

Resveratrol–Maltol and Resveratrol–Thiophene Hybrids as Cholinesterase Inhibitors and Antioxidants: Synthesis, Biometal Chelating Capability and Crystal Structure

 , , , ,
, , , ,  , ,
, ,
Abstract
:
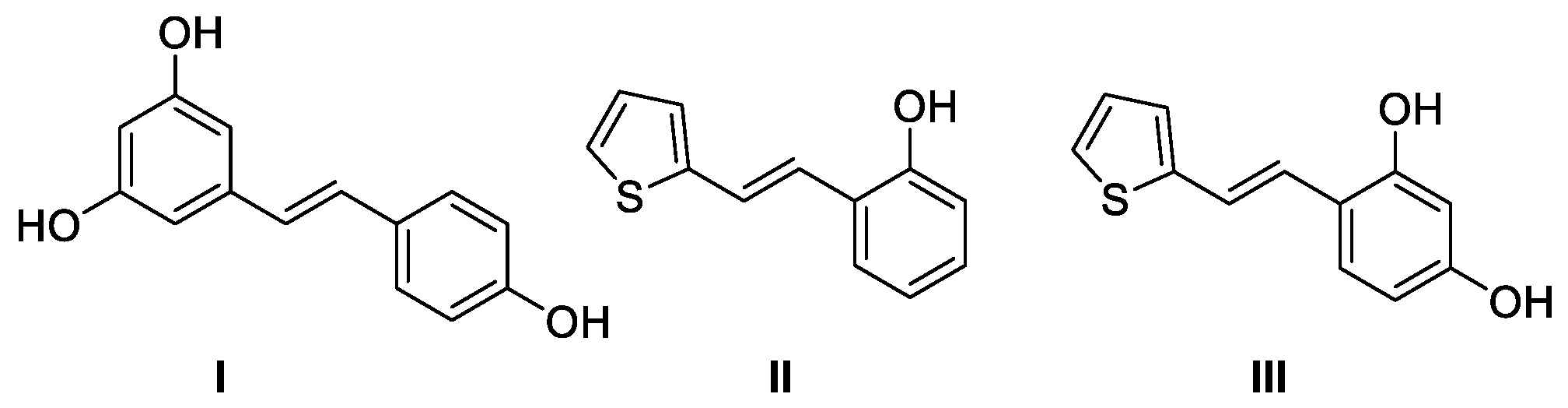
1. Introduction
2. Results and Discussion
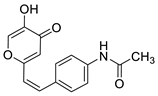
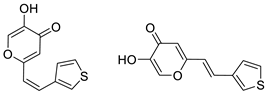
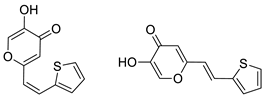
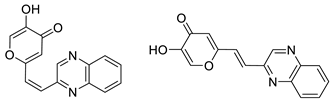
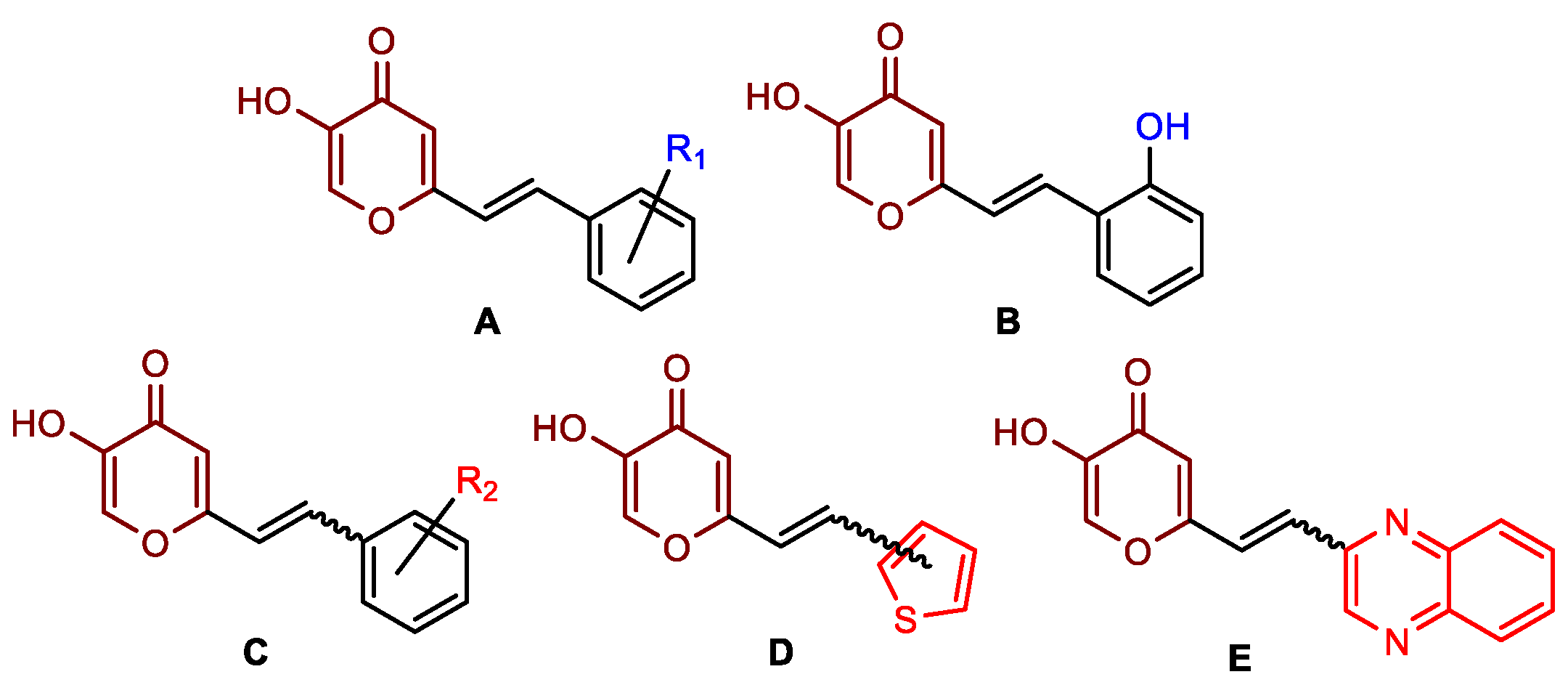
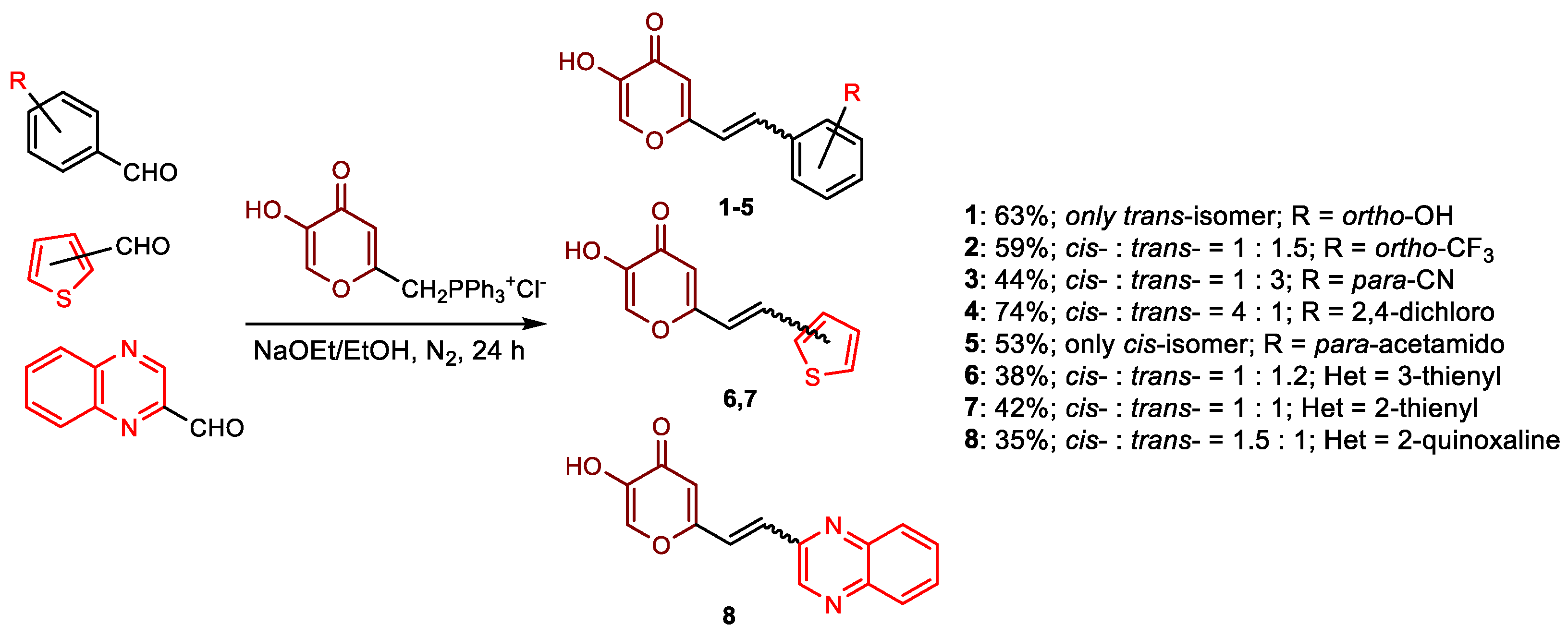
2.1. Synthesis and Characterization of Resveratrol–Thiophene II and III and Resveratrol–Maltol Hybrids 1–8
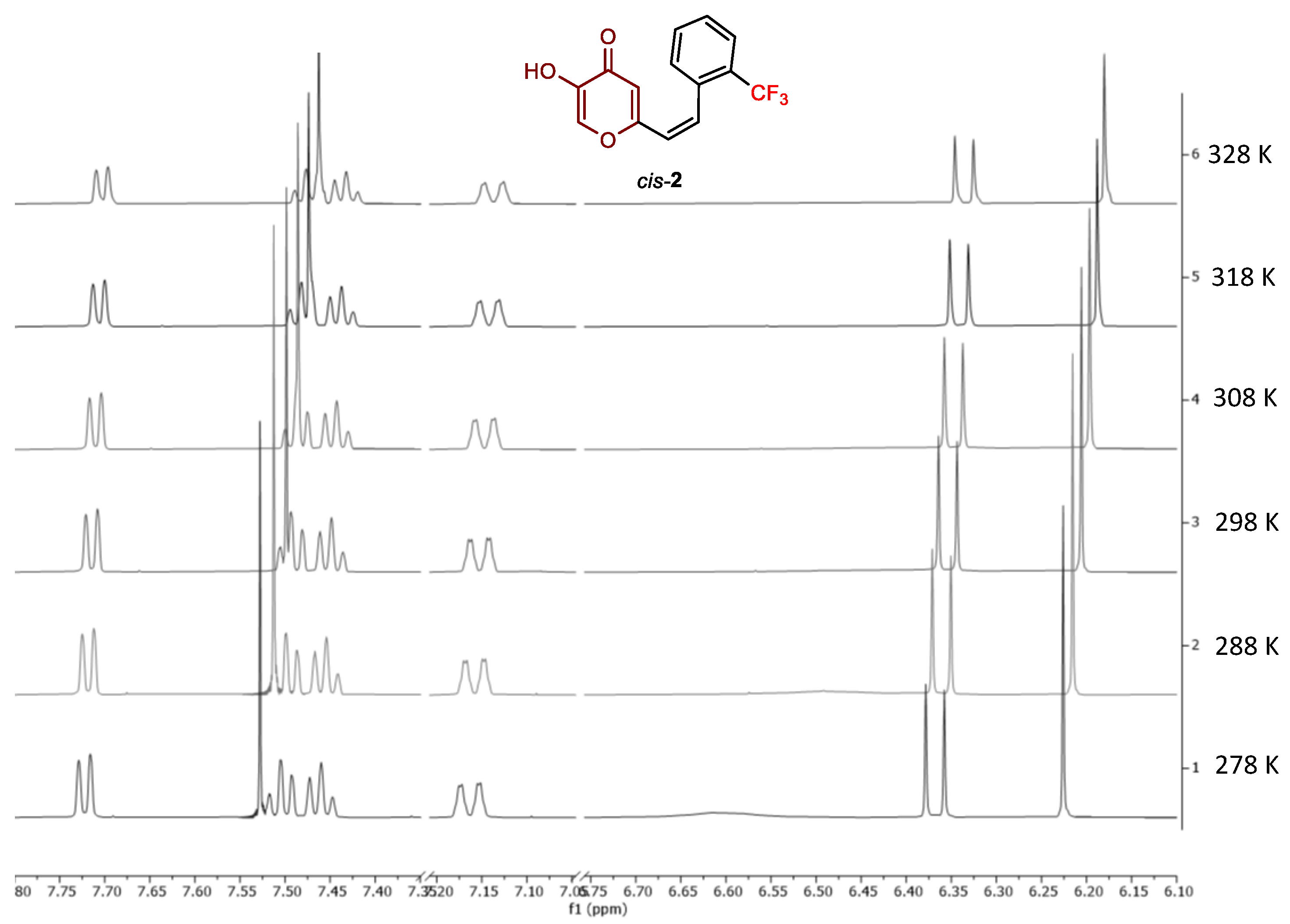
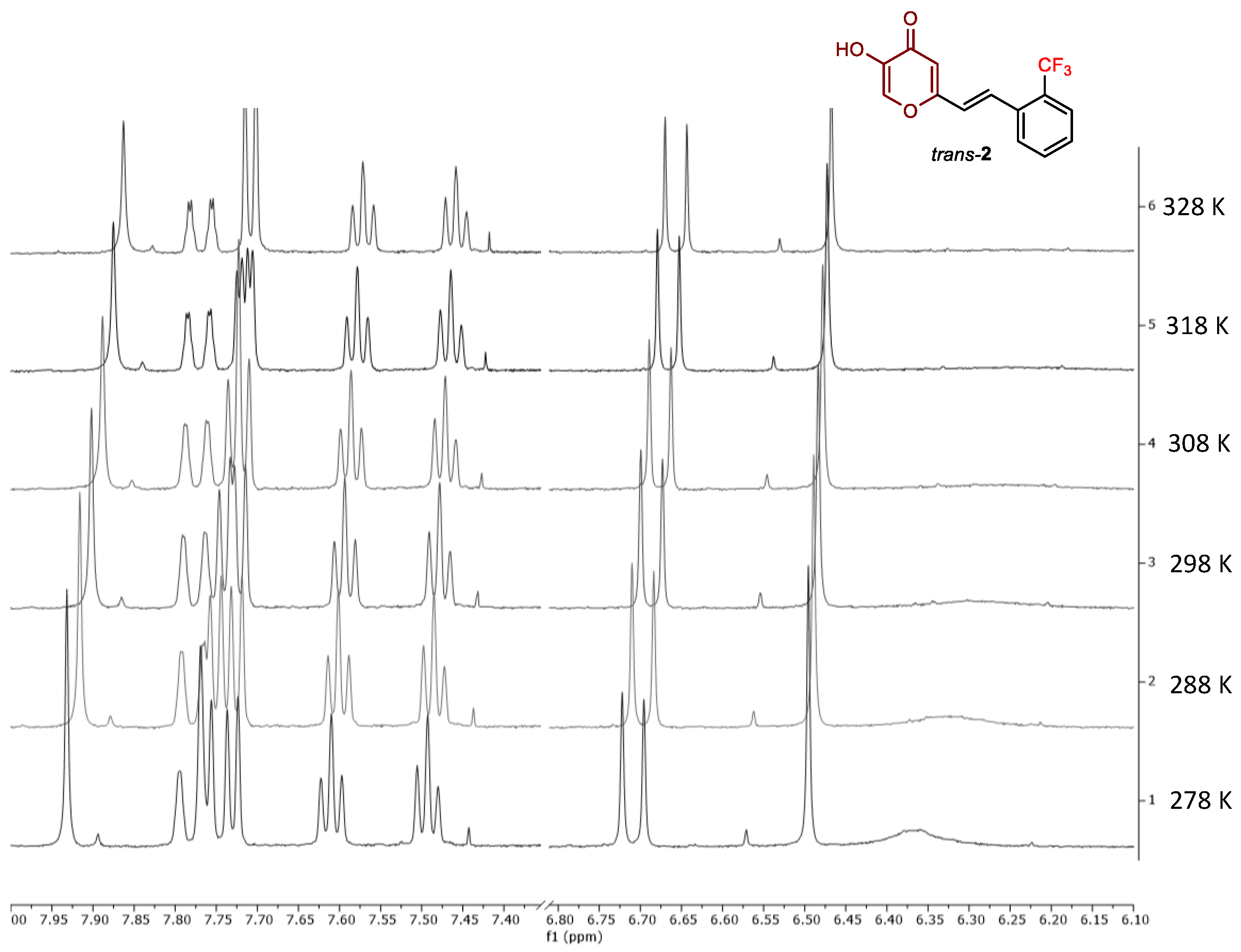
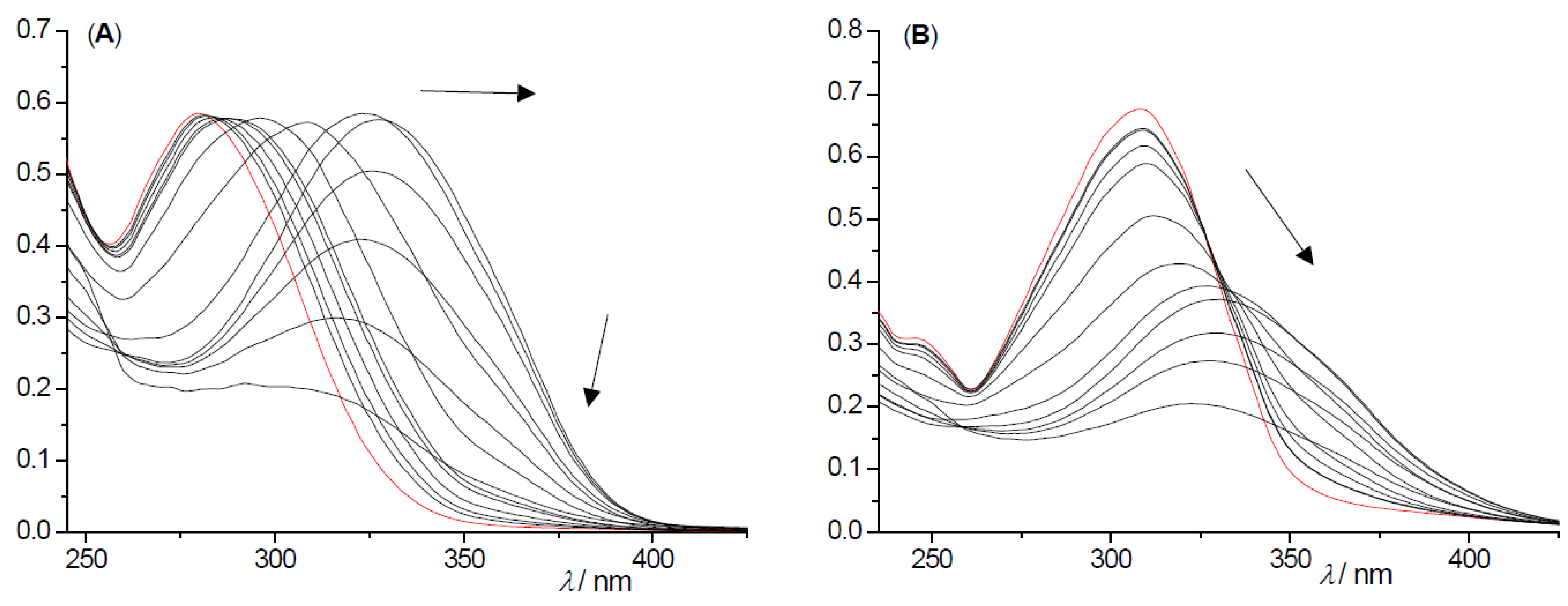
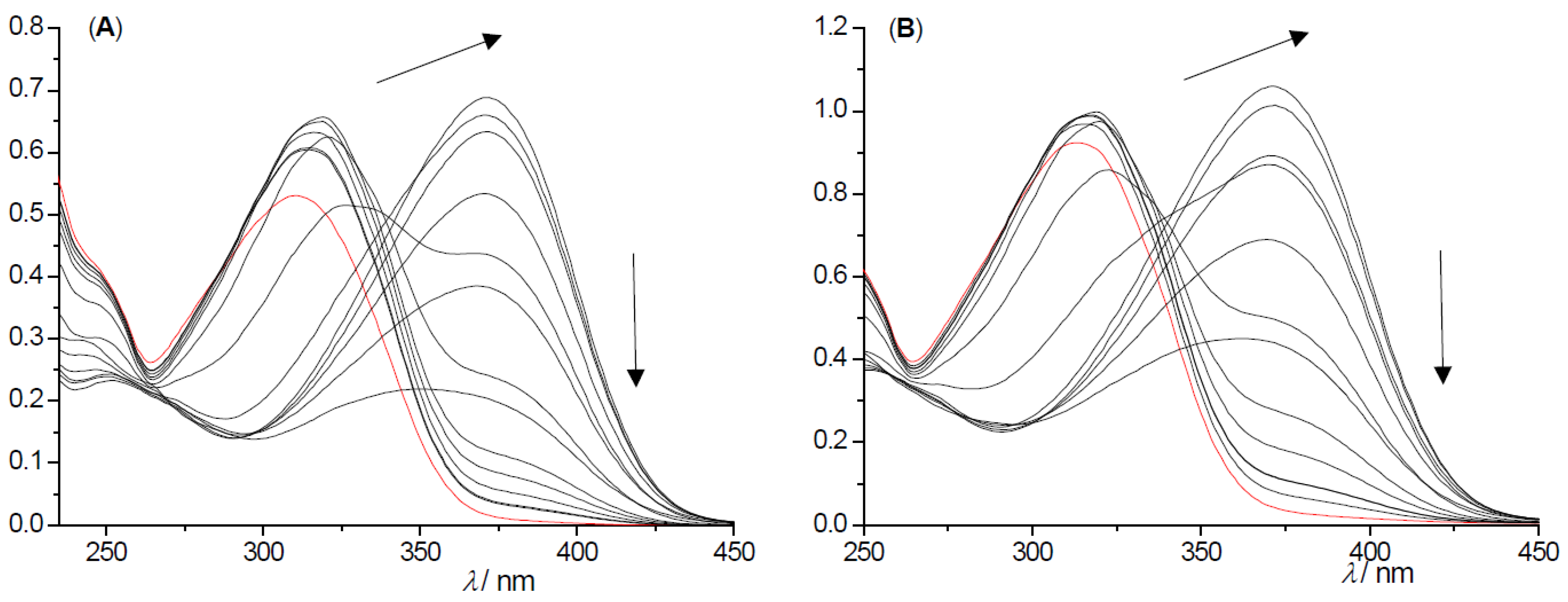
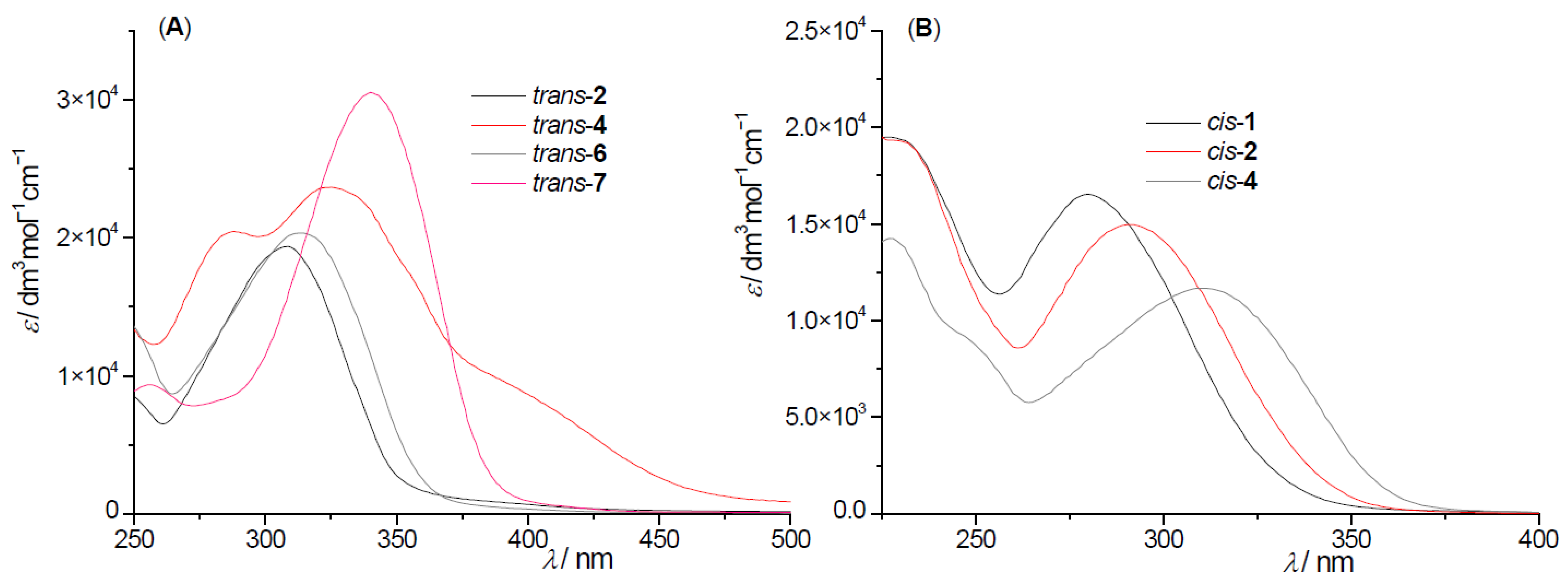
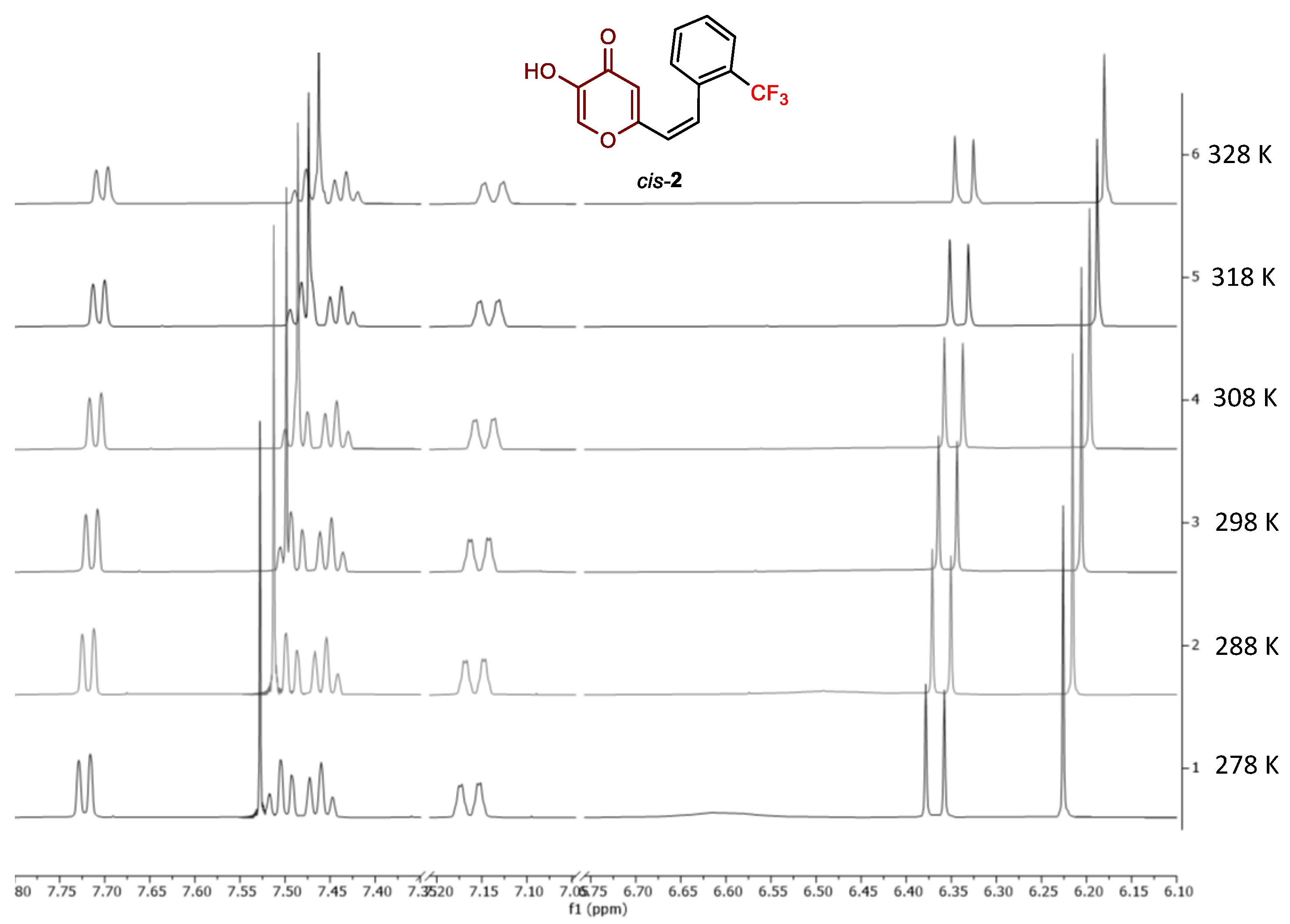
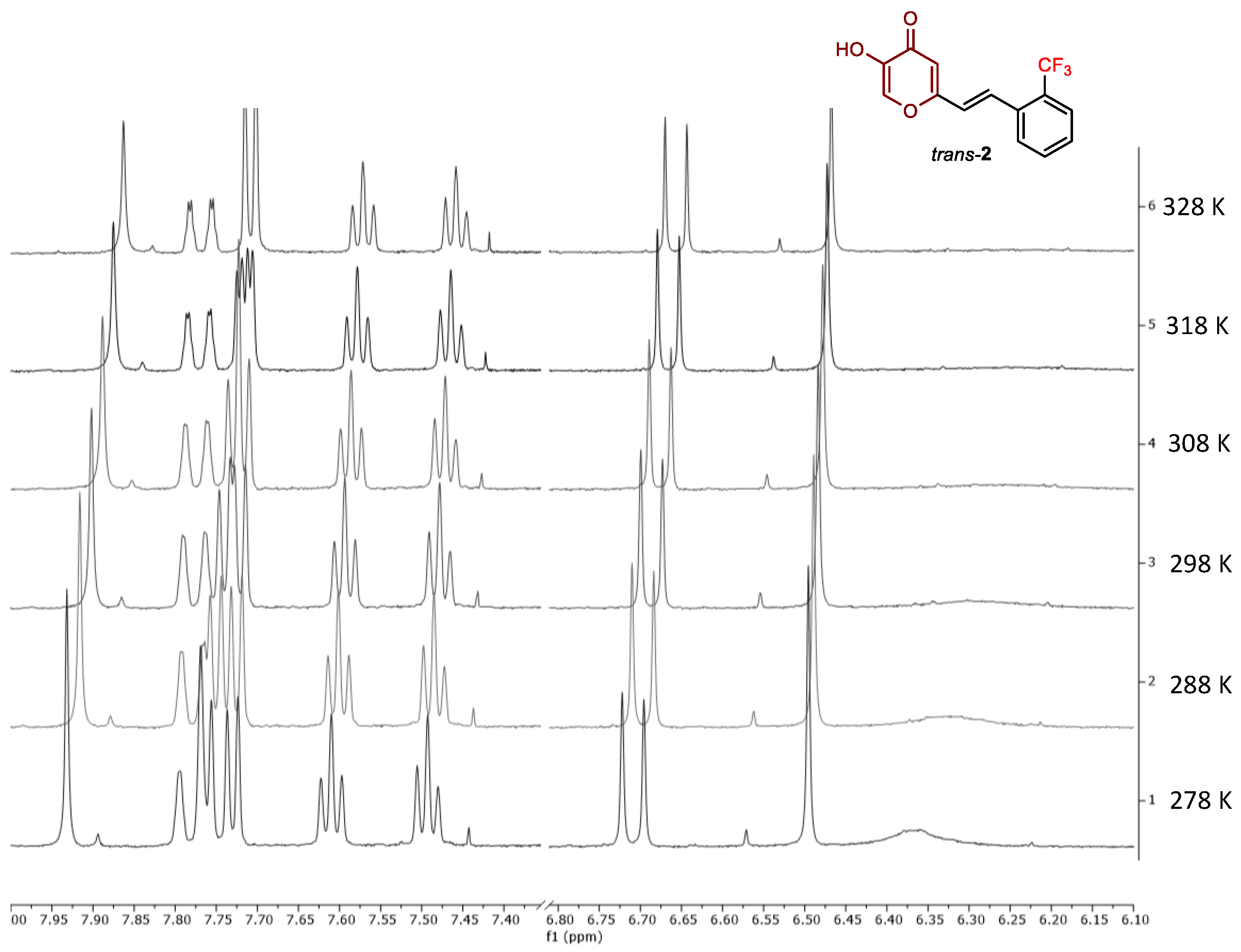
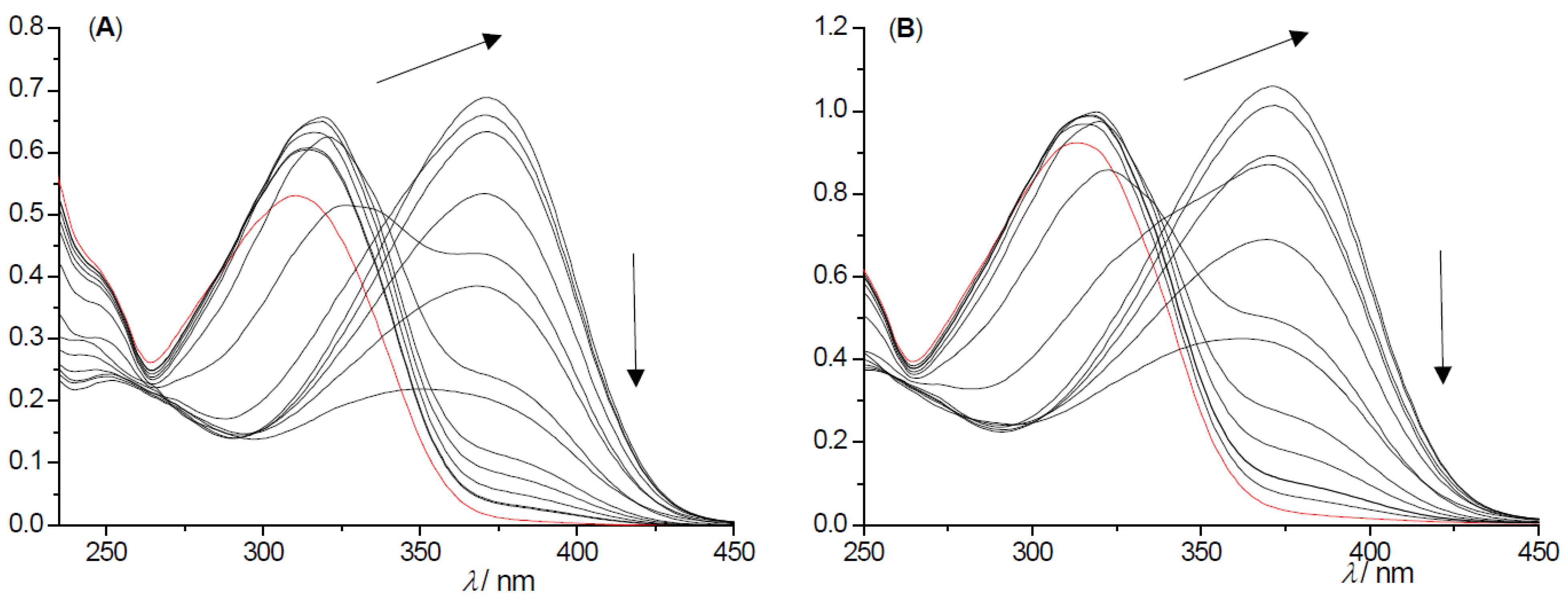
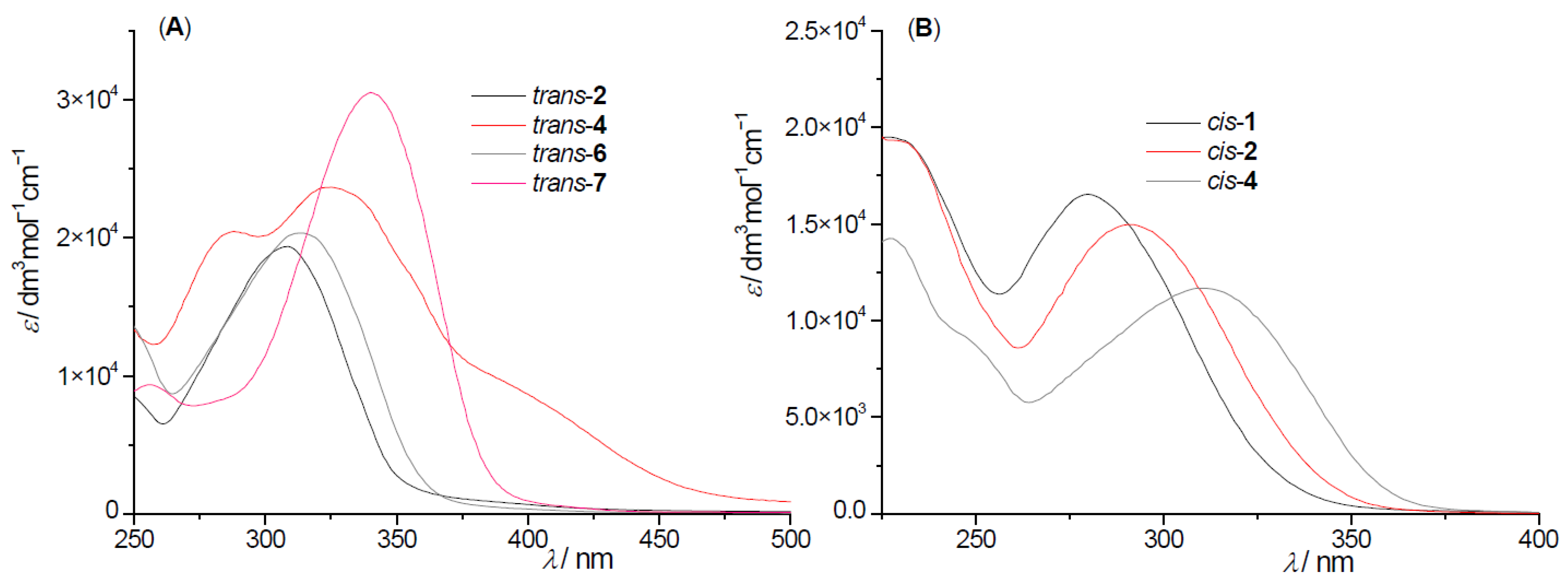
2.2. Photochemical Reactivity and Photophysical Properties of New Styryl-Pyranones 1–8
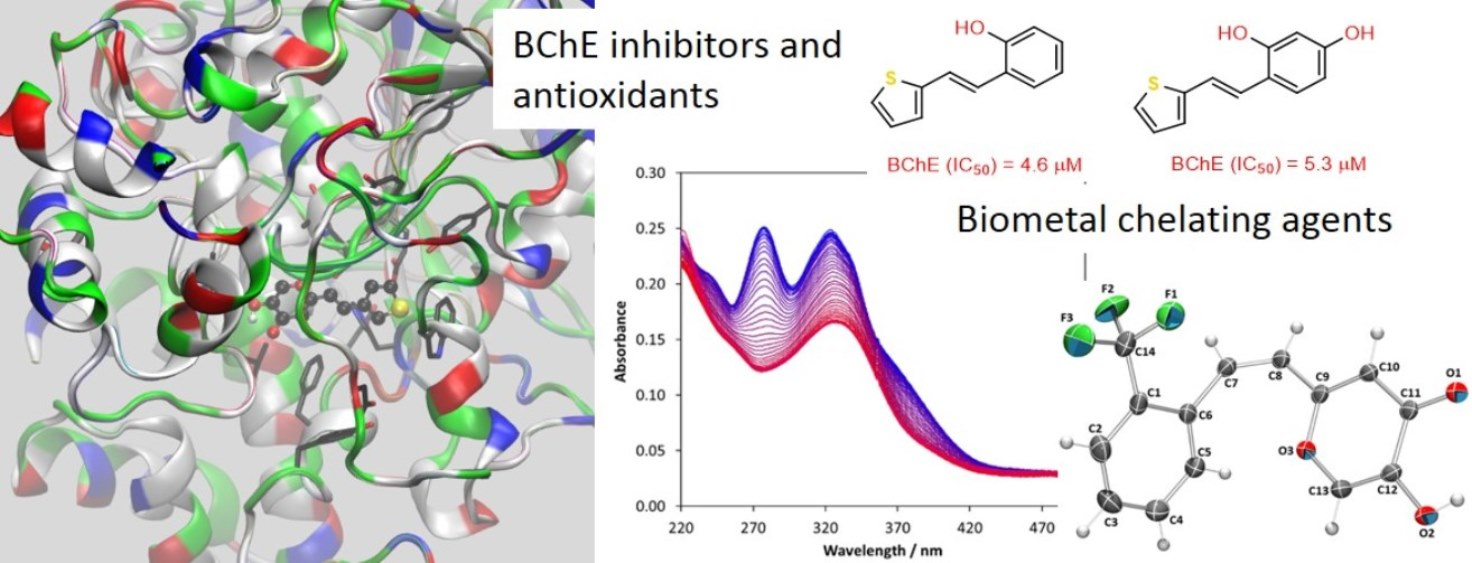
2.3. Cholinesterases Inhibitory and Antioxidative Activity of Resveratrol–Maltol and Resveratrol–Thiophene Hybrids
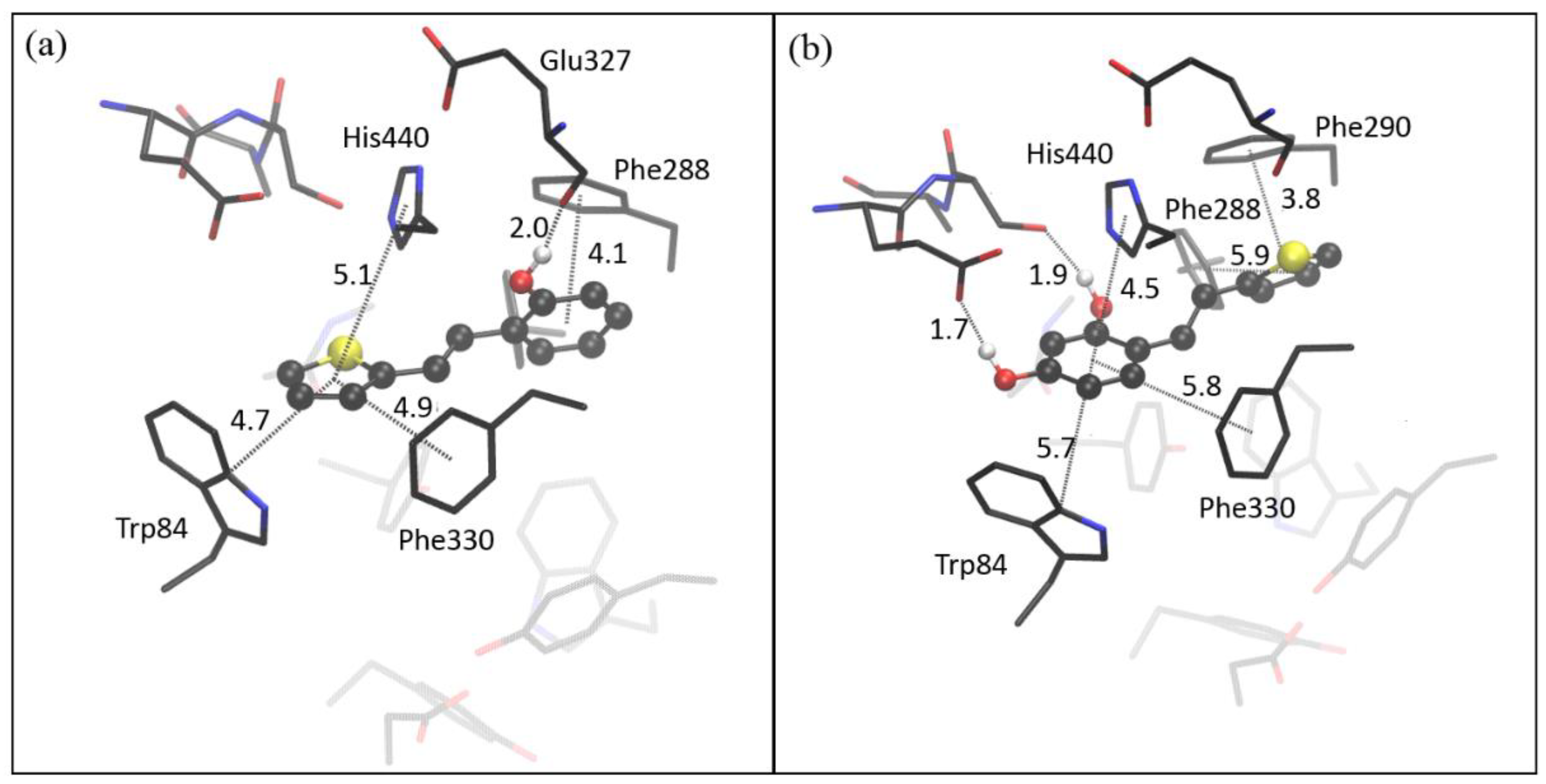
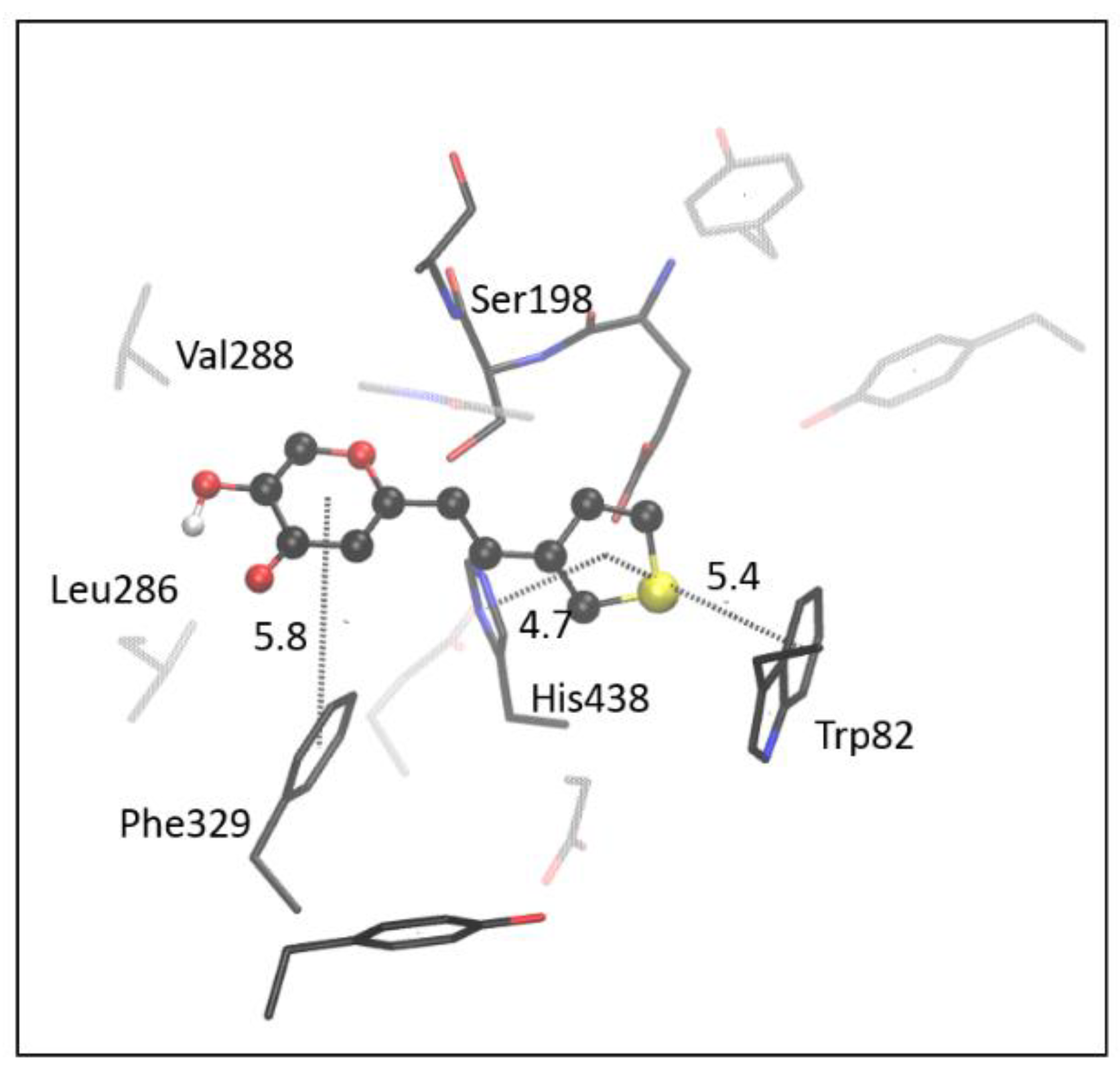
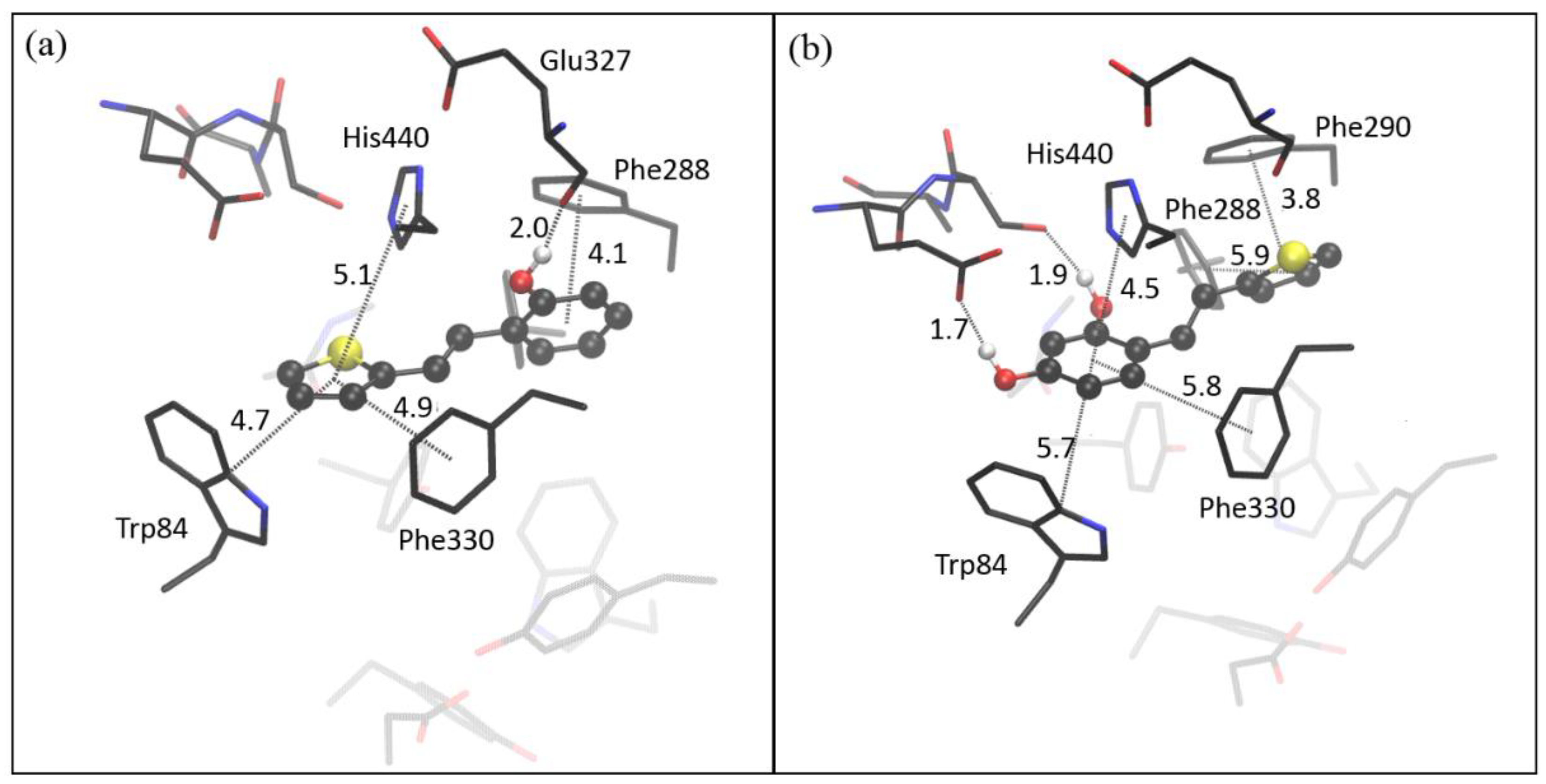
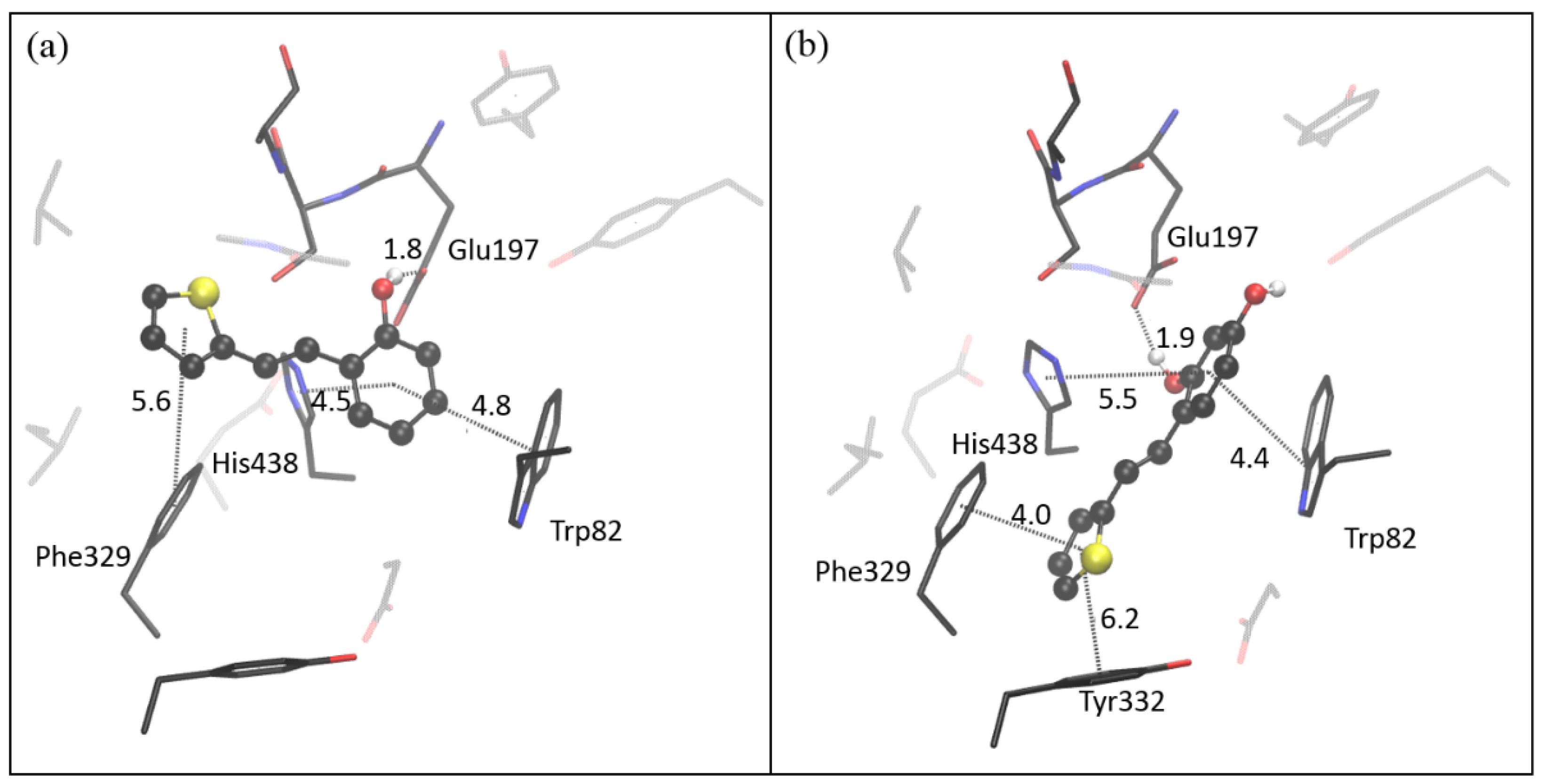
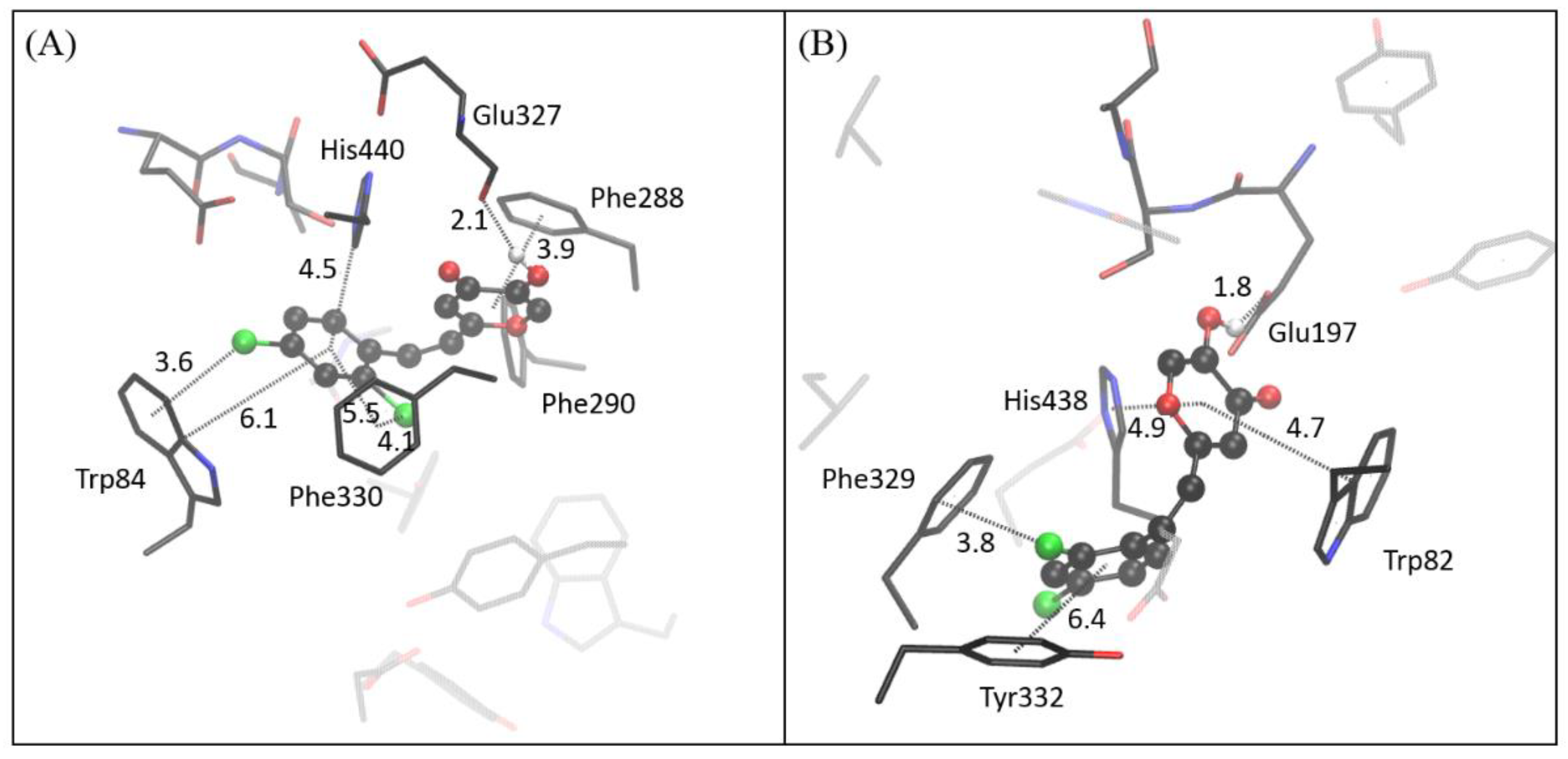
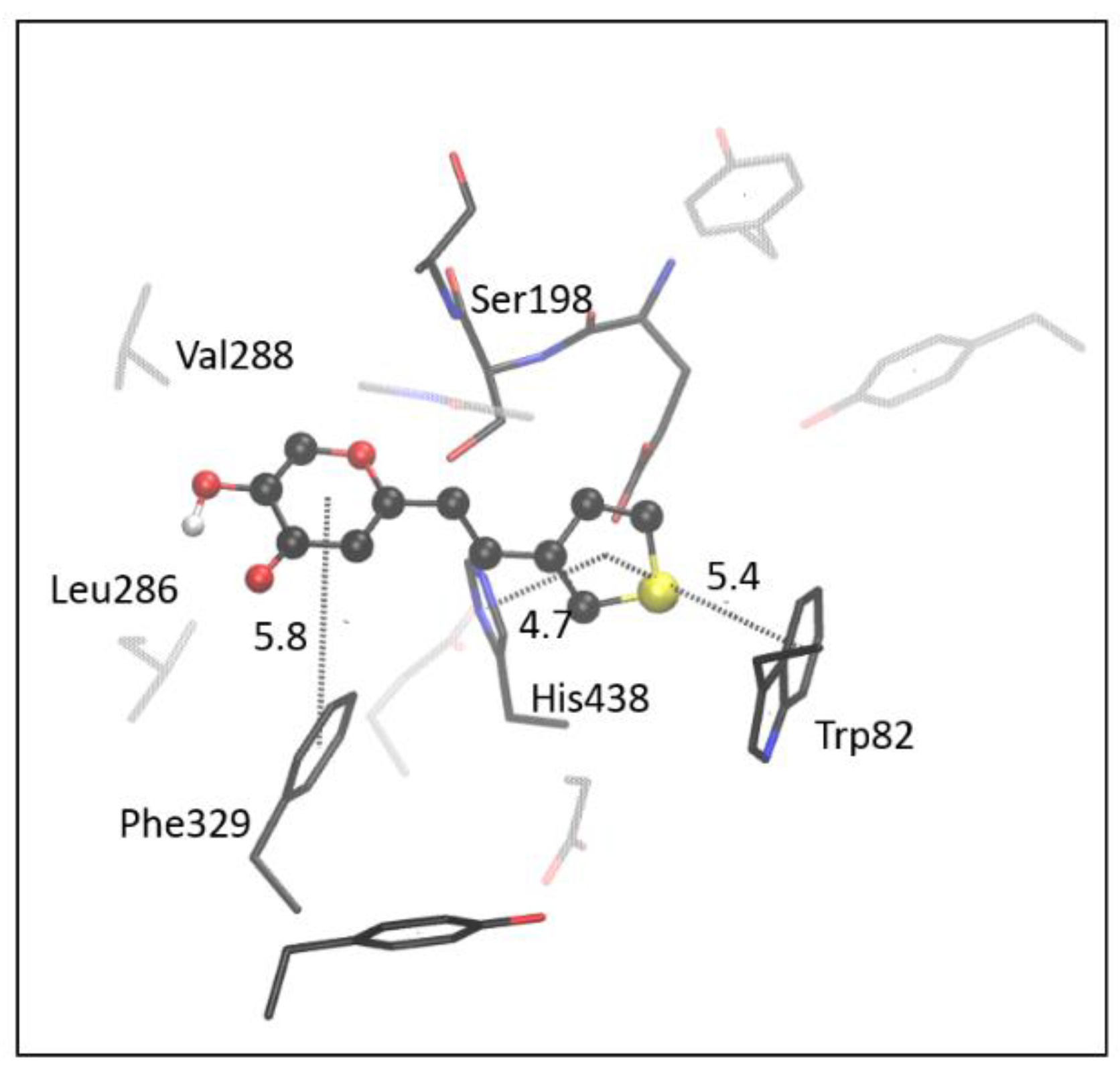
2.4. Computational Study of Resveratrol–Thiophene and Resveratrol–Maltol Hybrids as Cholinesterase Inhibitors
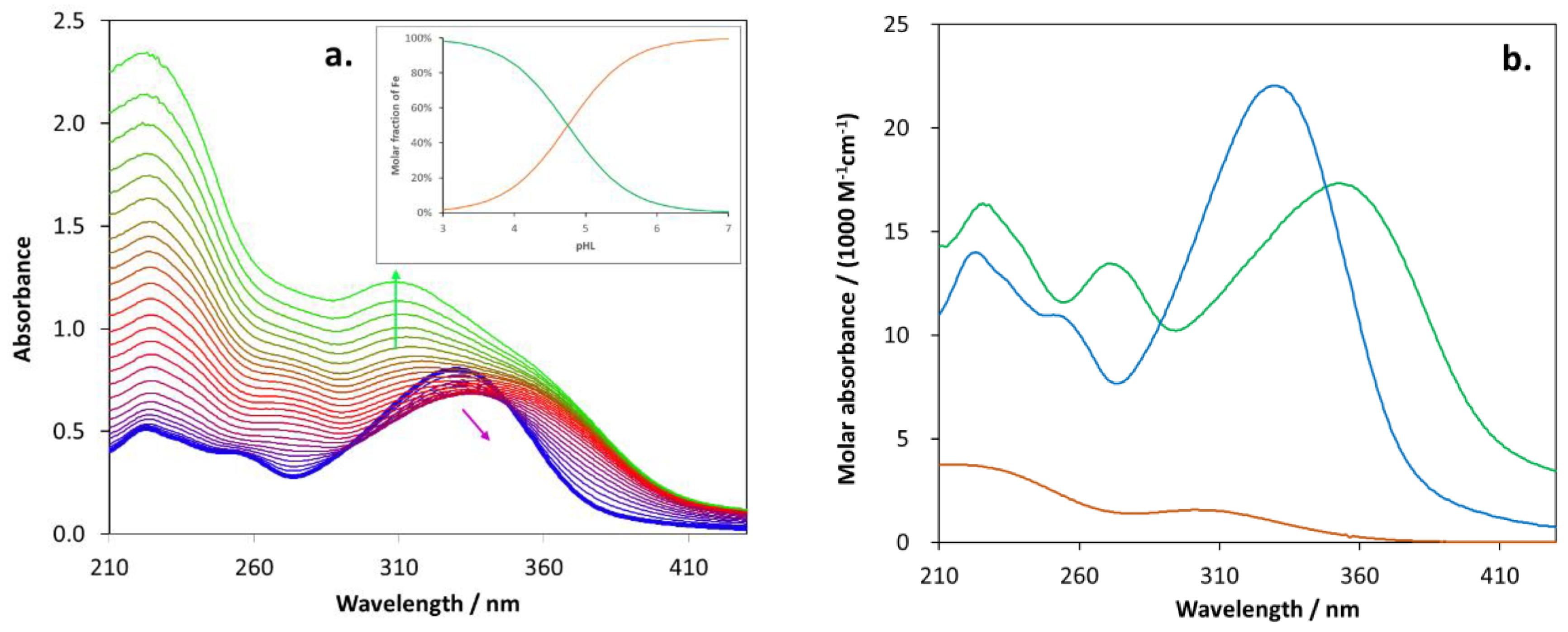
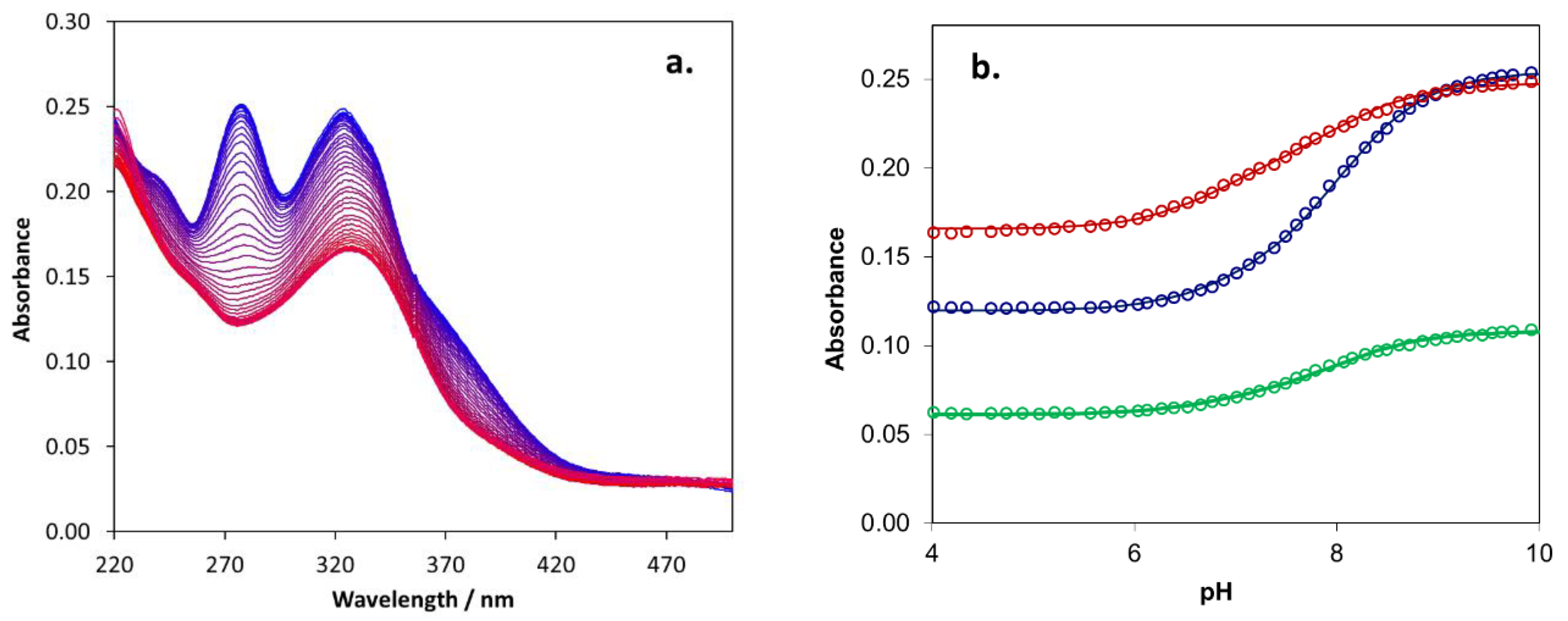
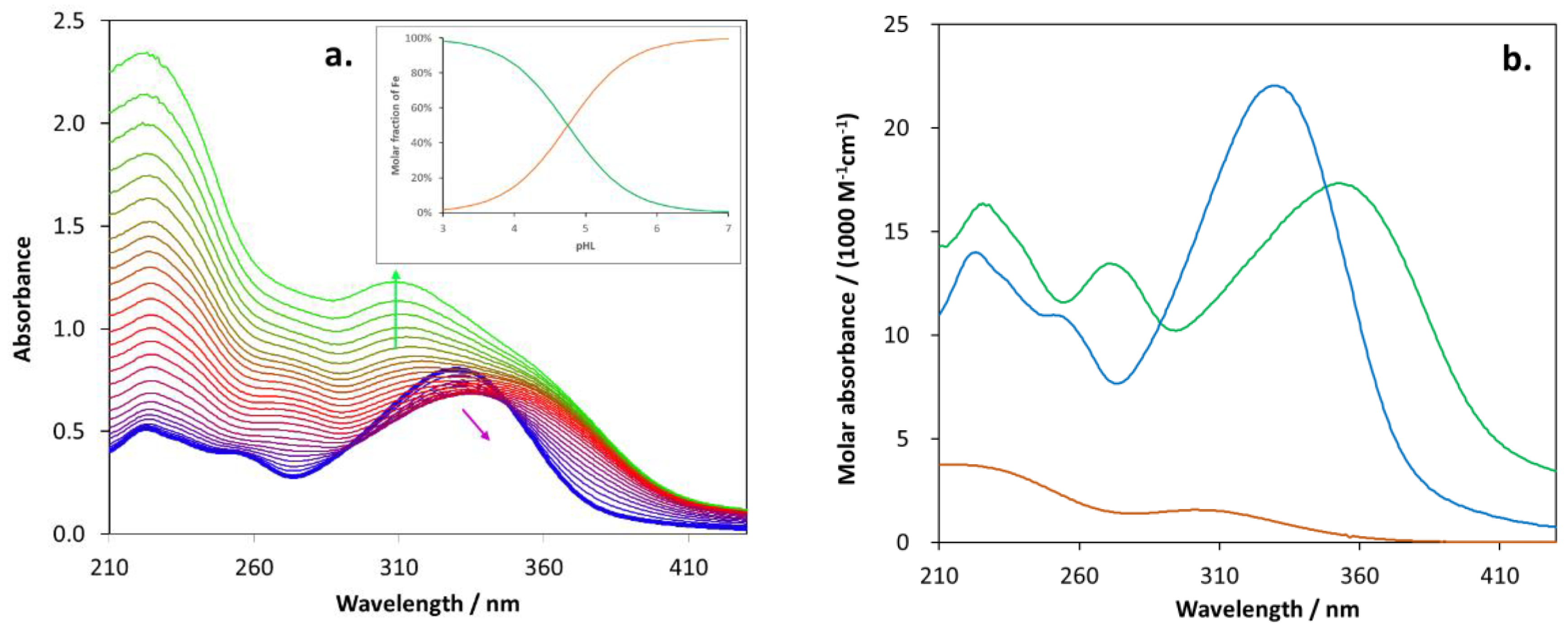
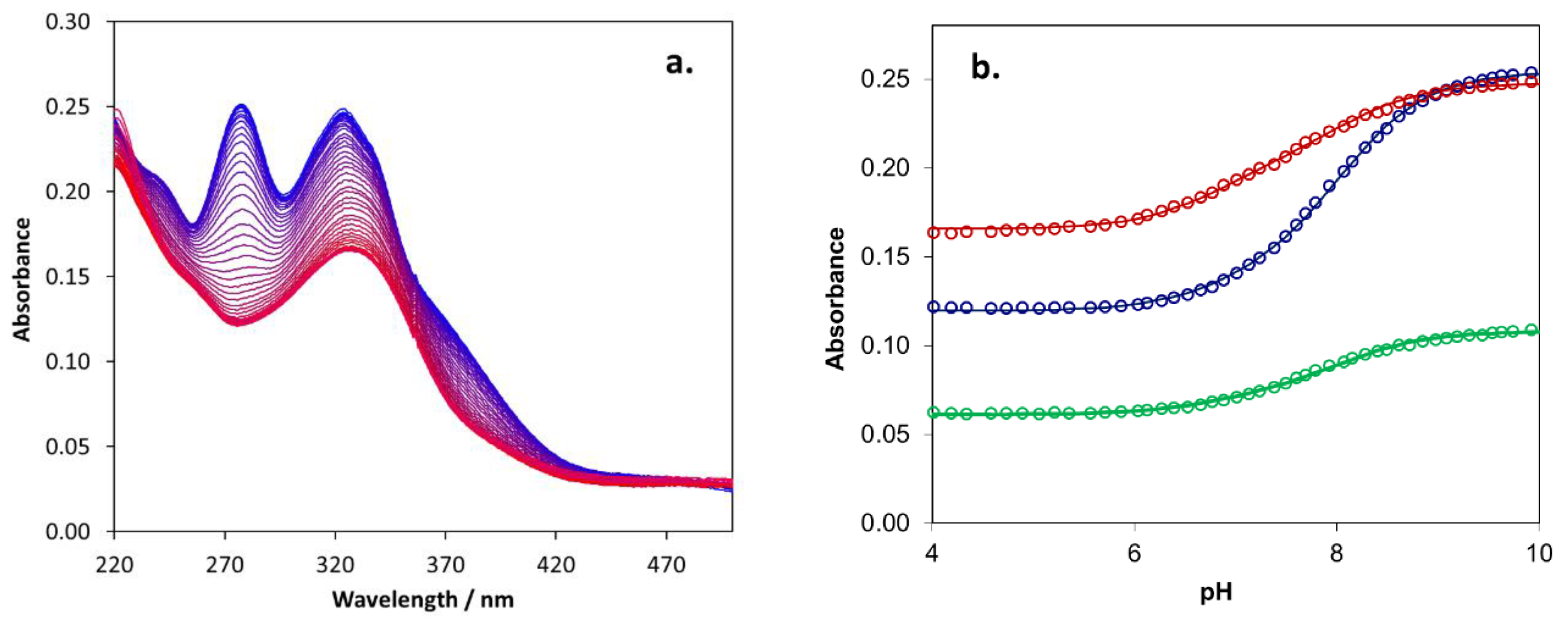
2.5. Biometal Chelating Capability of Cholinesterase Inhibitory Active Pyranones
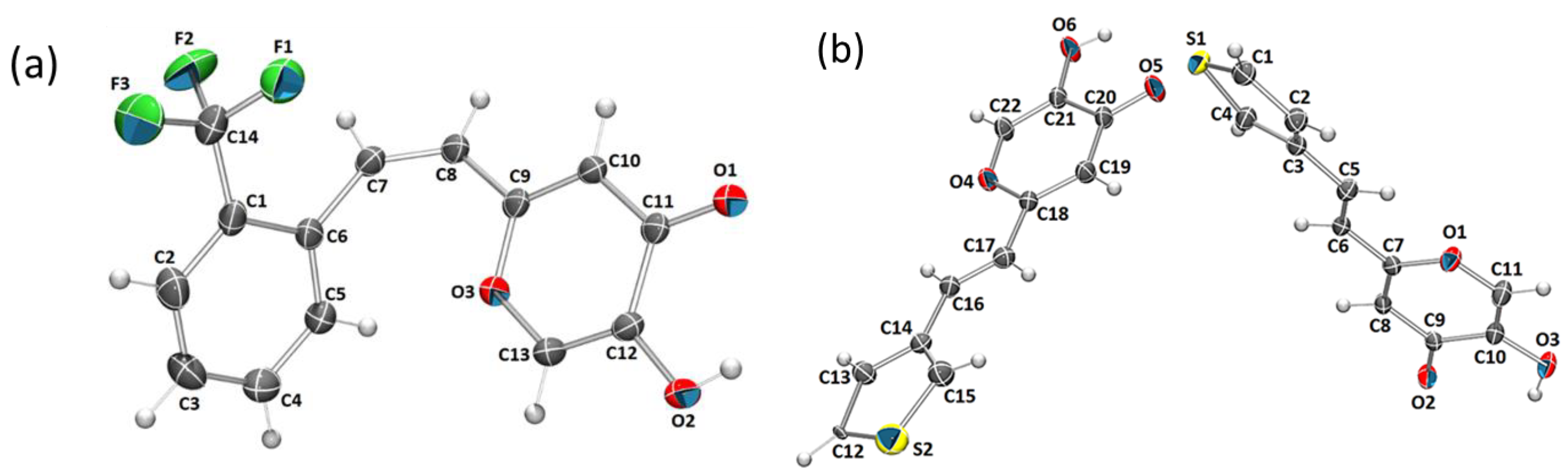
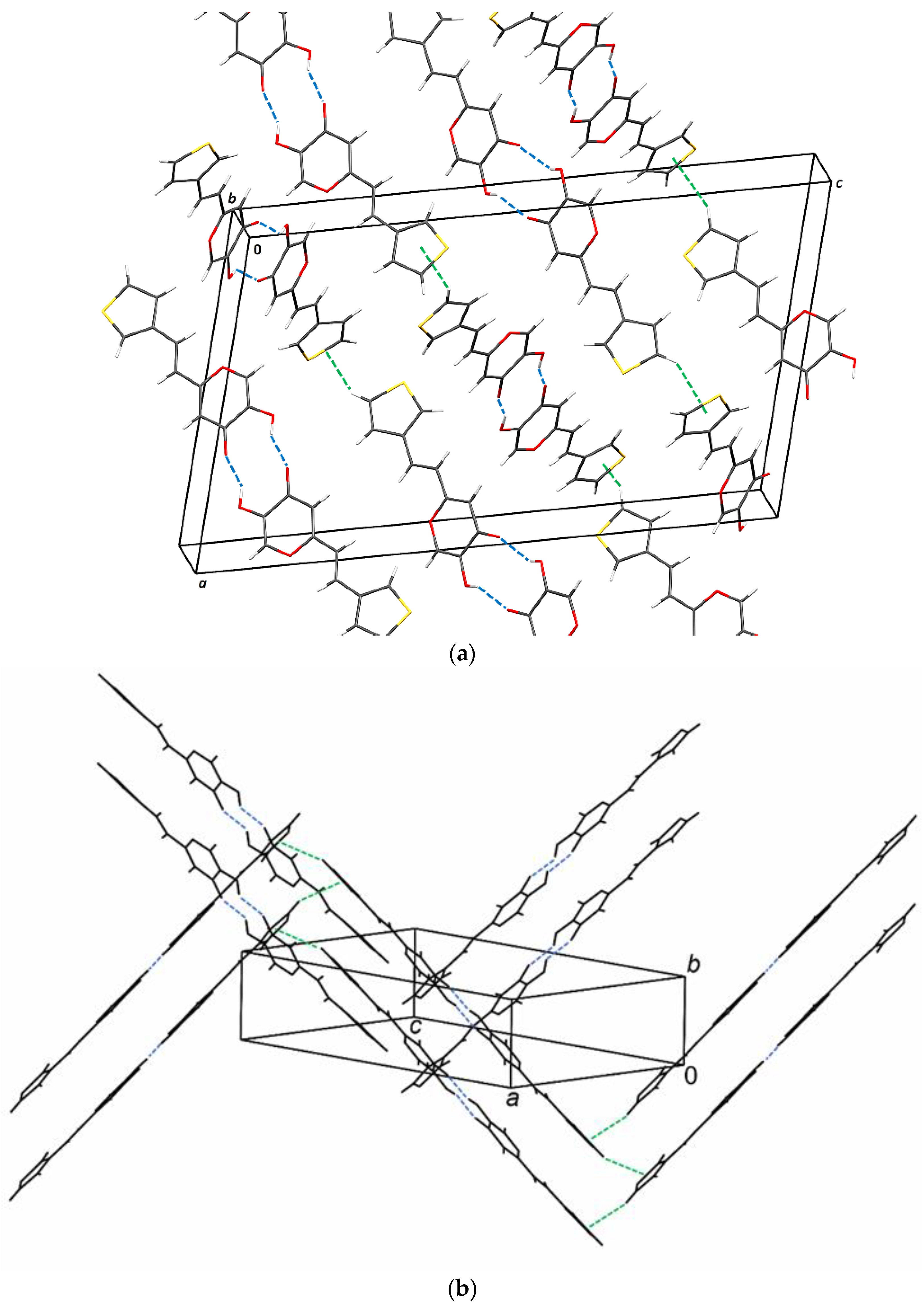
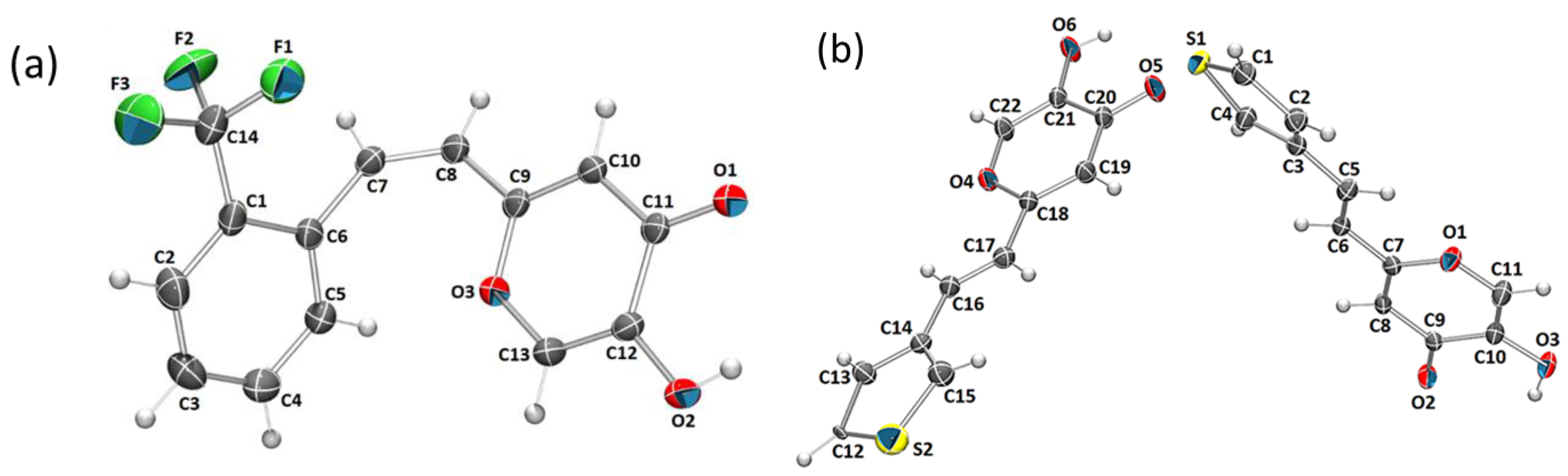
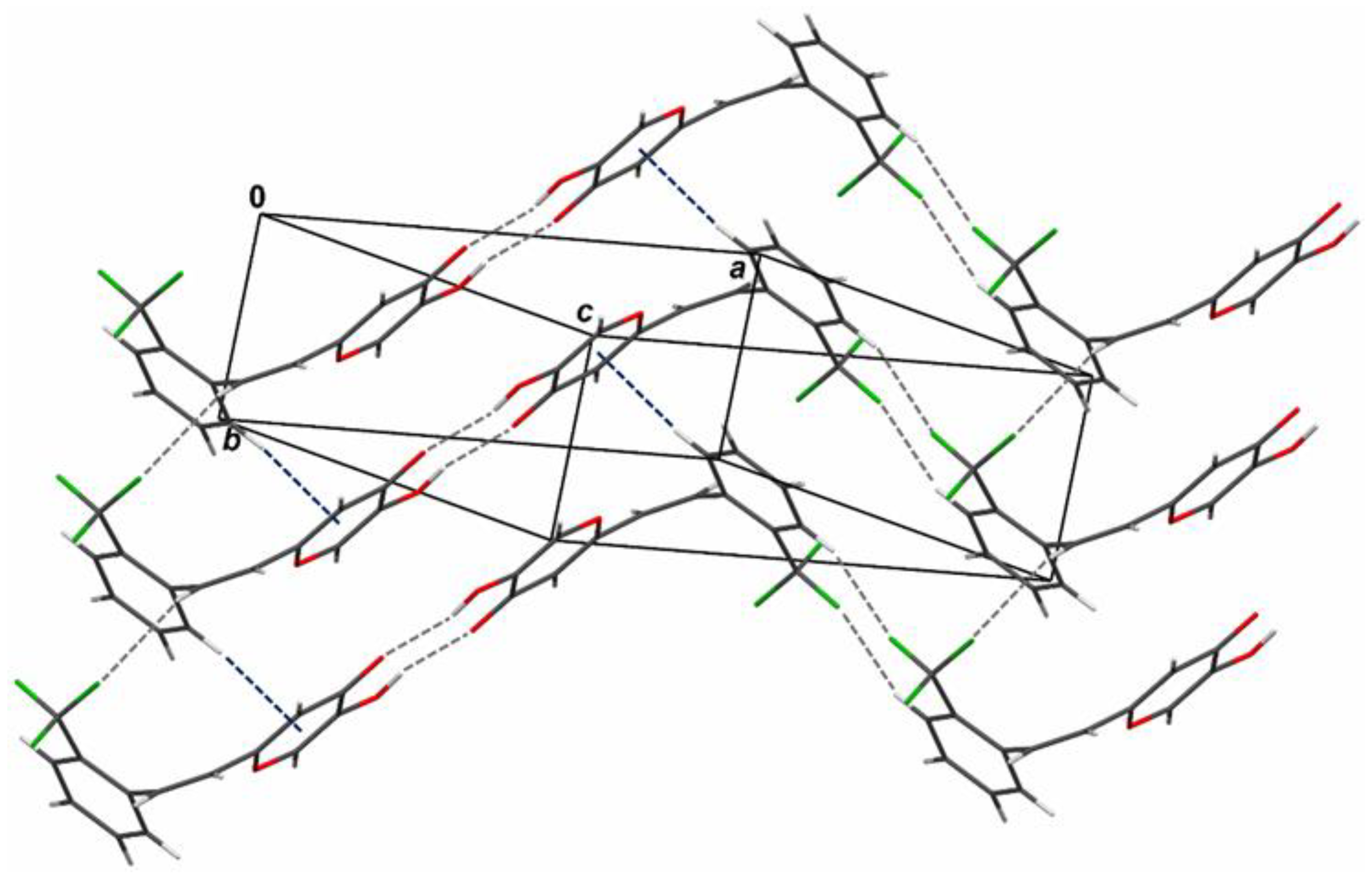
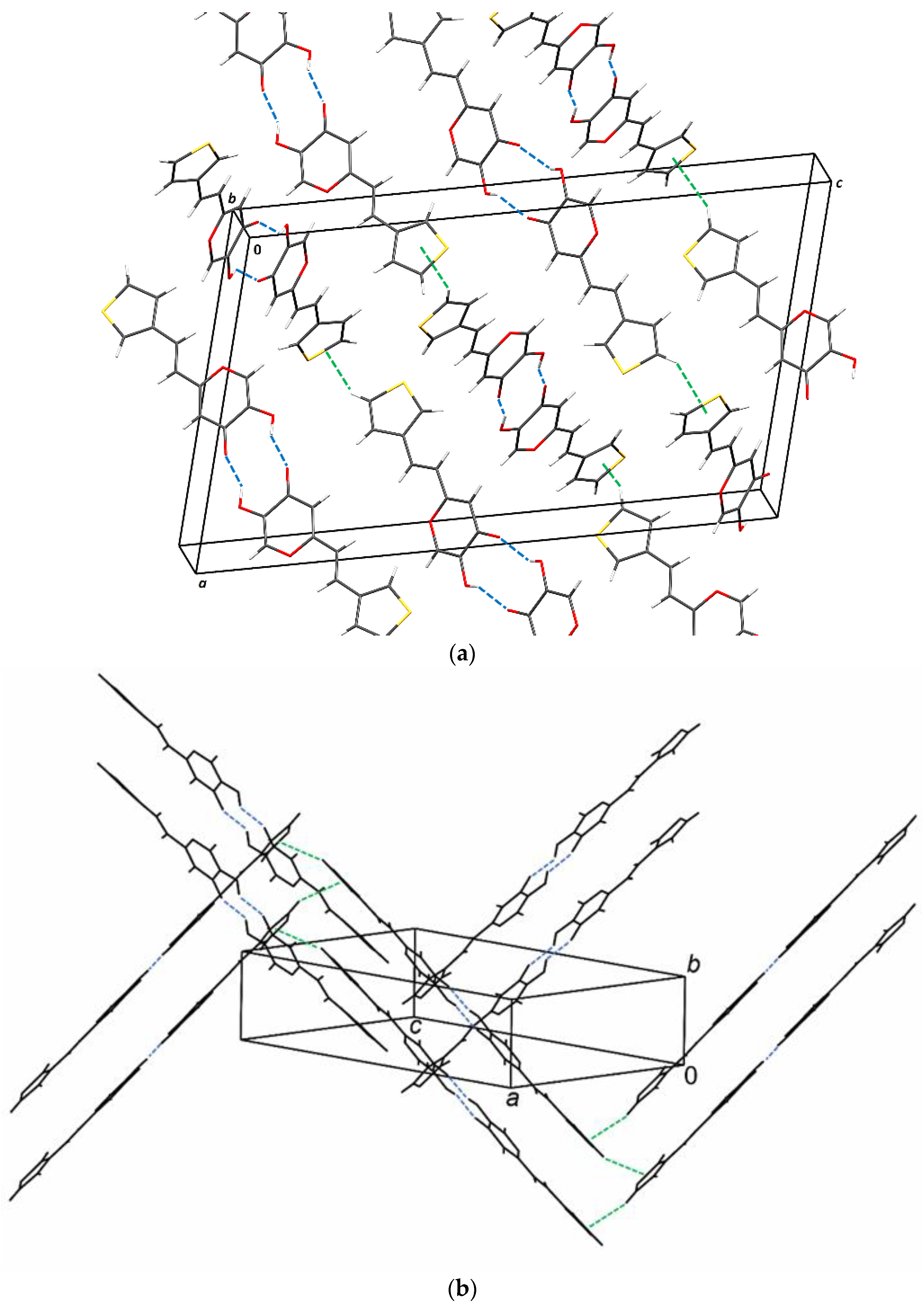
2.6. Crystal Structure and Packing of Resveratrol–Maltol Hybrids
