
JND4135, a New Type II TRK Inhibitor, Overcomes TRK xDFG and Other Mutation Resistance In Vitro and In Vivo

 ,
, 
Abstract
1. Introduction
2. Results
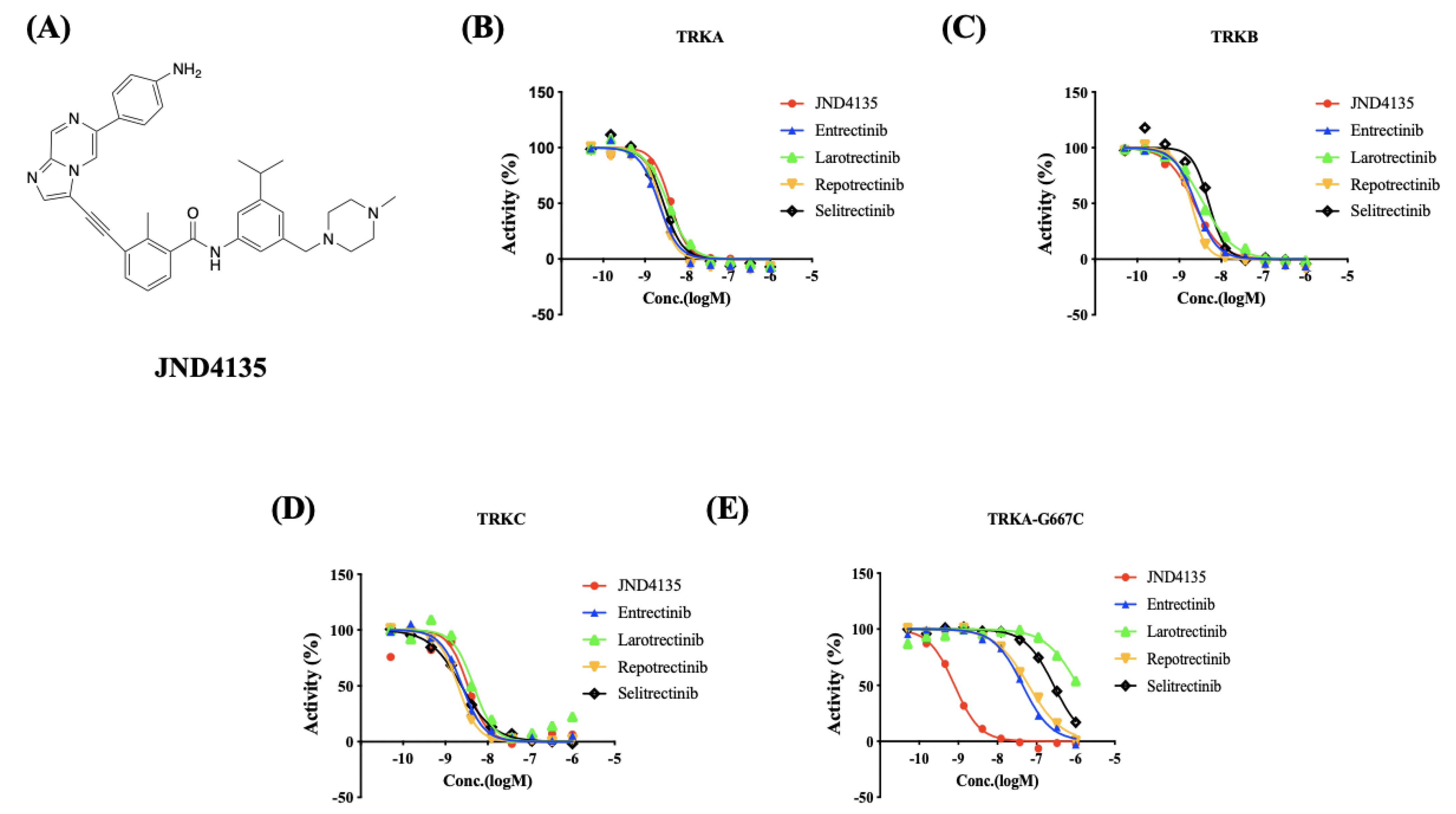
2.1. JND4135 Is a Strong TRK Inhibitor against TRK WT and TRKA-G667C
2.2. JND4135 Overcomes TRK xDFG and Other Mutant Resistance in BaF3 Stable Models
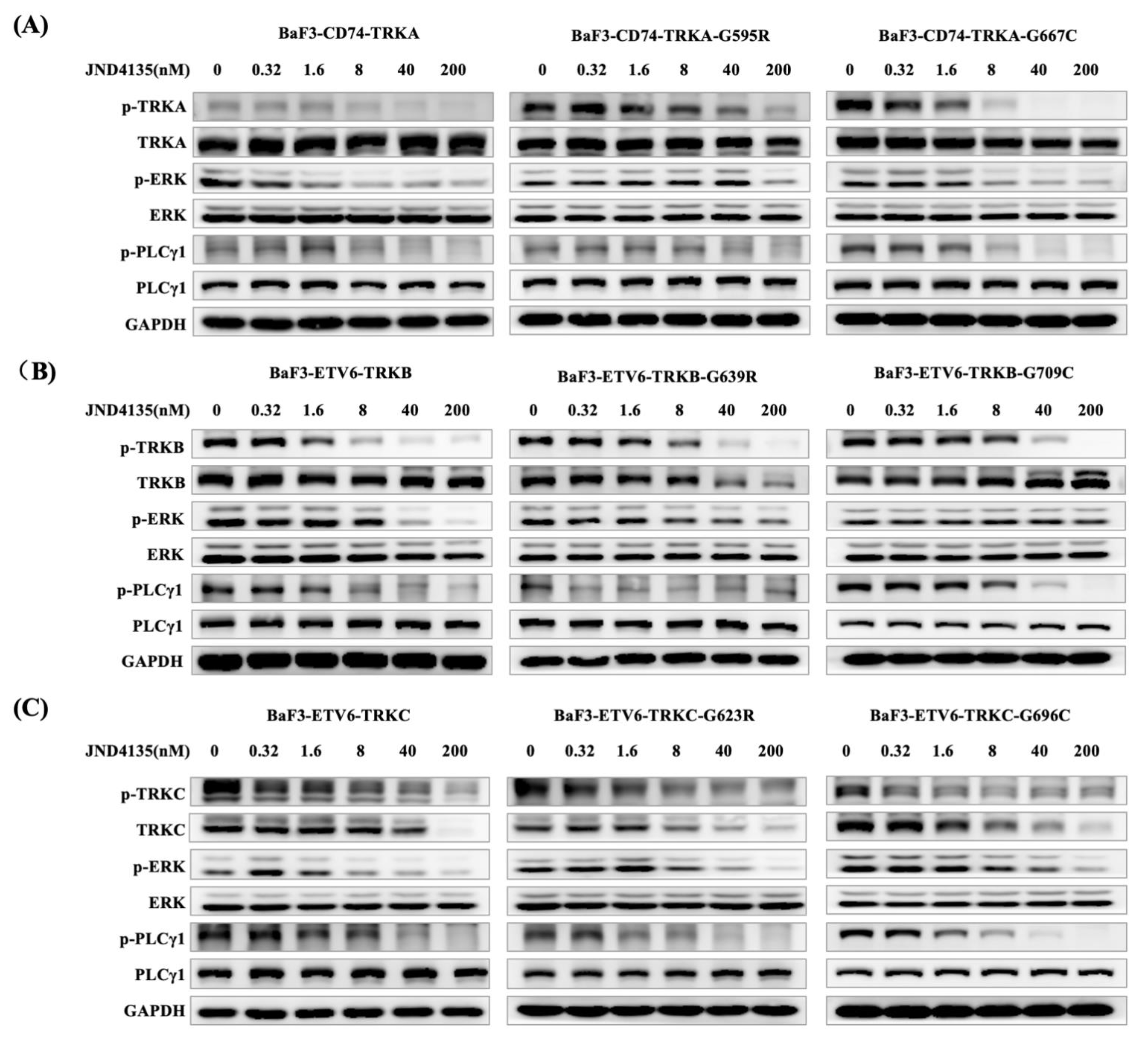
2.3. JND4135 Dose-Dependently Inhibits the Phosphorylation of TRKs WT and xDFG Mutation and Its Downstream Signal Molecules
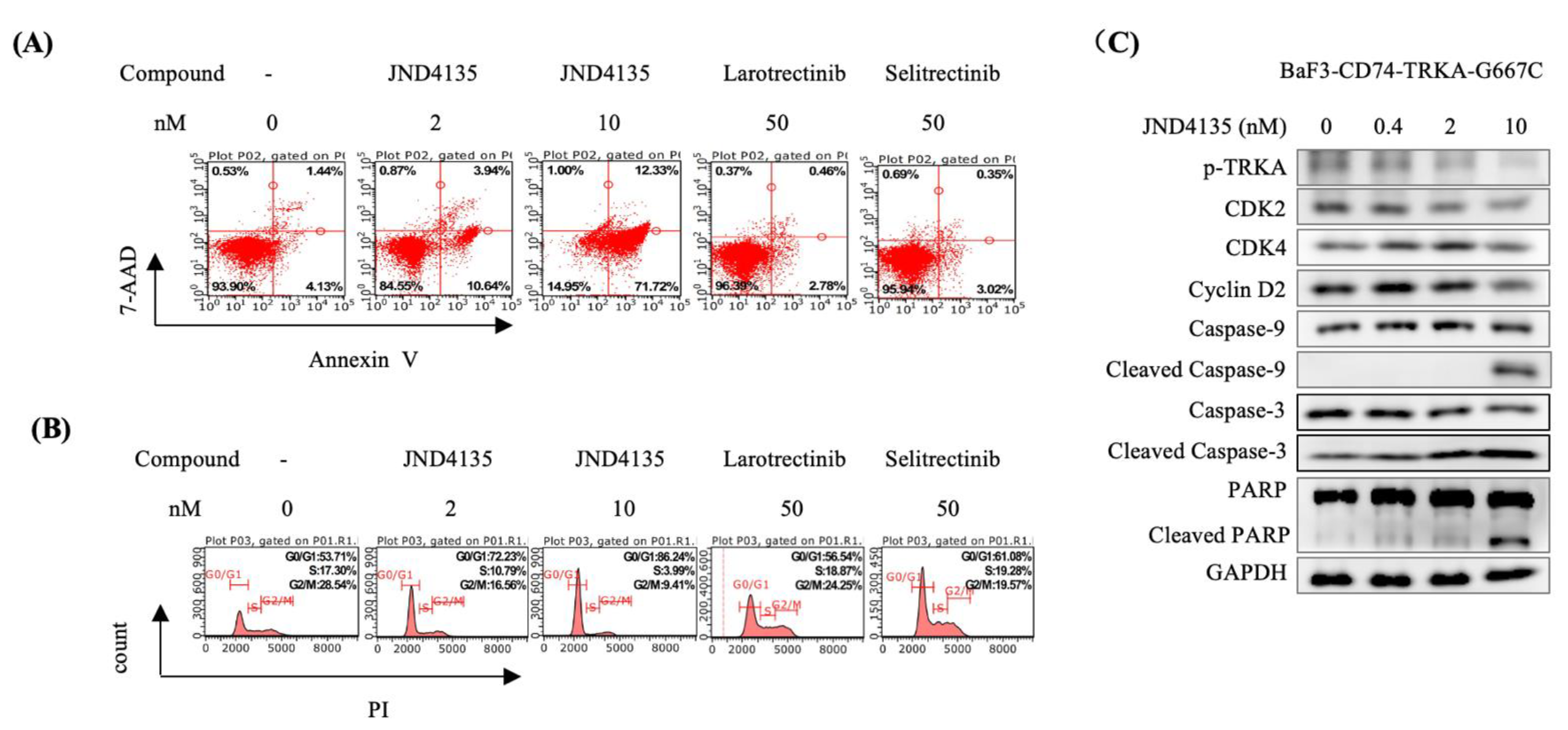
2.4. JND4135 Induces G0/G1 Phase Arrest and Apoptosis in BaF3–CD74-TRKA-G667C Cells
2.5. Prolonged Occupation of JND4135 in TRKs Protein
2.6. JND4135 Is a Type II TRK Inhibitor
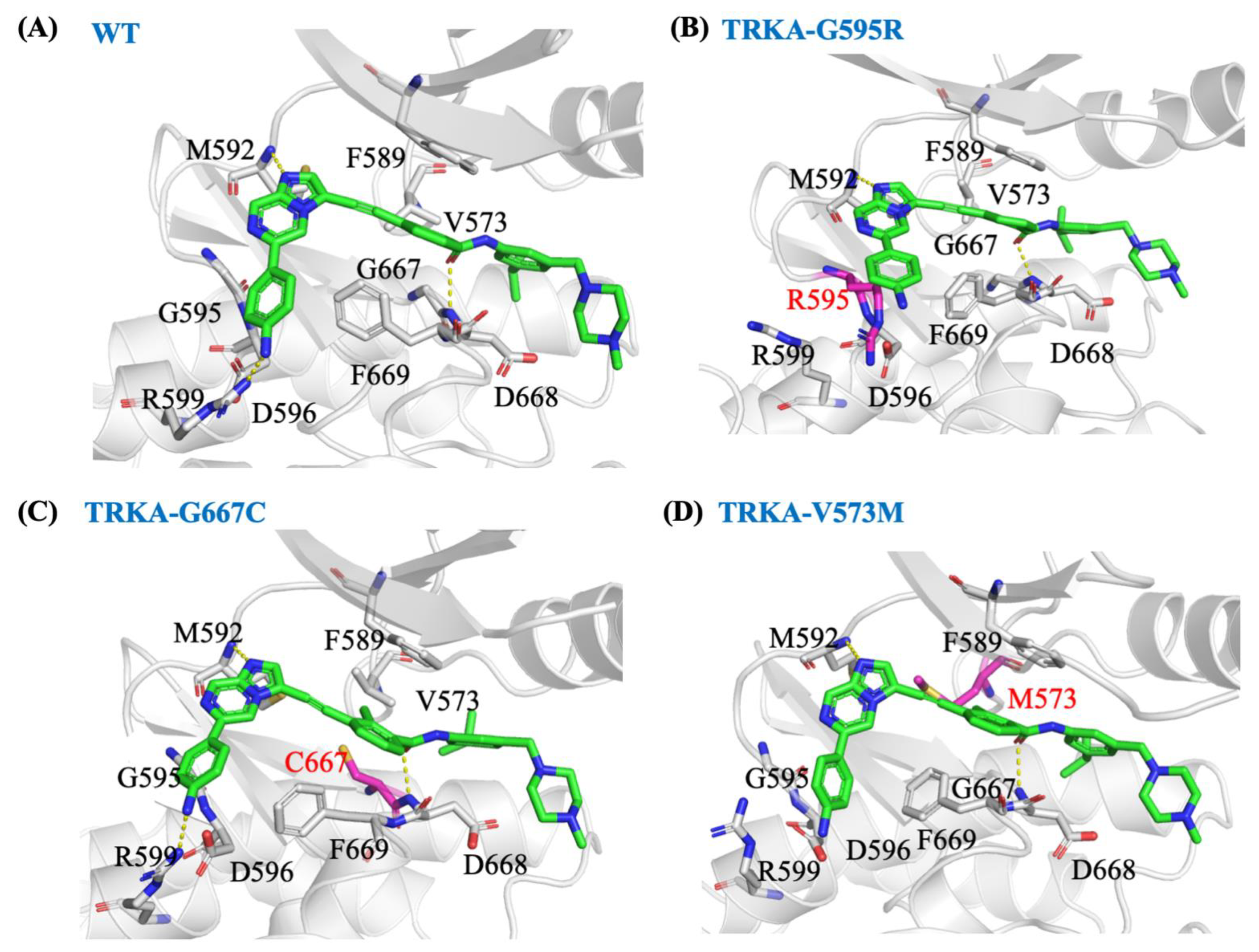
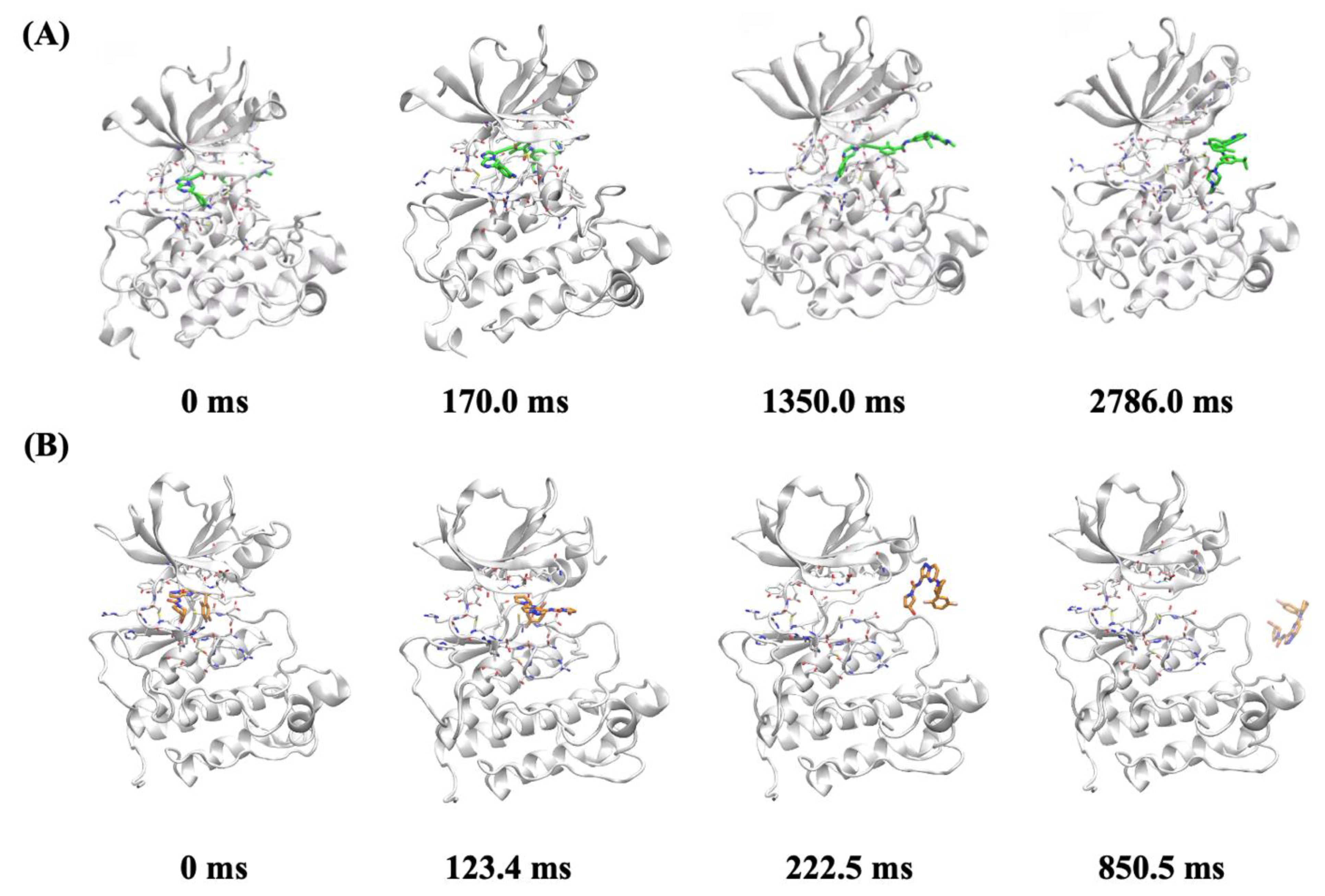
2.7. In Silico Molecular Mechanism Supporting the Long Occupation Time of JND4135
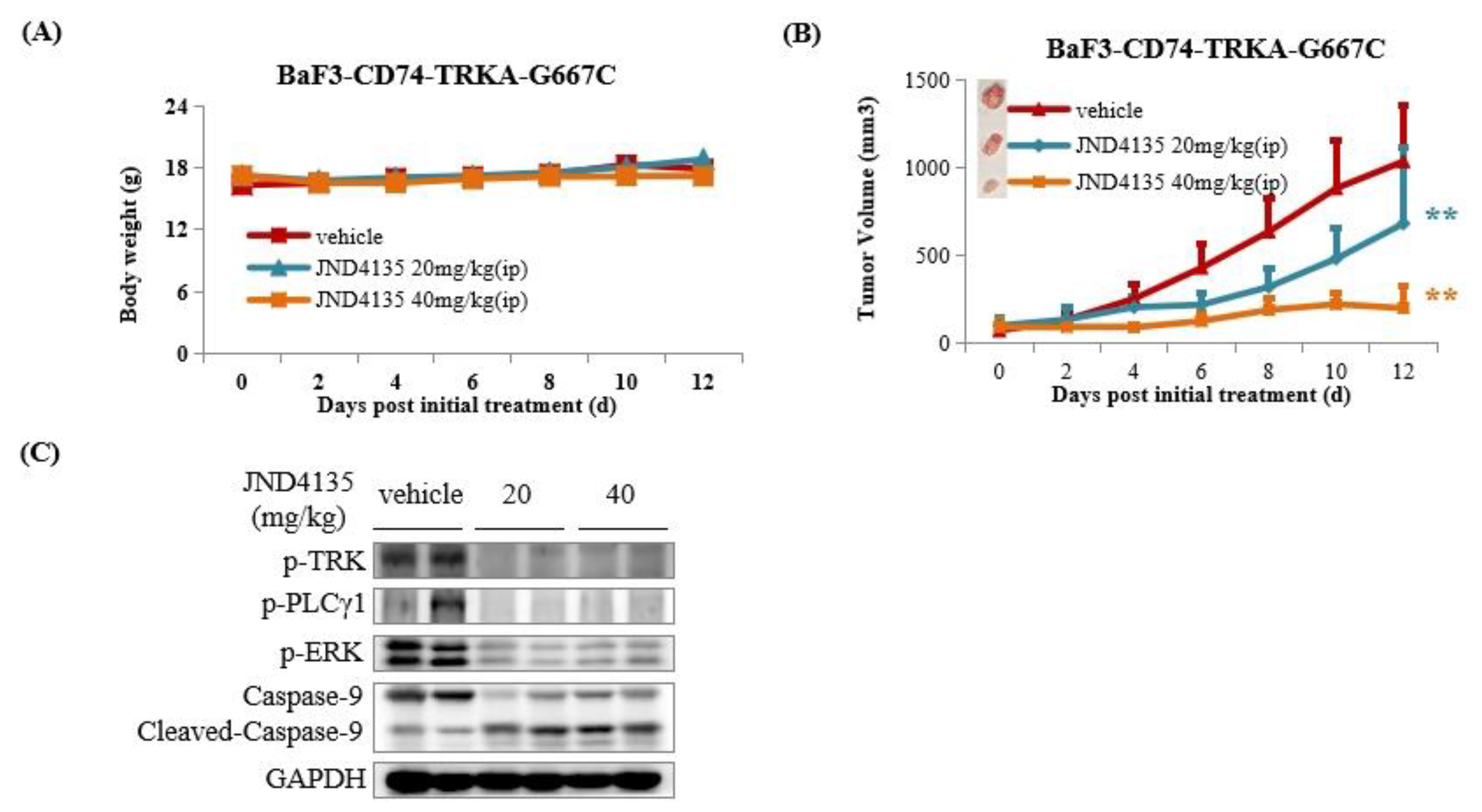
2.8. JND4135 Suppresses Tumor Growth in BaF3–CD74-TRKA-G667C Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Agents
4.3. Anti-Proliferation Assay In Vitro
4.4. In Vitro Enzymatic Activity Assay
4.5. Analysis of Cell Cycle and Cell Apoptosis by Flow Cytometry
4.6. Molecular Docking
4.7. Molecular Dynamic Simulations
4.8. Infrequent Metadynamics Simulations
4.9. Western Blot Analysis
4.10. Octet Binding Assay
4.11. In Vivo Experiments
