

Natural Phytocompounds from Common Indian Spices for Identification of Three Potential Inhibitors of Breast Cancer: A Molecular Modelling Approach



Abstract
1. Introduction
2. Result and Discussion
2.1. Molecular Docking
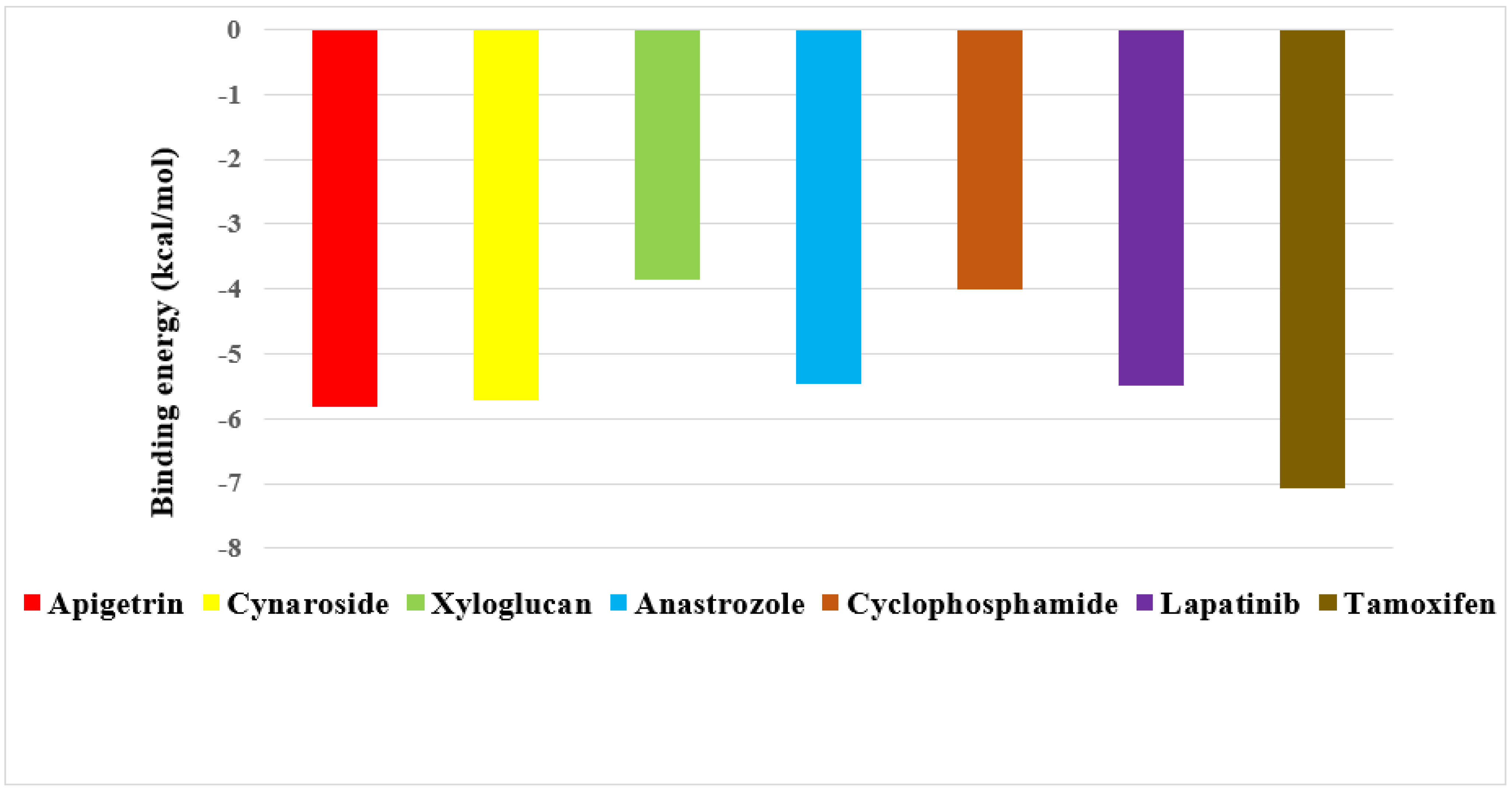