3. Materials and Methods
3.1. General Remarks
3.2. General Procedure for the Synthesis of 4-Pyranone Phosphonium Salt
3.3. General Procedure for the Synthesis of 4-Pyranone Heterostilbenes 1–8
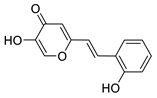
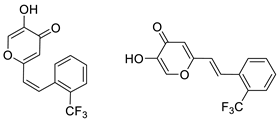
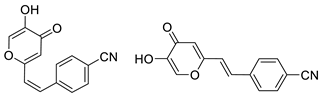
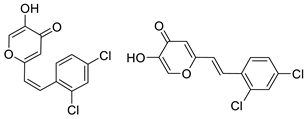
3.4. Cholinesterase Inhibitory Activity
3.5. Molecular Docking
3.6. Metal Chelating Affinity
3.7. X-Ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | aggregation of β-amyloid |
| AChE | Acetylcholinesterase |
| AD | Alzheimer’s disease |
| ACN | acetonitrile |
| AS | anionic site |
| BChE | butyrylcholinesterase |
| DCM | dichloromethane |
| DPPH | 2,2′-diphenyl-1-picrylhydrazyl |
| HB | hydrogen bond |
| E | ether |
| EtOH | ethanol |
| NMR | nuclear magnetic resonance |
| EtOAc | ethyl acetate |
| MeOH | methanol |
| PAS | peripheric anionic site |
| PE | petroleum ether, |
| UV | ultraviolet spectrophotometry |
| s | singlet |
| d | doublet |
| t | triplet |
| q | quartet |
| dd | doublet of doublets |
| m | multiplet |
References
- Belwal, T.; Nabavi, S.M.; Nabavi, S.F.; Dehpour, A.R.; Shirooie, S. Naturally Occurring Chemicals against Alzheimer’s Disease; Andre Gerhard Wolff: Cambridge, MA, USA, 2020; pp. 33–42. [Google Scholar]
- Anekonda, T.S. Resveratrol—A boon for treating Alzheimer’s disease? Brain Res. Rev. 2006, 52, 316–326. [Google Scholar] [CrossRef]
- Anekonda, T.S.; Wadsworth, T.L.; Sabin, R.; Frahler, K.; Harris, C.; Petriko, B.; Ralle, M.; Woltjer, R.; Quinn, J.F. Phytic acid as a potential treatment for Alzheimer’s pathology: Evidence from animal and in vitro models. J. Alzheimer’s Dis. 2011, 23, 21–35. [Google Scholar] [CrossRef]
- Li, J.; Qiu, Z.; Zhao, G.R. Modular engineering of E. coli coculture for efficient production of resveratrol from glucose and arabinose mixture. Synth. Syst. Biotechnol. 2022, 7, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C. Alzheimer’s disease and the amyloid cascade hypothesis: A critical review. Int. J. Alzheimer’s Dis. 2012, 2012, 369808. [Google Scholar] [CrossRef] [PubMed]