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Vaishnavi, A.; Le, A.T.; Doebele, R.C. TRKing Down an Old Oncogene in a New Era of Targeted Therapy. Cancer Discov. 2015, 5, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A. TRK inhibitors in TRK fusion-positive cancers. Ann. Oncol. 2019, 30 (Suppl. S8), viii23–viii30. [Google Scholar] [CrossRef]
- Haratake, N.; Seto, T. NTRK Fusion-positive Non–small-cell Lung Cancer: The Diagnosis and Targeted Therapy. Clin. Lung Cancer 2021, 22, 1–5. [Google Scholar] [CrossRef]
- Joshi, S.K.; Davare, M.A.; Druker, B.J.; Tognon, C.E. Revisiting NTRKs as an emerging oncogene in hematological malignancies. Leukemia 2019, 33, 2563–2574. [Google Scholar] [CrossRef]
- Doebele, R.C.; Davis, L.E.; Vaishnavi, A.; Le, A.T.; Estrada-Bernal, A.; Keysar, S.; Jimeno, A.; Varella-Garcia, M.; Aisner, D.L.; Li, Y.; et al. An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101. Cancer Discov. 2015, 5, 1049–1057. [Google Scholar] [CrossRef]
- Rolfo, C.; Ruiz, R.; Giovannetti, E.; Gil-Bazo, I.; Russo, A.; Passiglia, F.; Giallombardo, M.; Peeters, M.; Raez, L. Entrectinib: A potent new TRK, ROS1, and ALK inhibitor. Expert Opin. Investig. Drugs 2015, 24, 1493–1500. [Google Scholar] [CrossRef]
- Thein, K.Z.; Lemery, S.J.; Kummar, S. Tissue-Agnostic Drug Development: A New Path to Drug Approval. Cancer Discov. 2021, 11, 2139–2144. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Misale, S.; Wei, G.; Siravegna, G.; Crisafulli, G.; Lazzari, L.; Corti, G.; Rospo, G.; Novara, L.; Mussolin, B.; et al. Acquired Resistance to the TRK Inhibitor Entrectinib in Colorectal Cancer. Cancer Discov. 