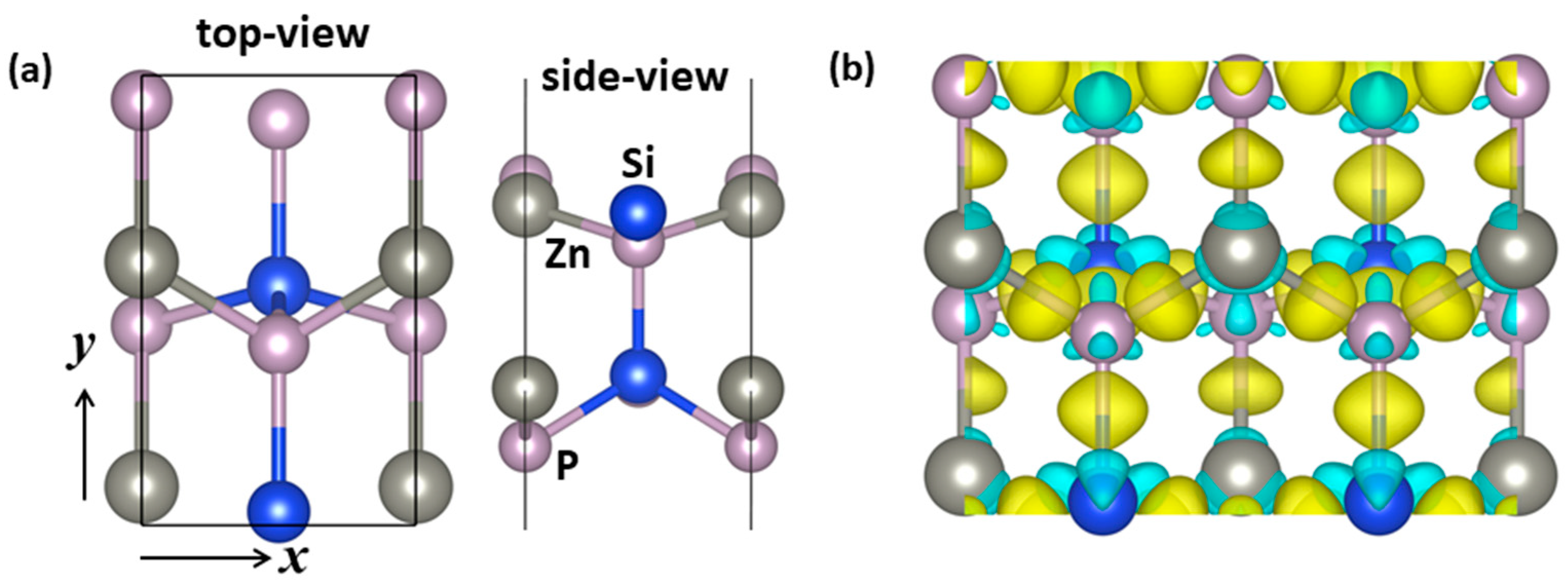
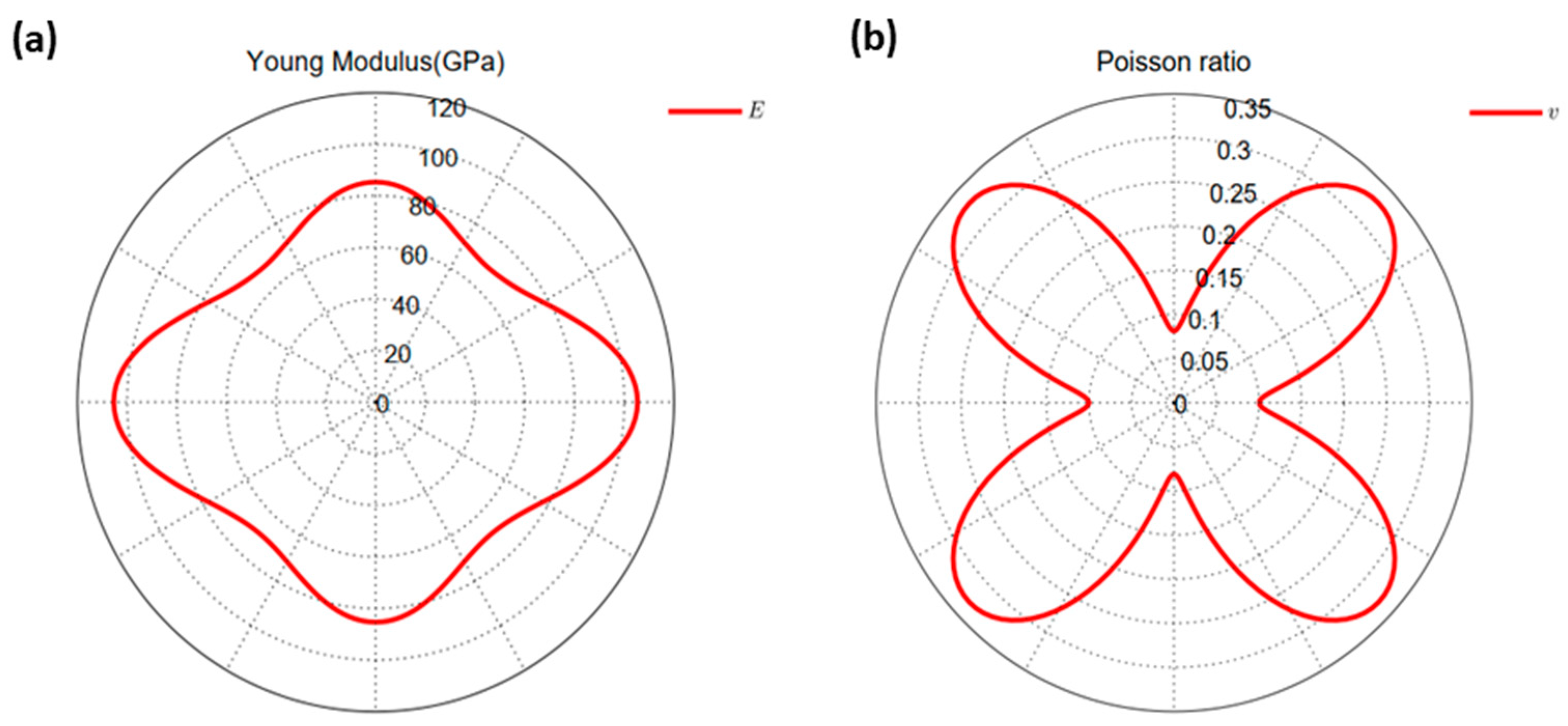
2.1. Structure and Stability
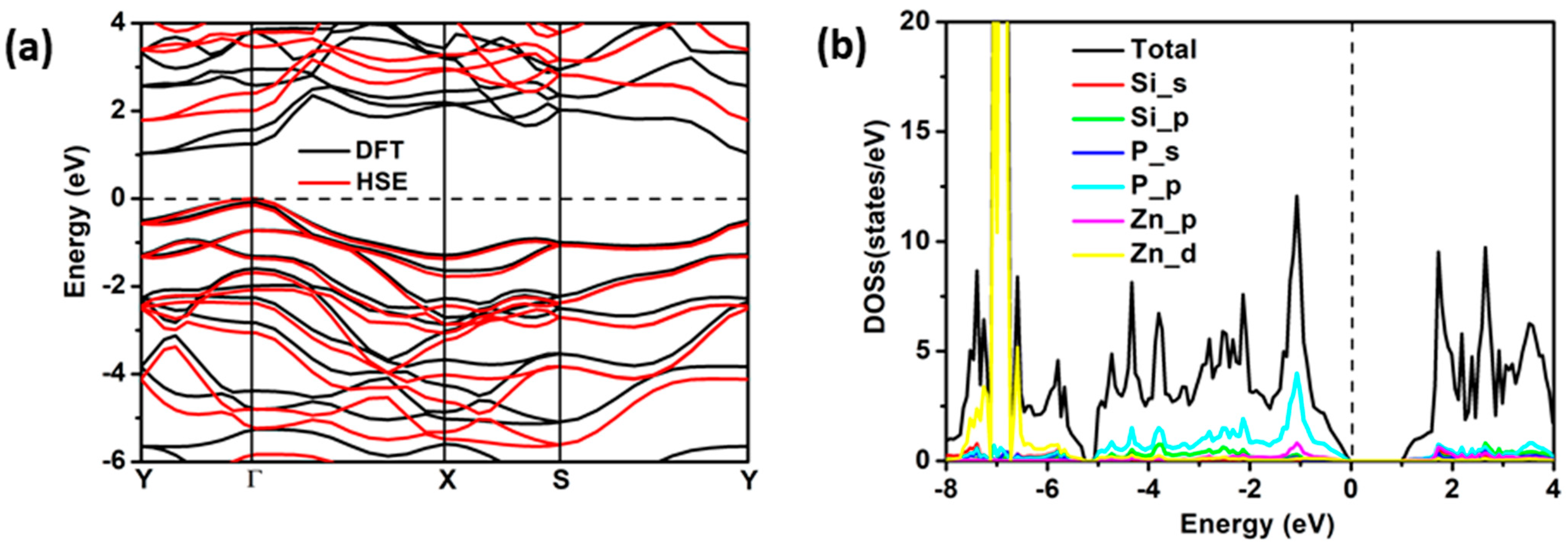
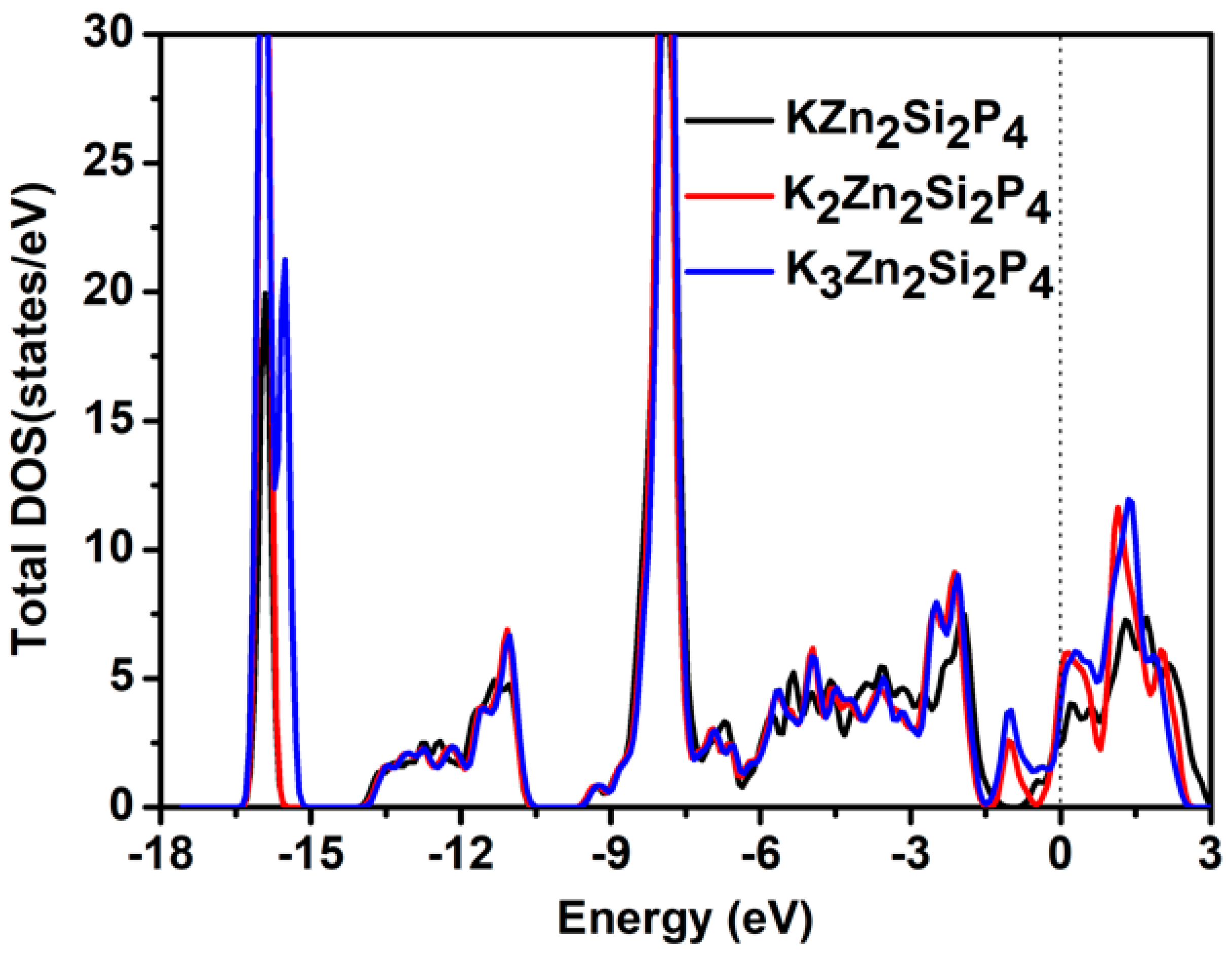
2.2. Electronic and Adsorption Properties
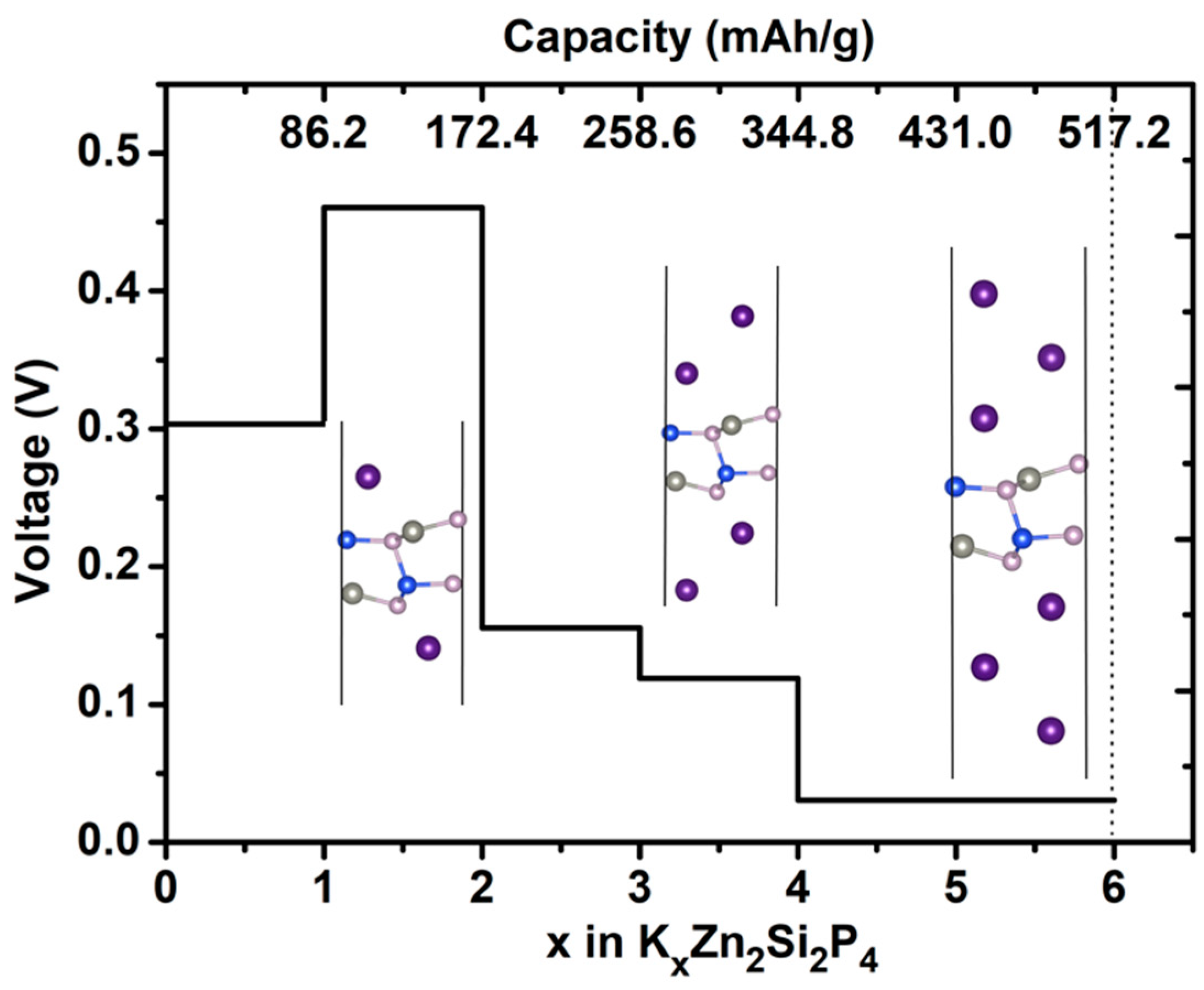
2.3. Capacity and Open-Circuit Voltage
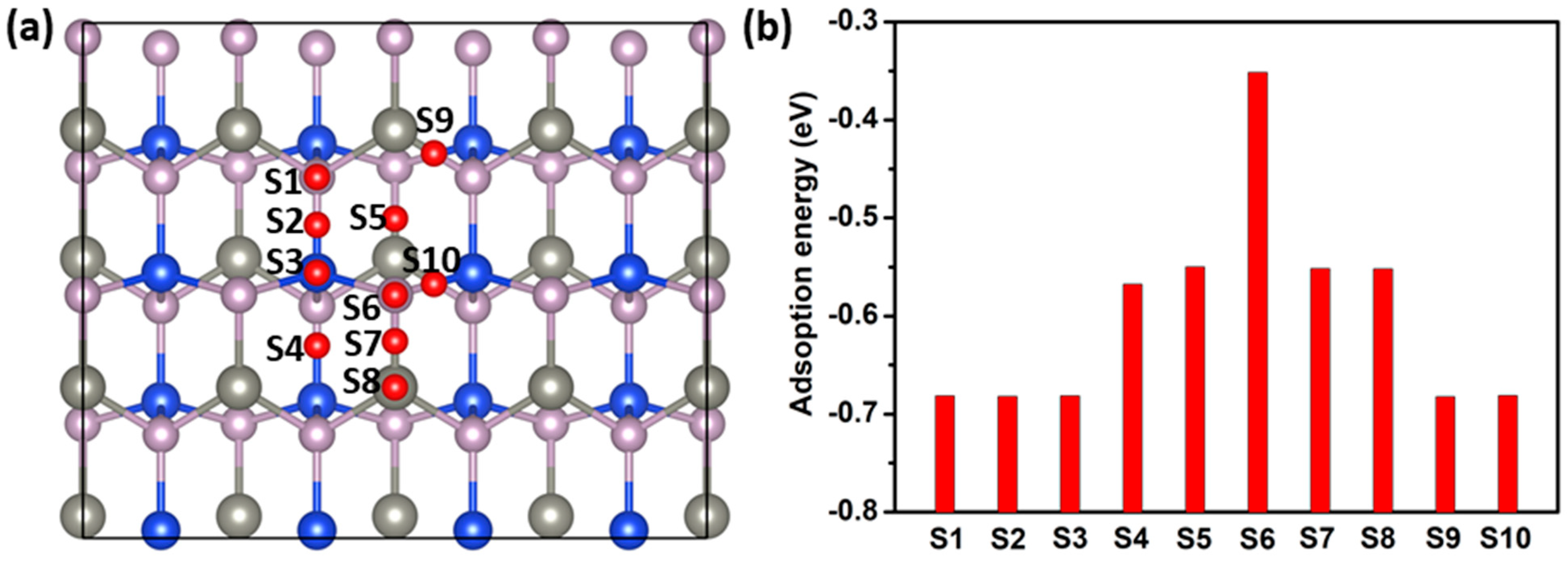
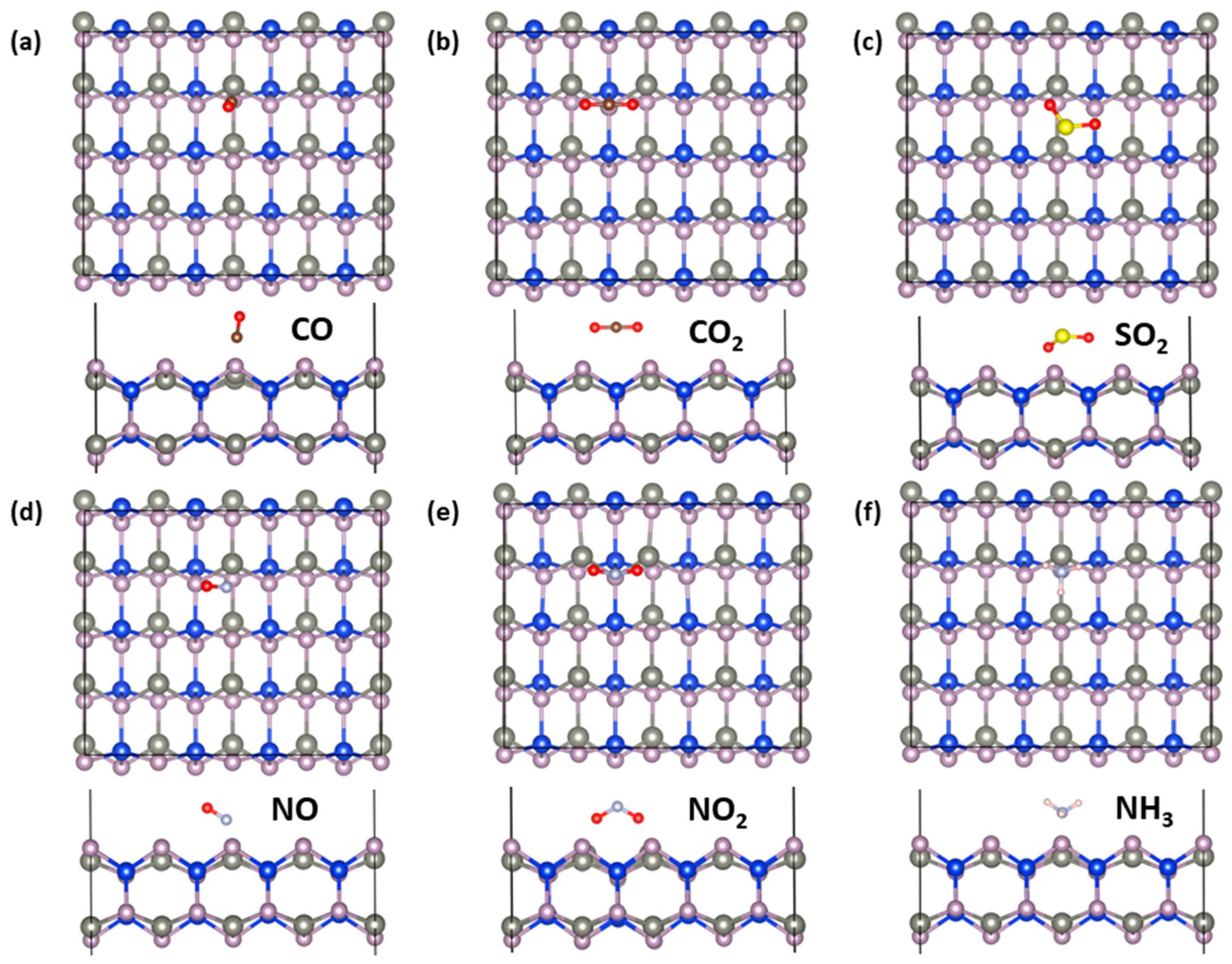
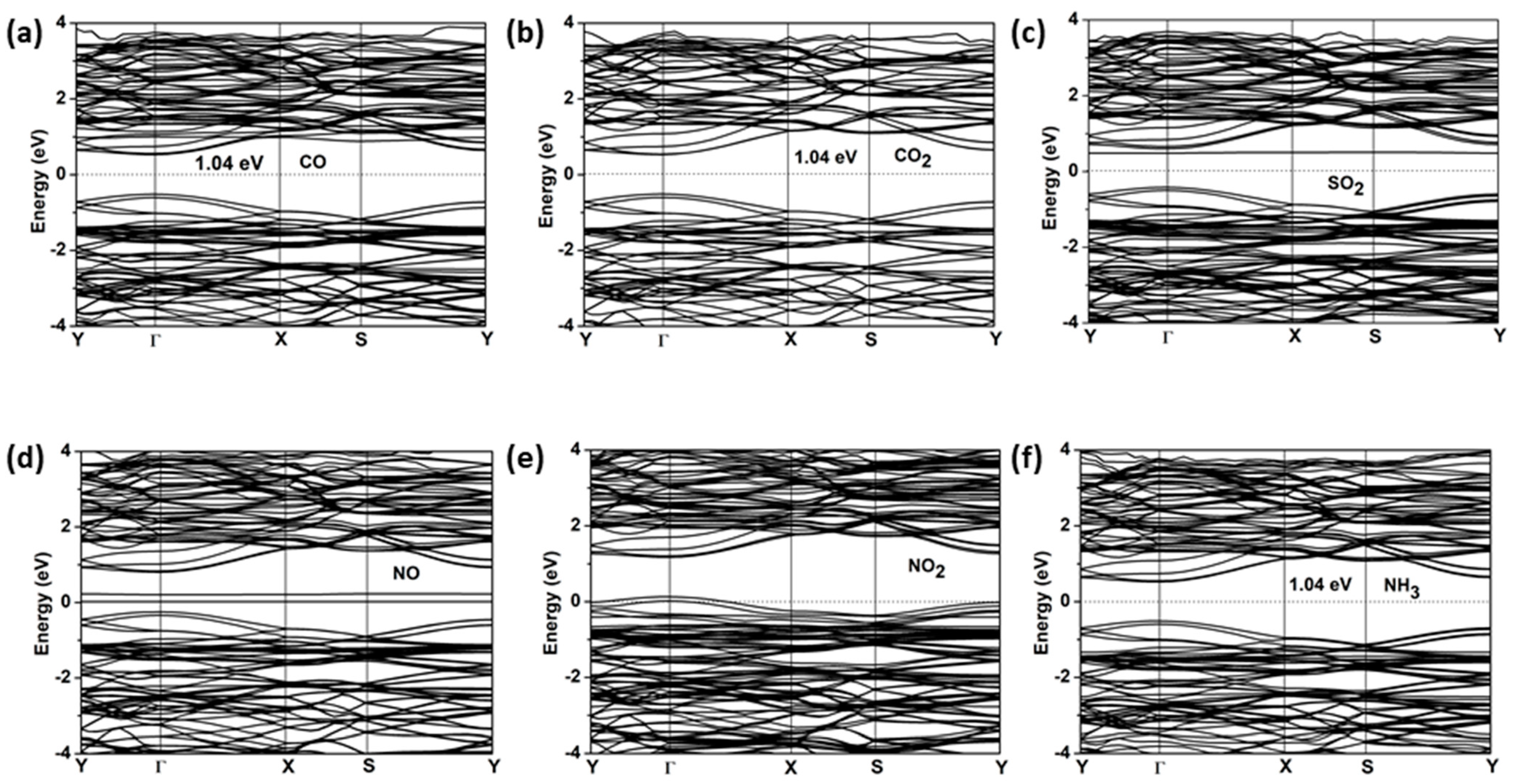
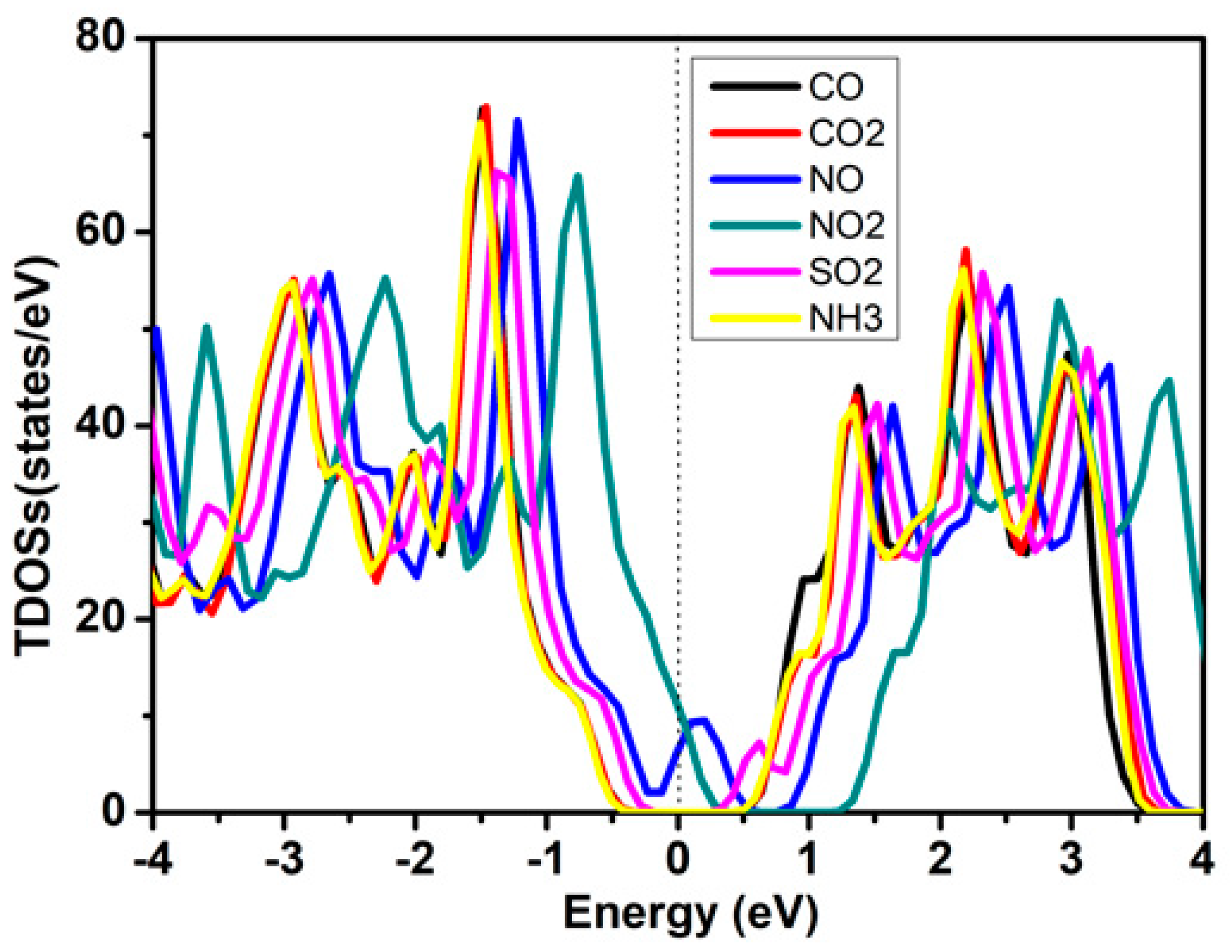
2.4. Gas-Sensing Properties
3. Computational Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wang, Q.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 2012, 7, 699–712. [Google Scholar] [CrossRef]
- Jana, S.; Thomas, S.; Chi, H.L.; Jun, B.; Sang, U.L. Rational design of a PC3 monolayer: A high-capacity, rapidly charging anode material for sodium-ion batteries. Carbon 2020, 157, 420–426. [Google Scholar] [CrossRef]
- Mayorga-Martinez, C.C.; Sofer, Z.; Pumera, M. Layered black phosphorus as a selective vapor sensor. Angew. Chem. 2015, 54, 14317–14320. [Google Scholar] [CrossRef]
- Kumar, R.; Goel, N.; Kumar, M. UV-Activated MoS2 based fast and reversible NO2 sensor at room temperature. ACS Sens. 2017, 2, 1744–1752. [Google Scholar] [CrossRef]
- Lin, J.; Yu, T.; Han, F.; Yang, G. Computational predictions of two-dimensional anode materials of metal-ion batteries. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, 1473. [Google Scholar] [CrossRef]
- Kumar, V.; Azhikodan, D.; Roy, D.R. 2D Sb2C3 monolayer: A promising material for recyclable gas sensor for environmentally toxic nitrogen-containing gases (NCGs). J. Hazard. Mater. 2021, 405, 124168. [Google Scholar] [CrossRef]