- Rauk, A. The chemistry of Alzheimer’s disease. Chem. Soc. Rev. 2009, 38, 2698. [Google Scholar] [CrossRef]
- Darvesh, A.S.; Carroll, R.T.; Bishayee, A.; Geldenhuys, W.J.; Schyf, C.J.V.D. Oxidative stress and Alzheimer’s disease: Dietary polyphenols as potential therapeutic agents. Expert Rev. Neurother. 2010, 10, 729–745. [Google Scholar] [CrossRef]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Lee, S.; Liu, Y.; Lim, M.H. Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem. Biol. 2013, 8, 856–865. [Google Scholar] [CrossRef]
- Lopez-Hernandez, J.; Paseiro-Losada, P.; Sanches-Silva, A.T.; Lage-Yusty, M.A. Study of the changes of trans-resveratrol caused by ultraviolet light and determination of trans- and cis-resveratrol in Spanish white wines. Eur. Food Res. Technol. 2007, 225, 789–796. [Google Scholar] [CrossRef]
- Montsko, G.; Nikfardjam, M.S.P.; Szabo, Z.; Boddi, K.; Lorand, T.; Ohmacht, R.; Mark, L. Determination of products derived from trans-resveratrol UV photoisomerisation by means of HPLC–APCI-MS. J. Photochem. Photobiol. A 2008, 196, 44–50. [Google Scholar] [CrossRef]
- Silva, C.G.; Monteiro, J.; Marques, R.R.N.; Silva, A.M.T.; Martínez, C.; Canle, M.; Faria, J.L. Photochemical and photocatalytic degradation of trans-resveratrol †‡. Photochem. Photobiol. Sci. 2013, 12, 638. [Google Scholar] [CrossRef]
- Cottart, C.H.; Nivet-Antoine, C.; Laguillier-Morizot, C.; Beaudeux, J.L. Resveratrol bioavailability and toxicity in humans. Mol. Nutr. Food Res. 2010, 54, 7–16. [Google Scholar] [CrossRef]
- Jie, L.; Wuliji, O.; Wei, L.; Zhi-Gang, J.; Hossein, A. Oxidative Stress and Neurodegenerative Disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef]
- Shaikh, S.; Dhavan, P.; Ramana, M.M.V.; Jadhav, B.L. Design, synthesis and evaluation of new chromone-derived aminophosphonates as potential acetylcholinesterase inhibitor. Mol. Divers. 2021, 25, 811–825. [Google Scholar] [CrossRef]