2016, 6, 36–44. [Google Scholar] [CrossRef]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion–Positive Solid Tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef]
- Doebele, R.C. Acquired Resistance Is Oncogene and Drug Agnostic. Cancer Cell 2019, 36, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Harada, G.; Santini, F.C.; Wilhelm, C.; Drilon, A. NTRK fusions in lung cancer: From biology to therapy. Lung. Cancer 2021, 161, 108–113. [Google Scholar] [CrossRef] [PubMed]
- MacFarland, S.P.; Naraparaju, K.; Iyer, R.; Guan, P.; Kolla, V.; Hu, Y.; Tan, K.; Brodeur, G.M. Mechanisms of Entrectinib Resistance in a Neuroblastoma Xenograft Model. Mol. Cancer Ther. 2020, 19, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Schram, A.M.; Kulick, A.; Misale, S.; Won, H.H.; Yaeger, R.; Razavi, P.; Ptashkin, R.; Hechtman, J.F.; Toska, E.; et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat. Med. 2019, 25, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Ou, S.-H.I.; Cho, B.C.; Kim, D.-W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.-J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef]
- Nishiyama, A.; Yamada, T.; Kita, K.; Wang, R.; Arai, S.; Fukuda, K.; Tanimoto, A.; Takeuchi, S.; Tange, S.; Tajima, A.; et al. Foretinib Overcomes Entrectinib Resistance Associated with the NTRK1 G667C Mutation in NTRK1 Fusion–Positive Tumor Cells in a Brain Metastasis Model. Clin. Cancer Res. 2018, 24, 2357–2369. [Google Scholar] [CrossRef]