2.2. Screening of Drug Candidates against CDK8
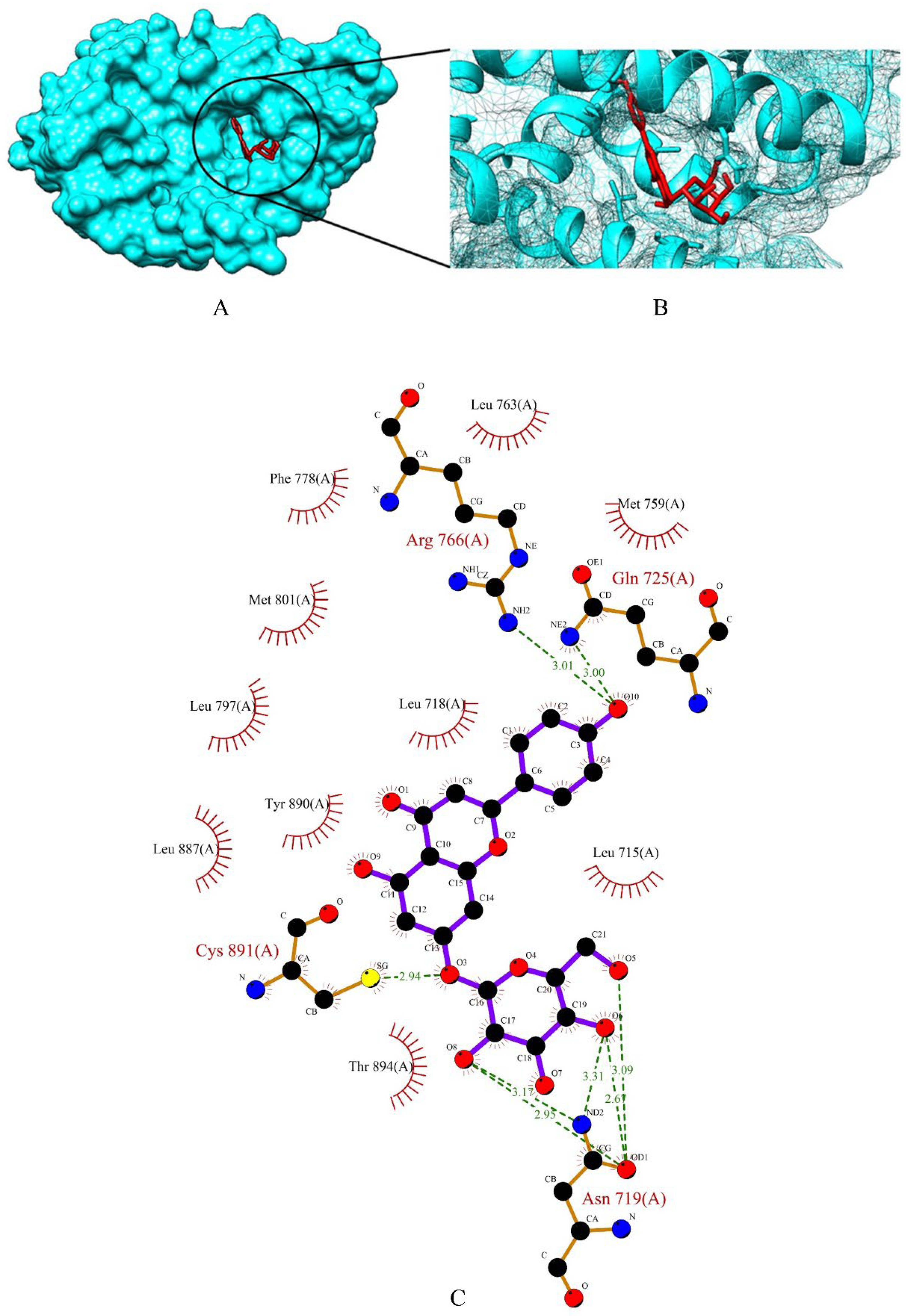
2.3. Screening of Drug Candidates against PR
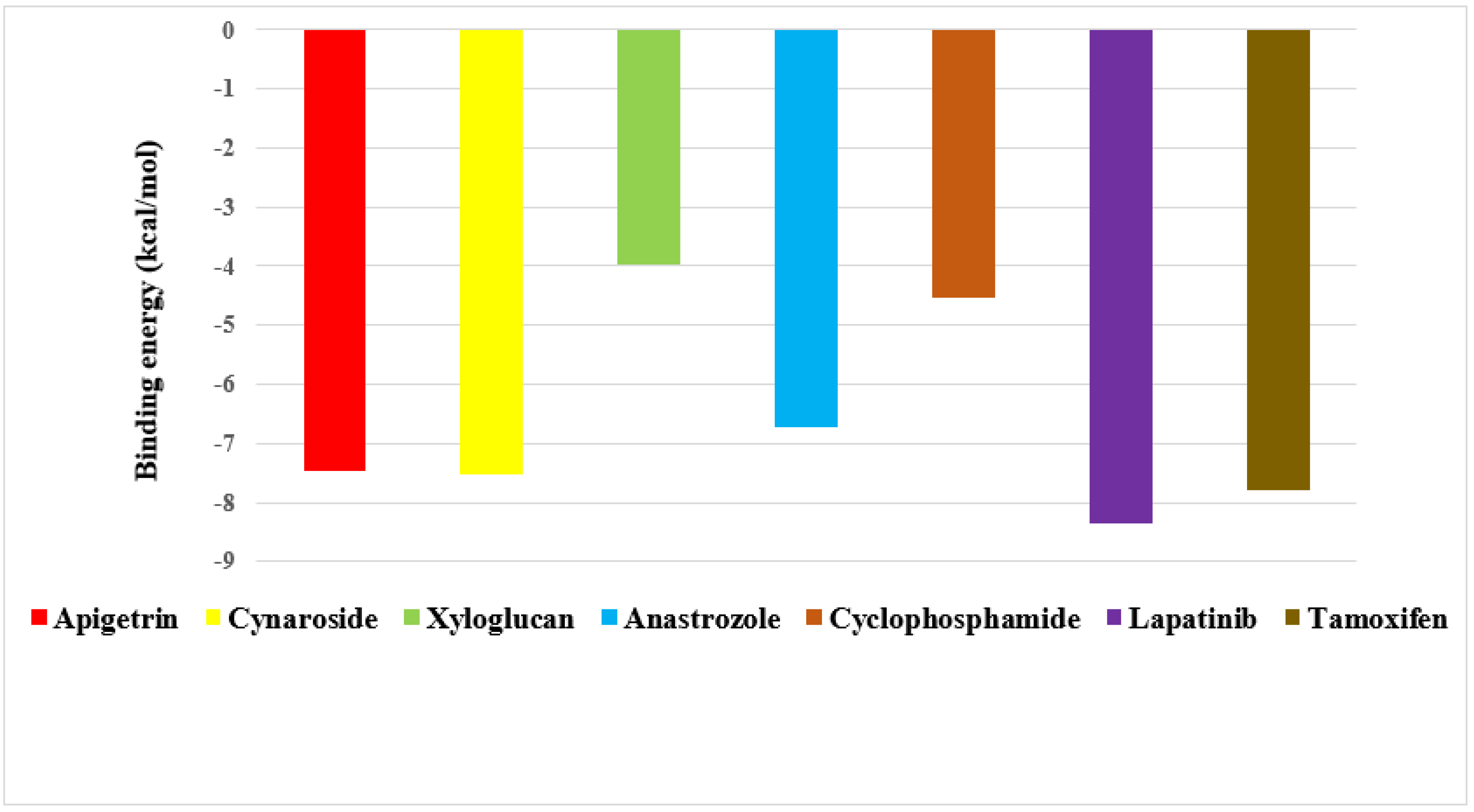
2.4. Screening of Drug Candidates against EGFR
2.5. Drug Likeness and Toxicity Prediction
2.6. Bioactivity Score
3. Materials and Methods
3.1. Protein Retrieval
3.2. Ligand Preparation
3.3. Molecular Docking
3.4. Drug Likeness and Toxicity Prediction
3.5. Bioactivity Score
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- WHO Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 1 August 2022).