- Jalili-Baleh, L.; Nadri, H.; Forootanfar, H.; Küçükkılınç, T.T.; Ayazgök, B.; Sharifzadeh, M.; Rahimifard, M.; Baeeri, M.; Abdollahi, M.; Foroumadi, A.; et al. Chromone–lipoic acid conjugate: Neuroprotective agent having acceptable butyrylcholinesterase inhibition, antioxidant and copper-chelation activities. J. Pharm. Sci. 2021, 29, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Freitas, M.; Silva, M.S.; Fernandes, E.; Ribeiro, D. Styrylchromones: Biological Activities and Structure-Activity Relationship. Oxid. Med. Cell. Longev. 2021, 2021, 2804521. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Radić, Z. The cholinesterases: From genes to proteins. Annu. Rev. Pharmacol. Toxicol. 1994, 33, 281–320. [Google Scholar] [CrossRef]
- Soreq, H.; Seidman, S. Acetylcholinesterase—new roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef]
- Çokugras, A.N. Butyrylcholinesterase: Structure and physiological importance. Turk. J. Biochem. 2003, 28, 54–61. [Google Scholar]
- Mesulam, M.M.; Guillozet, A.; Shaw, P.; Levey, A.; Duysen, E.G.; Lockridge, O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. [Google Scholar] [CrossRef]
- Guillozet, J.A.L.; Smiley, F.; Mash, D.C. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann. Neurol. 1997, 42, 909–918. [Google Scholar] [CrossRef]
- Darvesh, S.; Walsh, R.; Kumar, R.; Caines, A.; Roberts, S.; Magee, D.; Rockwood, K.; Martin, E. Inhibition of human cholinesterases by drugs used to treat Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2003, 17, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Jiang, L.; Tang, L.; Zhang, M.; Sheng, R. Design, synthesis and biological evaluation of 2-styryl-5-hydroxy-4-pyrone derivatives and analogues as multiple functional agents with the potential for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2021, 44, 116306. [Google Scholar] [CrossRef]
- Guzior, N.; Wieckowska, A.; Panek, D.; Malawska, B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr. Med. Chem. 2014, 22, 373–404. [Google Scholar] [CrossRef]
- Mlakić, M.; Rajič, L.; Ljubić, A.; Vušak, V.; Zelić, B.; Gojun, M.; Odak, I.; Čule, I.; Šagud, I.; Šalić, A.; et al. Synthesis of new heterocyclic resveratrol analogues in milli- and microreactors: Intensification of the Wittig reaction. J. Flow Chem. 2022. [Google Scholar] [CrossRef]