- Wang, G.; Pandey, R.; Karna, S.P. Carbon phosphide monolayers with superior carrier mobility. Nanoscale 2016, 8, 8819–8825. [Google Scholar] [CrossRef]
- Guan, J.; Liu, D.; Zhu, Z.; Tomanek, D. Two-Dimensional phosphorus carbide: Competition between sp2 and sp3 bonding. Nano Lett. 2016, 16, 3247–3252. [Google Scholar] [CrossRef]
- Singh, D.; Kansara, S.; Gupta, S.K.; Sonvane, Y. Single layer of carbon phosphide as an efficient material for optoelectronic devices. J. Mater. Sci. 2018, 53, 8314–8327. [Google Scholar] [CrossRef]
- Li, F.; Liu, X.; Wang, J.; Zhang, X.; Yang, B.; Qu, Y.; Zhao, M. A promising alkali-metal ion battery anode material: 2D metallic phosphorus carbide (β0-PC). Electrochim. Acta 2017, 258, 582–590. [Google Scholar] [CrossRef]
- Qi, S.; Li, F.; Qu, Y.; Yang, Y.; Li, W.; Zhao, M. Prediction of a flexible anode material for Li/Na ion batteries: Phosphorous carbide monolayer (α-PC). Carbon 2019, 141, 444–450. [Google Scholar] [CrossRef]
- Wang, J.; Lei, J.; Yang, G.; Xue, J.; Cai, Q.; Chen, D.; Lu, H.; Zhang, R.; Zheng, Y. An Ultra-Sensitive and selective nitrogen dioxide sensor based on novel P2C2 monolayer from theoretical perspective. Nanoscale 2018, 10, 21936–21943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, L.; Yang, C.; Zhang, X.; Ma, L.; Zhang, M.; Lu, J. Two-dimensional single-layer PC6 as promising anode materials for Li-ion batteries: The first-principles calculations study. Appl. Surf. Sci. 2020, 510, 145493. [Google Scholar] [CrossRef]