- Sahayarayan, J.J.; Rajan, K.S.; Vidhyavathi, R.; Nachiappan, M.; Prabhu, D.; Alfarraj, S.; Arokiyaraj, S.; Daniel, A.N. In-silico protein-ligand docking studies against the estrogen protein of breast cancer using pharmacophore based virtual screening approaches. Saudi J. Biol. Sci. 2021, 28, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Ataollahi, M.R.; Sharifi, J.; Paknahad, M.R.; Paknahad, A. Breast cancer and associated factors: A review. J. Med. Life 2015, 8, 6–11. [Google Scholar] [PubMed]
- Kim, B.; Fatayer, H.; Hanby, A.M.; Horgan, K.; Perry, S.L.; Valleley, E.M.; Verghese, E.T.; Williams, B.J.; Thorne, J.L.; Hughes, T.A. Neoadjuvant chemotherapy induces expression levels of breast cancer resistance protein that predict disease-free survival in breast cancer. PLoS ONE 2013, 8, e62766. [Google Scholar] [CrossRef] [PubMed]
- Greenwell, M.; Rahman, P.K.S.M. Medicinal plants: Their use in anticancer treatment. Int. J. Pharm. Sci. Rev. Res. 2015, 6, 4103–4112. [Google Scholar] [CrossRef]
- Iqbal, J.; Abbasi, B.A.; Mahmood, T.; Kanwal, S.; Ali, B.; Shah, S.A.; Khalil, A.T. Plant-derived anticancer agents: A green anticancer approach. Asian Pac. J. Trop. Biomed. 2017, 7, 1129–1150. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef]
- Mullard, A. New drugs cost US$2.6 billion to develop. Nat. Rev. Drug. Discov. 2014, 13, 877. [Google Scholar] [CrossRef]
- Li, K.; Du, Y.; Li, L.; Wei, D.Q. Bioinformatics approaches for anti-cancer drug discovery. Curr. Drug Targets 2020, 21, 3–17. [Google Scholar] [CrossRef]
- Wang, G.; Zhu, W. Molecular docking for drug discovery and development: A widely used approach but far from perfect. Future Med. Chem. 2016, 8, 1707–1710. [Google Scholar] [CrossRef]
- Blundell, T.L.; Jhoti, H.; Abell, C. High-throughput crystallography for lead discovery in drug design. Nat. Rev. Drug. Discov. 2002, 1, 45–54. [Google Scholar] [CrossRef]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug. Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef]
- Issa, N.T.; Badiavas, E.V.; Schürer, S. Research techniques made simple: Molecular docking in dermatology-A foray into in silico drug discovery. J. Investig. Dermatol. 2019, 139, 2400–2408. [Google Scholar] [CrossRef]
- Gohlke, H.; Klebe, G. Approaches to the description and prediction of the binding affinity of small-molecule ligands to macromolecular receptors. Angew. Chem. Int. Ed. 2002, 41, 2644–2676. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.A.; Targowska-Duda, K.M.; Matosiuk, D. Recent advances and applications of molecular docking to G protein-coupled receptors. Molecules 2017, 22, 340. [Google Scholar] [CrossRef]
- DesJarlais, R.L.; Dixon, J.S. A shape-and chemistry-based docking method and its use in the design of HIV-1 protease inhibitors. J. Comput. Aided Mol. 1994, 8, 231–242. [Google Scholar] [CrossRef]
- Venhorst, J.; ter Laak, A.M.; Commandeur, J.N.; Funae, Y.; Hiroi, T.; Vermeulen, N.P. Homology modeling of rat and human cytochrome P450 2D (CYP2D) isoforms and computational rationalization of experimental ligand-binding specificities. J. Med. Chem. 2003, 46, 74–86. [Google Scholar] [CrossRef]