- Somwar, R.; Hofmann, N.E.; Smith, B.; Odintsov, I.; Vojnic, M.; Linkov, I.; Tam, A.; Khodos, I.; Mattar, M.S.; de Stanchina, E.; et al. NTRK kinase domain mutations in cancer variably impact sensitivity to type I and type II inhibitors. Commun. Biol. 2020, 3, 776. [Google Scholar] [CrossRef]
- Cocco, E.; Lee, J.E.; Kannan, S.; Schram, A.M.; Won, H.H.; Shifman, S.; Kulick, A.; Baldino, L.; Toska, E.; Arruabarrena-Aristorena, A.; et al. TRK xDFG Mutations Trigger a Sensitivity Switch from Type I to II Kinase Inhibitors. Cancer Discov. 2021, 11, 126–141. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, W.; Liu, X.; Zou, F.; Wang, J.; Liu, Q.; Wang, A.; Hu, Z.; Chen, Y.; Qi, S.; et al. Discovery of (E)-N-(4-methyl-5-(3-(2-(pyridin-2-yl)vinyl)-1H-indazol-6-yl)thiazol-2-yl)-2-(4-methylpiperazin-1-yl)acetamide (IHMT-TRK-284) as a novel orally available type II TRK kinase inhibitor capable of overcoming multiple resistant mutants. Eur. J. Med. Chem. 2020, 207, 112744. [Google Scholar] [CrossRef]
- Zhuo, L.-S.; Wang, M.-S.; Wu, F.-X.; Xu, H.-C.; Gong, Y.; Yu, Z.-C.; Tian, Y.-G.; Pang, C.; Hao, G.-F.; Huang, W.; et al. Discovery of Next-Generation Tropomyosin Receptor Kinase Inhibitors for Combating Multiple Resistance Associated with Protein Mutation. J. Med. Chem. 2021, 64, 15503–15514. [Google Scholar] [CrossRef]