- Lu, N.; Zhuo, Z.; Guo, H.; Wu, P.; Fa, W.; Wu, X.; Zeng, X.C. A new Two-dimensional functional material with desirable bandgap and ultrahigh carrier mobility. J. Phys. Chem. Lett. 2018, 9, 1728–1733. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Ma, Y.; Li, Y.; Heine, T. GeP3: A small indirect band gap 2D crystal with high carrier mobility and strong interlayer quantum confinement. Nano Lett. 2017, 17, 1833–1838. [Google Scholar] [CrossRef]
- Yi, W.; Chen, X.; Wang, Z.; Ding, Y.; Yang, B.; Liu, X. A novel two-dimensional δ-InP3 monolayer with high stability, tunable band gap, high carrier mobility, and gas sensing of NO2. J. Mater. Chem. C 2019, 7, 7352–7359. [Google Scholar] [CrossRef]
- Yu, T.; Zhao, Z.; Sun, Y.; Bergara, A.; Lin, J.; Zhang, S.; Xu, H.; Zhang, L.; Yang, G.; Liu, Y. Two-dimensional PC6 with direct band gap and anisotropic carrier mobility. J. Am. Chem. Soc. 2019, 141, 1599–1605. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, J.; Li, X.; Wang, Q. Two-Dimensional T-NiSe2 as a promising anode material for Potassium-ion batteries with low average voltage, high ionic conductivity, and superior carrier mobility. ACS Appl. Mater. Interfaces 2019, 11, 35661–35666. [Google Scholar] [CrossRef]
- Li, J.; Wu, J.; Yu, Y. DFT exploration of sensor performances of two-dimensional WO3 to ten small gases in terms of work function and band gap changes and I-V responses. Appl. Surf. Sci. 2021, 546, 149104. [Google Scholar] [CrossRef]
- Chen, R.; Luo, S.; Xie, D.; Yu, Y.; Xiang, L. Highly dispersive palladium loading on ZnO by galvanic replacements with improved methane sensing performances. Chemosensors 2022, 10, 329. [Google Scholar] [CrossRef]