- Gülçin, I. Antioxidant properties of resveratrol: A structure–activity insight. Innov. Food Sci. Emerg. Technol. 2010, 11, 210–218. [Google Scholar] [CrossRef]
- Fauconneau, B.; Waffo-Teguo, P.; Huguet, F.; Barrier, L.; Decendit, A.; Merillon, J.M. Comparative study of radical scavenger and antioxidant properties of phenolic compounds from vitis vinifera cell cultures using in vitro tests. Life Sci. 1997, 61, 2103–2110. [Google Scholar] [CrossRef]
- Francioso, A.; Mastromarino, P.; Masci, A.; d’Erme, M.; Mosca, L. Chemistry, stability and bioavailability of resveratrol. Med. Chem. 2014, 10, 237–245. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Canto, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef]
- Cheng, G.; Xu, P.; Zhang, M.; Chen, J.; Sheng, R.; Ma, Y. Resveratrol-maltol hybrids as multi-target-directed agents for Alzheimer’s disease. Bioorganic Med. Chem. 2018, 26, 5759–5765. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, M.; Sheng, R.; Ma, Y. Synthesis and biological evaluation of deferiprone-resveratrol hybrids as antioxidants, Aβ1–42 aggregation inhibitors and metal-chelating agents for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 127, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Mlakić, M.; Faraho, I.; Odak, I.; Talić, S.; Vukovinski, A.; Raspudić, A.; Bosnar, M.; Zadravec, R.; Ratković, A.; Lasić, K.; et al. Synthesis, photochemistry and computational study of novel 1,2,3-triazole heterostilbenes: Expressed biological activity of their electrocyclization photoproducts. Bioorganic Chem. 2022, 121, 105701. [Google Scholar] [CrossRef] [PubMed]
- Šagud, I.; Maček Hrvat, N.; Grgičević, A.; Čadež, T.; Hodak, J.; Dragojević, M.; Lasić, K.; Kovarik, Z.; Škorić, I. Design, synthesis and cholinesterase inhibitory properties of new oxazole benzylamine derivatives. J. Enzym. Inhib. Med. Chem. 2020, 35, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Škorić, I.; Flegar, I.; Marinić, Ž.; Šindler-Kulyk, M. Synthesis of the novel conjugated ω,ω′-diaryl/heteroaryl hexatriene system with the central double bond in a heteroaromatic ring: Photochemical transformation of 2,3-divinylfuran derivatives. Tetrahedron 2006, 62, 7396–7407. [Google Scholar] [CrossRef]