- Bailey, J.J.; Jaworski, C.; Tung, D.; Wängler, C.; Wängler, B.; Schirrmacher, R. Tropomyosin receptor kinase inhibitors: An updated patent review for 2016–2019. Expert. Opin. Ther. Pat. 2020, 30, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Wang, Y.; Wang, Y.; Tang, X.; Ren, X.; Zhang, L.; Xu, Y.; Zhang, Z.; Zhang, Z.-M.; Lu, X.; et al. Design, synthesis and biological evaluation of 3-(imidazo[1,2-a]pyrazin-3-ylethynyl)-2-methylbenzamides as potent and selective pan-tropomyosin receptor kinase (TRK) inhibitors. Eur. J. Med. Chem. 2019, 179, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wang, J.; Zhu, S.; Tu, Z.-C.; Zhang, Z.; Chan, S.; Ding, K. Design, synthesis, and Structure–Activity Relationships (SAR) of 3-vinylindazole derivatives as new selective tropomyosin receptor kinases (Trk) inhibitors. Eur. J. Med. Chem. 2020, 203, 112552. [Google Scholar] [CrossRef]
- Zou, J.; Zhang, Z.; Xu, F.; Cui, S.; Qi, C.; Luo, J.; Wang, Z.; Lu, X.; Tu, Z.; Ren, X.; et al. GZD2202, a novel TrkB inhibitor, suppresses BDNF-mediated proliferation and metastasis in neuroblastoma models. J. Drug Target. 2019, 27, 442–450. [Google Scholar] [CrossRef]
- Tanaka, H.; Sase, H.; Tsukaguchi, T.; Hasegawa, M.; Tanimura, H.; Yoshida, M.; Sakata, K.; Fujii, T.; Tachibana, Y.; Takanashi, K.; et al. Selective TRK Inhibitor CH7057288 against TRK Fusion-Driven Cancer. Mol. Cancer Ther. 2018, 17, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Zhang, L.; Luo, X.; Nan, J.; Yang, W.; Bin, H.; Li, Y.; Huang, Q.; Wang, T.; Pan, Z.; et al. Structural