- Beniwal, S.; Hooper, J.; Miller, D.P.; Costa, P.S.; Chen, G.; Liu, S.Y.; Dowben, P.A.; Sykes, E.C.H.; Zurek, E.; Enders, A. Graphene-like boron-carbon-nitrogen monolayers. ACS Nano 2017, 11, 2486–2493. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.; Das, S.; Pathak, B. BCN monolayer for high capacity Al-based dual-ion batteries. Mater. Adv. 2020, 1, 2418–2425. [Google Scholar] [CrossRef]
- Pu, C.; Li, C.; Lv, L.; Zhou, D.; Tang, X. Structure and optoelectronic properties for two dimensional BCN from first-principles calculations. Chin. J. Lumin. 2020, 41, 48–55. [Google Scholar] [CrossRef]
- Fu, X.; Guo, J.; Li, L.; Dai, T. Structural and electronic properties of predicting two-dimensional BC2P and BC3P3 monolayers by the global optimization method. Chem. Phys. Lett. 2019, 726, 69–76. [Google Scholar] [CrossRef]
- Tang, M.; Wang, C.; Schwingenschlogl, U.; Yang, G. BC6P Monolayer: Isostructural and isoelectronic analogues of graphene with desirable properties for K-Ion batteries. Chem. Mater. 2021, 33, 9262–9269. [Google Scholar] [CrossRef]
- Popov, V.P.; Pamplin, B.R. Epitaxial growth of solid solutions of ZnSiP2 in Si. J. Cryst. Growth 1972, 15, 129–132. [Google Scholar] [CrossRef]
- Martinez, A.D.; Miller, E.M.; Norman, A.G.; Schnepf, R.R.; Leick, N.; Perkins, C.; Stradins, P.; Toberer, E.S.; Tamdoli, A.C. Growth of amorphous and epitaxial ZnSiP2–Si alloys on Si. J. Mater. Chem. C 2018, 6, 2696–2703. [Google Scholar] [CrossRef]
- Martinez, A.D.; Warren, E.L.; Gorai, P.; Borup, K.A.; Kuciauskas, D.; Dippo, P.C.; Ortiz, B.R.; Macaluso, R.T.; Nguyen, S.D.; Greenaway, A.L.; et al. Solar energy conversion properties and defect physics of ZnSiP2. Energy Environ. Sci. 2016, 9, 1031–1041. [Google Scholar] [CrossRef]