- Williams, P.A.; Cosme, J.; Ward, A.; Angove, H.C.; Matak Vinković, D.; Jhoti, H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003, 424, 464–468. [Google Scholar] [CrossRef]
- Padma, V.V. An overview of targeted cancer therapy. BioMedicine 2015, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Acharya, R.; Chacko, S.; Bose, P.; Lapenna, A.; Pattanayak, S.P. Structure based multitargeted molecular docking analysis of selected furanocoumarins against breast cancer. Sci. Rep. 2019, 9, 15743. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Devkar, S.; Gharat, K.; Kasbe, S.; Matharoo, D.K.; Pendse, S.; Bhosale, A.; Bhargava, A. Screening of Phytochemicals as Potential Inhibitors of Breast Cancer using Structure Based Multitargeted Molecular Docking Analysis. Phytomed. Plus. 2022, 2, 100227. [Google Scholar] [CrossRef]
- Daniel, A.R.; Hagan, C.R.; Lange, C.A. Progesterone receptor action: Defining a role in breast cancer. Expert Rev. Endocrinol. Metab. 2011, 6, 359–369. [Google Scholar] [CrossRef]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Donner, A.J.; Espinosa, J.M. CDK8: A positive regulator of transcription. Transcription 2010, 1, 4–12. [Google Scholar] [CrossRef]
- Crown, J. CDK8: A new breast cancer target. Oncotarget 2017, 8, 14269–14270. [Google Scholar] [CrossRef]
- Rajagopal, K.; Byran, G.; Jupudi, S.; Vadivelan, R. Activity of phytochemical constituents of black pepper, ginger, and garlic against coronavirus (COVID-19): An in silico approach. Int. J. Health Allied Sci. 2020, 9, 43–50. [Google Scholar] [CrossRef]
- Aftab, A.; Khan, R.; Shah, W.; Azhar, M.; Unar, A.; Jafar Hussain, H.M.; Waqas, A. Computational analysis of Cyclin D1 gene SNPs and association with breast cancer. Biosci. Rep. 2021, 41, BSR20202269. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Ashfaq, U.A.; Mumtaz, A.; ul Qamar, T.; Fatima, T. MAPS database: Medicinal plant activities, phytochemical and structural database. Bioinformation 2013, 9, 993–995. [Google Scholar] [CrossRef]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In silico methods for design of kinase inhibitors as anticancer drugs. Front. Chem. 2020, 7, 873. [Google Scholar] [CrossRef]
- Iheagwam, F.N.; Ogunlana, O.O.; Ogunlana, O.E.; Isewon, I.; Oyelade, J. Potential anti-cancer flavonoids isolated from Caesalpinia bonduc young twigs and leaves: Molecular docking and in silico studies. Bioinform. Biol. Insights 2019, 13, 1177932218821371. [Google Scholar] [CrossRef]
- Kim, S.M.; Vetrivel, P.; Ha, S.E.; Kim, H.H.; Kim, J.A.; Kim, G.S. Apigetrin induces extrinsic apoptosis, autophagy and G2/M phase cell cycle arrest through PI3K/AKT/mTOR pathway in AGS human gastric cancer cell. J. Nutr. Biochem. 2020, 83, 108427. [Google Scholar] [CrossRef]
- Ji, J.; Wang, Z.; Sun, W.; Li, Z.; Cai, H.; Zhao, E.; Cui, H. Effects of Cynaroside on Cell Proliferation, Apoptosis, Migration and Invasion though the MET/AKT/mTOR Axis in Gastric Cancer. Int. J. Mol. Sci. 2021, 22, 12125. [Google Scholar] [CrossRef]
- Liu, H.T.; Ho, Y.S. Anticancer effect of curcumin on breast cancer and stem cells. Food Sci. Hum. Wellness 2018, 7, 134–137. [Google Scholar] [CrossRef]