- Horspool, W.M.; Song, P.S. CRC Handbook of Organic Photochemistry and Photobiology; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Griesbeck, A.; Oelgemöller, M.; Ghetti, F. CRC Handbook of Organic Photochemistry and Photobiology; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Ellman, G.L.; Courtnex, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Imai, Y.N.; Inoue, Y.; Nakanishi, I.; Kitaura, K. Cl–π interactions in protein–ligand complexes. Protein Sci. 2008, 17, 1129–1137. [Google Scholar] [CrossRef]
- Koellner, G.; Kryger, G.; Millard, C.B.; Silman, I.; Sussman, J.L.; Steiner, T. Active-site Gorge and Buried Water Molecules in Crystal Structures of Acetylcholinesterase from Torpedo californica. Mol. Biol. 2000, 296, 713–735. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, X.; Yang, H.; Chen, Y.; Wang, F.; Li, J.; Tang, Z.; Cheng, X.; Yang, Y.; Xu, L.; et al. Discovery of Selective Butyrylcholinesterase (BChE) Inhibitors through a Combination of Computational Studies and Biological Evaluations. Molecules 2019, 24, 4217. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDock-823 Tools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef]
- Raves, M.L.; Giles, K.; Schrag, J.D.; Schmid, M.F.; Phillips, G.N., Jr.; Wah, C.; Howard, A.J.; Silman, I.; Sussman, J.L.; Doctor, B.P.; et al. Quaternary Structure of Tetrameric Acetylcholinesterase. In Structure and Function of Cholinesterases and Related Proteins; Springer: Berlin/Heidelberg, Germany, 1998. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Zékány, L.; Nagypál, I.; Peintler, G. PSEQUAD for Chemical Equilibria; Technical Distributors: Baltimore, MD, USA, 1991. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. It ORTEP-3 for Windows—A Version of It ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst. 1997, 30, 565. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Košak, U.; Brus, B.; Knez, D.; Šink, R.; Žakel, S.; Trontelj, J.; Pišlar, A.; Šlenc, J.; Gobec, M.; Živin, M.; et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci. Rep. 2016, 6, 39495. [Google Scholar] [CrossRef] [PubMed]