- Scanlon, D.O.; Walsh, A. Bandgap engineering of ZnSnP2 for high-efficiency solar cells. Appl. Phys. Lett. 2012, 100, 251911. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhu, X.; Zhou, Y.; Chen, X.; An, C.; Zhou, Y.; Zhang, R.; Gu, C.; Zhang, L.; Li, X.; et al. Pressure-engineered optical properties and emergent superconductivity in chalcopyrite semiconductor ZnSiP2. NPG Asia Mater. 2021, 13, 15. [Google Scholar] [CrossRef]
- Sreeparvathy, P.C.; Kanchana, V.; Vaitheeswaran, G. Thermoelectric properties of zinc based pnictide semiconductors. J. Appl. Phys. 2016, 119, 085701. [Google Scholar] [CrossRef]
- Liu, J.; Emrys, T.; Miao, J.; Huang, Y.; Rondinelli, J.M.; Hendrik, H. Understanding chemical bonding in alloys and the representation in atomistic simulations. J. Phys. Chem. C 2018, 122, 14996–15009. [Google Scholar] [CrossRef]
- Guan, J.; Zhu, Z.; Tománek, D. Phase coexistence and metal-insulator transition in few-layer phosphorene: A computational study. Phys. Rev. Lett. 2014, 113, 046804. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.M.; Bačić, V.; Popov, I.A.; Boldyrev, A.I.; Heine, T.; Frauenheim, T.; Ganz, E. Two-dimensional Cu2Si monolayer with planar hexacoordinate copper and silicon bonding. J. Am. Chem. Soc. 2015, 137, 2757–2762. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, J.; Yu, Y. A theoretical analysis on the oxidation and water dissociation resistance on group-IV phosphide monolayers. ChemPhysChem 2020, 21, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhuang, H.; Yoon, M.; Sumpter, B.G.; Wei, S. Highly stable two-dimensional silicon phosphides: Different stoichiometries and exotic electronic properties. Phys. Rev. B 2015, 91, 121401. [Google Scholar] [CrossRef]