Optimization and Structure–Activity Relationship Studies of 6,6-Dimethyl-4-(phenylamino)-6H-pyrimido[5,4-b][1,4]oxazin-7(8H)-one Derivatives as A New Class of Potent Inhibitors of Pan-Trk and Their Drug-Resistant Mutants. J. Med. Chem. 2022, 65, 2035–2058. [Google Scholar] [CrossRef]
- Smith, K.M.; Fagan, P.C.; Pomari, E.; Germano, G.; Frasson, C.; Walsh, C.; Silverman, I.M.; Bonvini, P.; Li, G. Antitumor Activity of Entrectinib, a Pan-TRK, ROS1, and ALK Inhibitor, in ETV6-NTRK3–Positive Acute Myeloid Leukemia. Mol. Cancer Ther. 2018, 17, 455–463. [Google Scholar] [CrossRef]
- Casasnovas, R.; Limongelli, V.; Tiwary, P.; Carloni, P.; Parrinello, M. Unbinding Kinetics of a p38 MAP Kinase Type II Inhibitor from Metadynamics Simulations. J. Am. Chem. Soc. 2017, 139, 4780–4788. [Google Scholar] [CrossRef]
- Tiwary, P.; Mondal, J.; Berne, B.J. How and when does an anticancer drug leave its binding site? Sci. Adv. 2017, 3, e1700014. [Google Scholar] [CrossRef]
- Zhou, Y.; Zou, R.; Kuang, G.; Långström, B.; Halldin, C.; Ågren, H.; Tu, Y. Enhanced Sampling Simulations of Ligand Unbinding Kinetics Controlled by Protein Conformational Changes. J. Chem. Inf. Model. 2019, 59, 3910–3918. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Wickstrom, L.; Okur, A.; Simmerling, C. Evaluating the Performance of the ff99SB Force Field Based on NMR Scalar Coupling Data. Biophys. J. 2009, 97, 853–856. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50(nM ± SD) | JND4135 | Entrectinib | Larotrectinib | Selitrectinib | Repotrectinib | |