- Raves, M.L.; Harel, M.; Pang, Y.P.; Silman, I.; Kozikowski, A.P.; Sussman, J.L. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (−)-huperzine A. Nat. Struct. Biol. 1997, 4, 57. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resveratrol-Thiophene/ Resveratrol–Maltol Hybrids | AChE IC50 (μM) | BChE IC50 (μM) | DPPH IC50 (μM) |
|---|---|---|---|
| II | 15.7 ± 1.0 | 4.6 ± 0.1 | 158.8 * |
| cis-II | ~500 | 18.9 ± 3.4 | 782.1 * |
| III | 46.6 ± 8.3 | 5.3 ± 1.8 | 26.8 * |
| trans-1 | - | 600.7 ± 34.5 | 251.3 ± 45.6 |
| cis-2 | - | 458.1 ± 30.2 | - |
| cis-4 | 1003.0 ± 10.1 | 638.1 ± 25.8 | - |
| trans-6 | - | 426.3 ± 41.8 | - |
| Galantamine | 0.15 | 7.9 |
| Compounds | Protonation Constants | Formation Constants (lgKFeL or lgKFeHLa, b) | ||
|---|---|---|---|---|
| (HL or H2L) | lgK1 | lgK2 | Fe3+ + HL ⇌ FeL2+ + H+ | Fe3+ + L− ⇌ FeL2+ |
| trans-1 | 8.9 ± 0.20 | 7.25 ± 0.20 | 4.38 ± 0.20 a | 11.6 ± 0.2 b |
| cis-2 | 7.95 ± 0.10 | 4.38 ± 0.20 | 11.8 ± 0.2 | |
| cis-4 | 7.99 ± 0.15 | 4.16 ± 0.10 | 12.15 ± 0.15 | |
| trans-6 | 7.81 ± 0.15 | 4.75 ± 0.10 | 12.56 ± 0.15 | |
| Compound | cis-2 | trans-6 |
|---|---|---|
| Empirical formula | C14H9F3O3 | C11H8O3S |
| Formula wt./g mol−1 | 282.21 | 220.23 |
| Crystal dimensions/mm | 0.4 × 0.25 × 0.2 | 0.4 × 0.3 × 0.25 |
| Space group | P21/c | P21/c |
| a/Å | 18.0019 (10) | 15.3582 (2) |
| b/Å | 4.8342 (3) | 5.14770 (10) |
| c/Å | 14.7412 (8) | 25.9362 (5) |
| α/° | 90 | 90 |
| β/° | 107.425 (6) | 102.554 (2) |
| γ/° | 90 | 90 |
| Z | 4 | 8 |
| V/Å3 | 1223.98 (13) | 2001.48 (6) |
| Dcalc/g cm−3 | 1.531 | 1.462 |
| μ/mm−1 | 1.2 | 2.748 |
| Θ range/° | 5.15–79.384 | 3.492–79.837 |
| T(K) | 293 (2) | 299 (2) |
| Radiation wavelength | 1.54184 (CuKα) | 1.54184 (CuKα) |
| Diffractometer type | XtaLAB Synergy, Dualflex, HyPix | XtaLAB Synergy, Dualflex, HyPix |