- Mouhat, F.; Couder, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Cadelano, E.; Palla, P.L.; Giordano, S.; Colombo, L. Elastic properties of hydrogenated grapheme. Phys. Rev. B 2010, 82, 235414. [Google Scholar] [CrossRef]
- Wang, L.; Kutana, A.; Zou, X.; Yakobson, B.I. Electro-mechanical anisotropy of phosphorene. Nanoscale 2015, 7, 9746–9751. [Google Scholar] [CrossRef]
- Cooper, R.C.; Lee, C.; Marianetti, C.A.; Wei, X.; Hone, J.; Kysar, J.W. Nonlinear elastic behavior of two-dimensional molybdenum disulfide. Phys. Rev. B 2013, 87, 035423. [Google Scholar] [CrossRef]
- Yu, Y. High storage capacity and small volume change of potassium-intercalation into novel vanadium oxychalcogenide monolayers V2S2O, V2Se2O and V2Te2O: An ab initio DFT investigation. Appl. Surf. Sci. 2021, 546, 149062. [Google Scholar] [CrossRef]
- Zhang, S.H.; Liu, B.G. Superior ionic and electronic properties of ReN2 monolayers for Na-ion battery electrodes. Nanotechnology 2018, 29, 325401. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Shyy, W.; Liu, M.; Wei, L.; Wu, M.; Zhao, T.S. Boron phosphide monolayer as a potential anode material for alkali metal-based batteries. J. Mater. Chem. A 2017, 5, 672–679. [Google Scholar] [CrossRef]
- Dou, K.; Ma, Y.; Zhang, T.; Huang, B.; Dai, Y. Prediction of two-dimensional PC6 as a promising anode material for potassium-ion batteries. Phys. Chem. Chem. Phys. 2019, 21, 26212–26218. [Google Scholar] [CrossRef]
- Rehman, J.; Fan, X.; Laref, A.; Zheng, W.T. Adsorption and diffusion of potassium on 2D SnC sheets for potential high-performance anodic applications of potassium-ion batteries. ChemElectroChem 2020, 7, 3832–3838. [Google Scholar] [CrossRef]