|---|---|---|---|---|---|---|
| Parental | BaF3 (+IL3) | 1946 ± 25.1 | 2344 ± 202 | >10,000 | >10,000 | 906 ± 69.0 |
| WT | MPRIP-TRKA | 7.1 ± 2.3 | 1.6 ± 0.4 | 4.8 ± 1.2 | 1.1 ± 0.4 | 1.1 ± 0.2 |
| CD74-TRKA | 10.4 ± 3.2 | 4.4 ± 1.0 | 8.0 ± 0.8 | 2.5 ± 0.2 | 1.6 ± 0.3 | |
| QKI-TRKB | 2.4 ± 0.6 | 4.8 ± 1.6 | 11.4 ± 4.2 | 1.5 ± 0.4 | 0.8 ± 0.7 | |
| ETV6-TRKB | 23.1 ± 5.9 | 8.0 ± 3.1 | 24.0 ± 4.2 | 4.9 ± 1.4 | 2.3 ± 0.5 | |
| EML4-TRKC | 13.2 ± 2.6 | 5.2 ± 1.6 | 21.7 ± 6.9 | 2.1 ± 0.2 | 2.7 ± 1.6 | |
| ETV6-TRKC | 10.4 ± 4.0 | 7.8 ± 4.2 | 11.2 ± 2.8 | 1.7 ± 1.1 | 2.1 ± 0.9 | |
| Solvent Front | CD74-TRKA-G595R | 33.7 ± 7.1 | 321 ± 10.7 | 1110 ± 251 | 13.6 ± 3.3 | 12.0 ± 3.9 |
| ETV6-TRKB-G639R | 10.1 ± 3.9 | 529 ± 130 | 1896 ± 778 | 11.4 ± 2.2 | 9.7 ± 1.2 | |
| ETV6-TRKC-G623R | 9.4 ± 2.6 | 617 ± 69.8 | 1269 ± 165 | 9.6 ± 1.2 | 14.6 ± 4.7 | |
| ATP site | CD74-TRKA-V573M | 9.0 ± 4.8 | 114 ± 50.8 | 609 ± 195 | 26.2 ± 5.5 | 29.4 ± 1.3 |
| ETV6-TRKB-V617M | 3.9 ± 2.6 | 101 ± 4.8 | 1963 ± 203 | 45.9 ± 6.3 | 38.4 ± 4.4 | |
| ETV6-TRKC-V601M | 11.8 ± 5.7 | 125 ± 56.1 | 2983 ± 184 | 52.4 ± 36.9 | 65.6 ± 28.9 | |
| Gatekeeper | CD74-TRKA-F589L | 75.0 ± 12.7 | 2.1 ± 0.3 | 423 ± 92.6 | 17.3 ± 2.2 | 1.9 ± 0.1 |
| ETV6-TRKB-F633L | 178 ± 4.3 | 13.0 ± 3.6 | 1396 ± 214 | 55.8 ± 10.8 | 4.9 ± 2.2 | |
| ETV6-TRKC-F617L | 37.3 ± 14.0 | 4.6 ± 2.8 | 513 ± 156 | 20.8 ± 7.9 | 2.1 ± 1.1 | |