| Range of h, k, l | −22 > h > 19; −5 > k > 5; −18 > l > 18 | −19 > h > 16; −6 > k > 5; −31 > l > 33 |
| Reflections collected | 6269 | 13048 |
| Independent reflections | 2389 | 4146 |
| Observed reflections (I ≥ 2σ) | 1886 | 3366 |
| Rint | 0.0839 | 0.039 |
| R (F) | 0.1161 | 0.099 |
| Rw (F2) | 0.3576 | 0.3512 |
| No. of parameters, restraints | 181, 0 | 272, 0 |
| Goodness of fit | 1.495 | 1.539 |
| Δρmax, Δρmin (eÅ−3) | 0.654; −0.38 | 2.09; −1.034 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlakić, M.; Fodor, L.; Odak, I.; Horváth, O.; Lovrić, M.J.; Barić, D.; Milašinović, V.; Molčanov, K.; Marinić, Ž.; Lasić, Z.; et al. Resveratrol–Maltol and Resveratrol–Thiophene Hybrids as Cholinesterase Inhibitors and Antioxidants: Synthesis, Biometal Chelating Capability and Crystal Structure. Molecules 2022, 27, 6379. https://doi.org/10.3390/molecules27196379
Mlakić M, Fodor L, Odak I, Horváth O, Lovrić MJ, Barić D, Milašinović V, Molčanov K, Marinić Ž, Lasić Z, et al. Resveratrol–Maltol and Resveratrol–Thiophene Hybrids as Cholinesterase Inhibitors and Antioxidants: Synthesis, Biometal Chelating Capability and Crystal Structure. Molecules. 2022; 27(19):6379. https://doi.org/10.3390/molecules27196379
Chicago/Turabian StyleMlakić, Milena, Lajos Fodor, Ilijana Odak, Ottó Horváth, Marija Jelena Lovrić, Danijela Barić, Valentina Milašinović, Krešimir Molčanov, Željko Marinić, Zlata Lasić, and et al. 2022. "Resveratrol–Maltol and Resveratrol–Thiophene Hybrids as Cholinesterase Inhibitors and Antioxidants: Synthesis, Biometal Chelating Capability and Crystal Structure" Molecules 27, no. 19: 6379. https://doi.org/10.3390/molecules27196379
APA StyleMlakić, M., Fodor, L., Odak, I., Horváth, O., Lovrić, M. J., Barić, D., Milašinović, V., Molčanov, K., Marinić, Ž., Lasić, Z., & Škorić, I. (2022). Resveratrol–Maltol and Resveratrol–Thiophene Hybrids as Cholinesterase Inhibitors and Antioxidants: Synthesis, Biometal Chelating Capability and Crystal Structure. Molecules, 27(19), 6379. https://doi.org/10.3390/molecules27196379