- Li, F.; Qu, Y.; Zhao, M. Germanium sulfide nanosheet: A universal anode material for alkali metal ion batteries. J. Mater. Chem. A 2016, 4, 8905–8912. [Google Scholar] [CrossRef]
- Er, D.; Li, J.; Naguib, M.; Gogotsi, Y.; Shenoy, V. Ti3C2 MXene as a high capacity electrode material for metal (Li, Na, K, Ca) ion batteries. ACS Appl. Mater. Interfaces 2014, 6, 11173–11179. [Google Scholar] [CrossRef]
- Xiang, P.; Sharma, S.; Wang, Z.M.; Wu, J.; Schwingenschlogl, U. Flexible C6BN monolayers as promising anode materials for high-performance K-ion batteries. ACS Appl. Mater. Interfaces 2020, 12, 30731–30739. [Google Scholar] [CrossRef]
- Yuan, X.; Chen, Z.; Huang, B.; He, Y.; Zhou, N. Potential application of MoS2/M2CS2 (M = Ti, V) heterostructures as anode materials for metal-ion batteries. J. Phys. Chem. C 2021, 125, 10226–10234. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, Z.; Wang, S.S.; Guan, S.; Yang, H.Y.; Yao, Y.; Yang, S.A. Theoretical prediction of MoN2 monolayer as a high capacity electrode material for metal ion batteries. J. Mater. Chem. A 2016, 4, 15224–15231. [Google Scholar] [CrossRef]
- Joshi, R.; Ozdemir, B.; Peralta, J.; Barone, V. Hexagonal BC3: A robust electrode material for Li, Na, and K ion batteries. J. Phys. Chem. Lett. 2015, 6, 2728–2732. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 2010, 82, 094116–094118. [Google Scholar] [CrossRef]
- Liu, C.; Yang, X.; Liu, J.; Ye, X. Theoretical prediction of two-dimensional SnP3 as a promising anode material for Na-ion batteries. ACS Appl. Energy Mater. 2018, 1, 3850–3859. [Google Scholar] [CrossRef]