- Lombardo, F.; Lipinski, C.A.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Setting. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Verma, A. Lead finding from Phyllanthus debelis with hepatoprotective potentials. Asian Pac. J. Trop. Biomed. 2012, 2, S1735–S1737. [Google Scholar] [CrossRef]
- Klatt, F.; Leitner, A.; Kim, I.V.; Ho-Xuan, H.; Schneider, E.V.; Langhammer, F.; Weinmann, R.; Müller, M.R.; Huber, R.; Meister, G.; et al. A precisely positioned MED12 activation helix stimulates CDK8 kinase activity. Proc. Natl. Acad. Sci. USA 2020, 117, 2894–2905. [Google Scholar] [CrossRef]
- Petit-Topin, I.; Fay, M.; Resche-Rigon, M.; Ulmann, A.; Gainer, E.; Rafestin-Oblin, M.E.; Fagart, J. Molecular determinants of the recognition of ulipristal acetate by oxo-steroid receptors. J. Steroid Biochem. Mol. Biol. 2014, 144, 427–435. [Google Scholar] [CrossRef]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Schrodinger, L.; DeLano, W. PyMol. 2020. Available online: http://www.pymol.org/pymol (accessed on 1 June 2022).
- Hsu, K.C.; Chen, Y.F.; Lin, S.R.; Yang, J.M. iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinform. 2011, 12, S33. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics free web services. 2021. Available online: https://www.molinspiration.com (accessed on 21 June 2022).
- Fernandes, G.S.; de Moura Pereira, M.B.; Marinho, A.C.B.; Machado, B.; Moreira, A.C.; de Freitas, M.P.; Lang, K.L.; Antunes, J.E. In Silico Pharmacokinetics Studies for Quinazolines Proposed as EGFR Inhibitors. Open J. Med. Chem. 2015, 5, 106–115. [Google Scholar]
- Srivastava, R. Theoretical Studies on the Molecular Properties, Toxicity, and Biological Efficacy of 21 New Chemical Entities. ACS Omega 2021, 6, 24891–24901. [Google Scholar] [CrossRef]
- Ghosh, S.; Tripathi, P.; Talukdar, P.; Talapatra, S.N. In silico study by using ProTox-II webserver for oral acute toxicity, organ toxicity, immunotoxicity, genetic toxicity endpoints, nuclear receptor signalling and stress response pathways of synthetic pyrethroids. World Sci. News 2019, 132, 35–51. [Google Scholar]
- Bhat, V.; Chatterjee, J. The use of in silico tools for the toxicity prediction of potential inhibitors of SARS-CoV-2. Altern. Lab. Anim. 2021, 49, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.S.; Kumar, N.; Singh, H.P.; Ranjan, S.; Sharma, C.S. In silico pharmacokinetic, bioactivity and toxicity study of some selected anti-asthmatic agents. Int. J. Pharm. Sci. Drug. Res. 2018, 10, 278–282. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl No. | Compound Name | LogP (≤5) | TPSA (≤140 Å) | n Atoms | MW (≤500 da) | n ON (≤10) | n OHNH (≤5) | n Rotb | Volume | Violation of Rules |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. | Apigetrin | 2.46 | 90.89 | 20 | 270.24 | 5 | 3 | 1 | 224.05 | 0 |
| 2. | Cynaroside | 0.19 | 190.2 | 32 | 448.3 | 11 | 7 | 4 | 364.1 | 2 |
| 3. | Curcumin | 2.30 | 93.07 | 27 | 368.38 | 6 | 2 | 8 | 332.18 | 0 |
| 4. | Xyloglucan | −1.74 | 212.67 | 34 | 488.44 | 13 | 7 | 12 | 415.44 | 2 |
| Sl No. | Compound Name | LD50 (mg/kg) | Toxicity Class | Hepatotoxicity | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity |
|---|---|---|---|---|---|---|---|---|
| 1. | Apigetrin | 5000 | 5 | N (0.82) | N (0.86) | N (0.93) | Y (0.59) | N (0.69) |
| 2. | Cynaroside | 5000 | 5 | N (0.82) | N (0.85) | N (0.74) | N (0.76) | N (0.69) |