| xDFG | CD74-TRKA-G667C | 1.4 ± 0.2 | 33.2 ± 3.2 | 313 ± 48.5 | 66.3 ± 13 | 27.4 ± 5.6 |
| ETV6-TRKB-G709C | 18.7 ± 2.9 | 285 ± 92.4 | 1628 ± 821 | 168 ± 73.0 | 89.7 ± 38.8 | |
| ETV6-TRKC-G696C | 1.3 ± 0.7 | 124 ± 60.4 | 769 ± 325 | 98.4 ± 55.0 | 42.9 ± 33.6 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Zhou, Y.; Tang, X.; Yu, X.; Wang, Y.; Chan, S.; Song, X.; Tu, Z.; Zhang, Z.; Lu, X.; et al. JND4135, a New Type II TRK Inhibitor, Overcomes TRK xDFG and Other Mutation Resistance In Vitro and In Vivo. Molecules 2022, 27, 6500. https://doi.org/10.3390/molecules27196500
Wang J, Zhou Y, Tang X, Yu X, Wang Y, Chan S, Song X, Tu Z, Zhang Z, Lu X, et al. JND4135, a New Type II TRK Inhibitor, Overcomes TRK xDFG and Other Mutation Resistance In Vitro and In Vivo. Molecules. 2022; 27(19):6500. https://doi.org/10.3390/molecules27196500
Chicago/Turabian StyleWang, Jie, Yang Zhou, Xia Tang, Xiuwen Yu, Yongjin Wang, Shingpan Chan, Xiaojuan Song, Zhengchao Tu, Zhimin Zhang, Xiaoyun Lu, and et al. 2022. "JND4135, a New Type II TRK Inhibitor, Overcomes TRK xDFG and Other Mutation Resistance In Vitro and In Vivo" Molecules 27, no. 19: 6500. https://doi.org/10.3390/molecules27196500
APA StyleWang, J., Zhou, Y., Tang, X., Yu, X., Wang, Y., Chan, S., Song, X., Tu, Z., Zhang, Z., Lu, X., Zhang, Z., & Ding, K. (2022). JND4135, a New Type II TRK Inhibitor, Overcomes TRK xDFG and Other Mutation Resistance In Vitro and In Vivo. Molecules, 27(19), 6500. https://doi.org/10.3390/molecules27196500