- Lin, S.; Xu, M.; Hao, J.; Wang, X.; Wu, M.; Shi, J.; Cui, W.; Liu, D.; Lei, W.; Li, Y. Prediction of superhard B2N3 with two-dimensional metallicity. J. Mater. Chem. C 2019, 7, 4527–4532. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for Ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-Principles calculations of the ferroelastic transition between rutile-Type and CaCl2-Type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | CO | CO2 | SO2 | NO | NO2 | NH3 |
|---|---|---|---|---|---|---|
| Ead (eV) | −0.74 | −0.55 | −1.09 | −0.75 | −1.30 | −1.14 |
| d0 (Å) | 1.54 | 2.29 | 1.73 | 1.68 | 1.53 | 1.53 |
| Eg (eV) | 1.04 | 1.04 | 0.9 | metal | metal | 1.04 |
| Q (e) | 0 | 0 | −0.24 | 0.12 | 0.67 | −0.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pu, C.; Wang, Z.; Tang, X.; Zhou, D.; Cheng, J. A Novel Two-Dimensional ZnSiP2 Monolayer as an Anode Material for K-Ion Batteries and NO2 Gas Sensing. Molecules 2022, 27, 6726. https://doi.org/10.3390/molecules27196726
Pu C, Wang Z, Tang X, Zhou D, Cheng J. A Novel Two-Dimensional ZnSiP2 Monolayer as an Anode Material for K-Ion Batteries and NO2 Gas Sensing. Molecules. 2022; 27(19):6726. https://doi.org/10.3390/molecules27196726
Chicago/Turabian StylePu, Chunying, Zhuo Wang, Xin Tang, Dawei Zhou, and Jinbing Cheng. 2022. "A Novel Two-Dimensional ZnSiP2 Monolayer as an Anode Material for K-Ion Batteries and NO2 Gas Sensing" Molecules 27, no. 19: 6726. https://doi.org/10.3390/molecules27196726
APA StylePu, C., Wang, Z., Tang, X., Zhou, D., & Cheng, J. (2022). A Novel Two-Dimensional ZnSiP2 Monolayer as an Anode Material for K-Ion Batteries and NO2 Gas Sensing. Molecules, 27(19), 6726. https://doi.org/10.3390/molecules27196726