| 3. | Curcumin | 2000 | 4 | N (0.61) | N (0.84) | Y (0.92) | N (0.88) | N (0.88) |
| 4. | Xyloglucan | 5000 | 5 | N (0.86) | N (0.80) | N (0.99) | N (0.77) | N (0.80) |
| Sl No. | Compound Name | GPCR Ligand | Ion Channel Modulator | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor |
|---|---|---|---|---|---|---|---|
| 1. | Apigetrin | 0.10 | −0.01 | 0.14 | 0.31 | 0.02 | 0.43 |
| 2. | Cynaroside | 0.09 | −0.02 | 0.15 | 0.27 | −0.01 | 0.42 |
| 3. | Curcumin | −0.06 | −0.20 | −0.26 | 0.12 | −0.14 | 0.08 |
| 4. | Xyloglucan | 0.01 | −0.07 | −0.14 | −0.03 | 0.05 | 0.30 |
| Protein | Full Name of the Protein | Protein Function | PDB ID |
|---|---|---|---|
| CDK8 | Cyclin-dependent kinase 8 | The Notch intracellular domain, SREBP (Sterol regulatory-element binding proteins), and STAT1-S727 are all phosphorylated by CDK8. By modulating the turnover of subunits in the mediator complex tail module, CDK8 also represses transcriptional activity. CDK8 also affects interaction of RNA polymerase II with the mediator complex. | 6T41 |
| PR | Progesterone receptor | Proliferation and differentiation of cell, transcriptional activator and repressor. | 4OAR |
| EGFR | Epidermal growth factor receptor | When a ligand binds to an epidermal growth factor receptor, the receptor is able to attach (dimerize) with another epidermal growth factor receptor protein nearby, turning on (activating) the receptor complex. As a result, cell signalling cascades that promote cell growth and division (proliferation) as well as cell survival are activated. | 2J6M |
| Target Proteins | Active Site Residues | Grid Box Parameters | |
|---|---|---|---|
| x Centre × y Centre × z Centre | Number of Points in x-Dimension × Number of Points in y-Dimension × Number of Points in z-Dimension | ||
| CDK8 | ILE-79, TYR-99, LEU-158, ALA-100 | −5.797 × −8.769 × 12.8 | 60 × 60 × 60 |
| PR | GLY-722, GLN-725, ARG-766, THR-894 | 12.542 × 27.621 × 15.492 | 60 × 60 × 60 |
| EGFR | THR-854, THR-790, GLN-791, LEU-792, MET-793 | −56.344 × −0.481 × −24.16 | 60 × 60 × 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hazra, S.; Ray, A.S.; Rahaman, C.H. Natural Phytocompounds from Common Indian Spices for Identification of Three Potential Inhibitors of Breast Cancer: A Molecular Modelling Approach. Molecules 2022, 27, 6590. https://doi.org/10.3390/molecules27196590
Hazra S, Ray AS, Rahaman CH. Natural Phytocompounds from Common Indian Spices for Identification of Three Potential Inhibitors of Breast Cancer: A Molecular Modelling Approach. Molecules. 2022; 27(19):6590. https://doi.org/10.3390/molecules27196590
Chicago/Turabian StyleHazra, Samik, Anindya Sundar Ray, and Chowdhury Habibur Rahaman. 2022. "Natural Phytocompounds from Common Indian Spices for Identification of Three Potential Inhibitors of Breast Cancer: A Molecular Modelling Approach" Molecules 27, no. 19: 6590. https://doi.org/10.3390/molecules27196590
APA StyleHazra, S., Ray, A. S., & Rahaman, C. H. (2022). Natural Phytocompounds from Common Indian Spices for Identification of Three Potential Inhibitors of Breast Cancer: A Molecular Modelling Approach. Molecules, 27(19), 6590. https://doi.org/10.3390/molecules27196590