
Detection and Verification of a Key Intermediate in an Enantioselective Peptide Catalyzed Acylation Reaction

Abstract
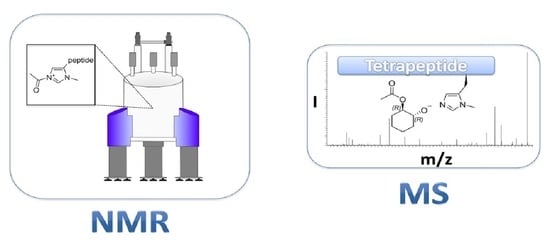
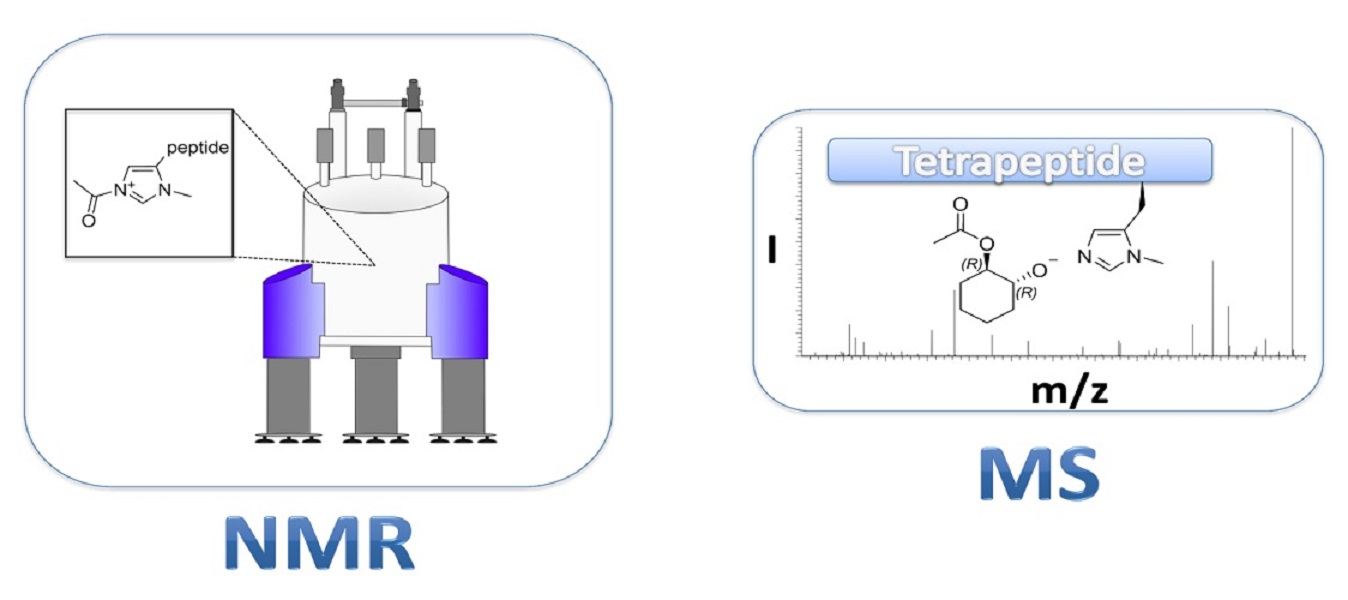
1. Introduction
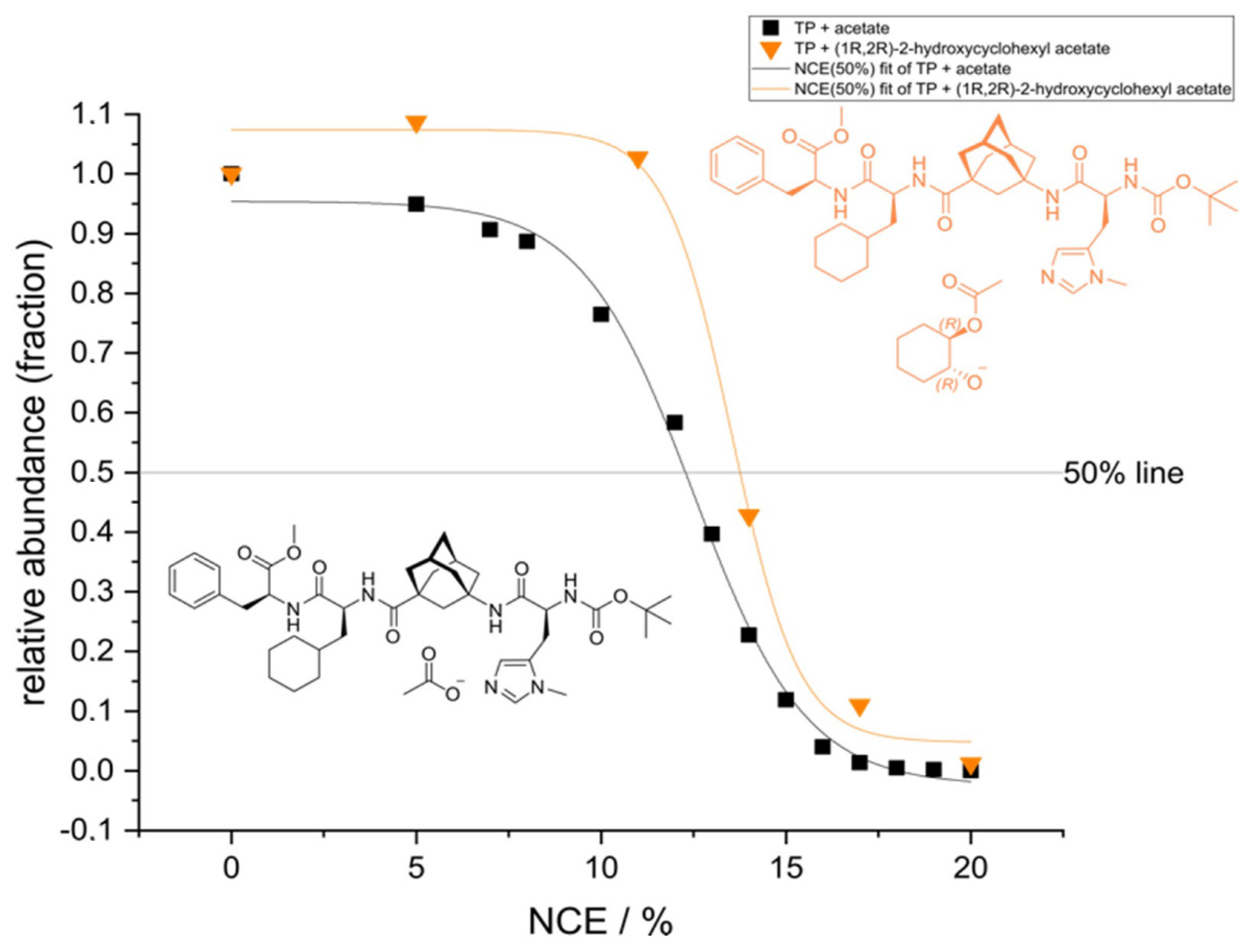
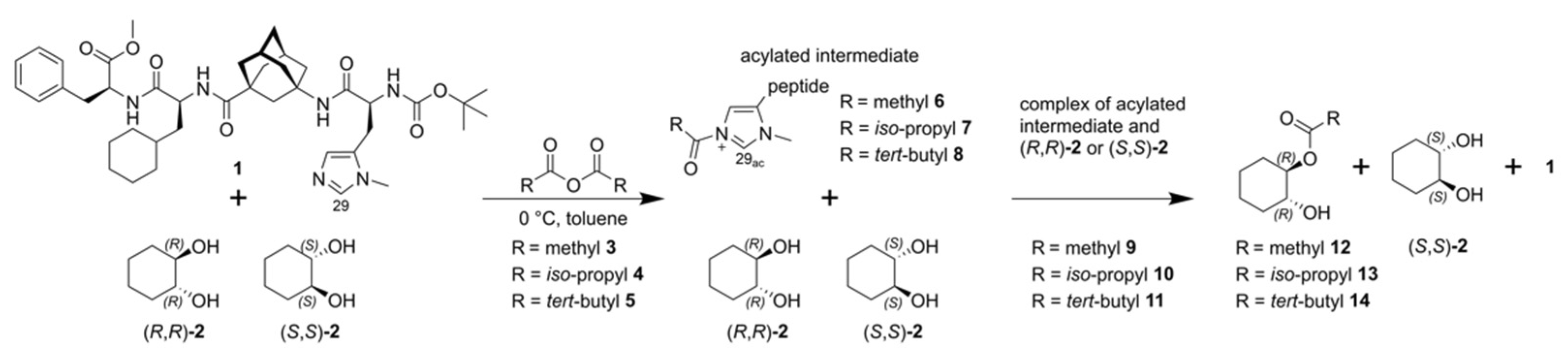
2. Results and Discussion
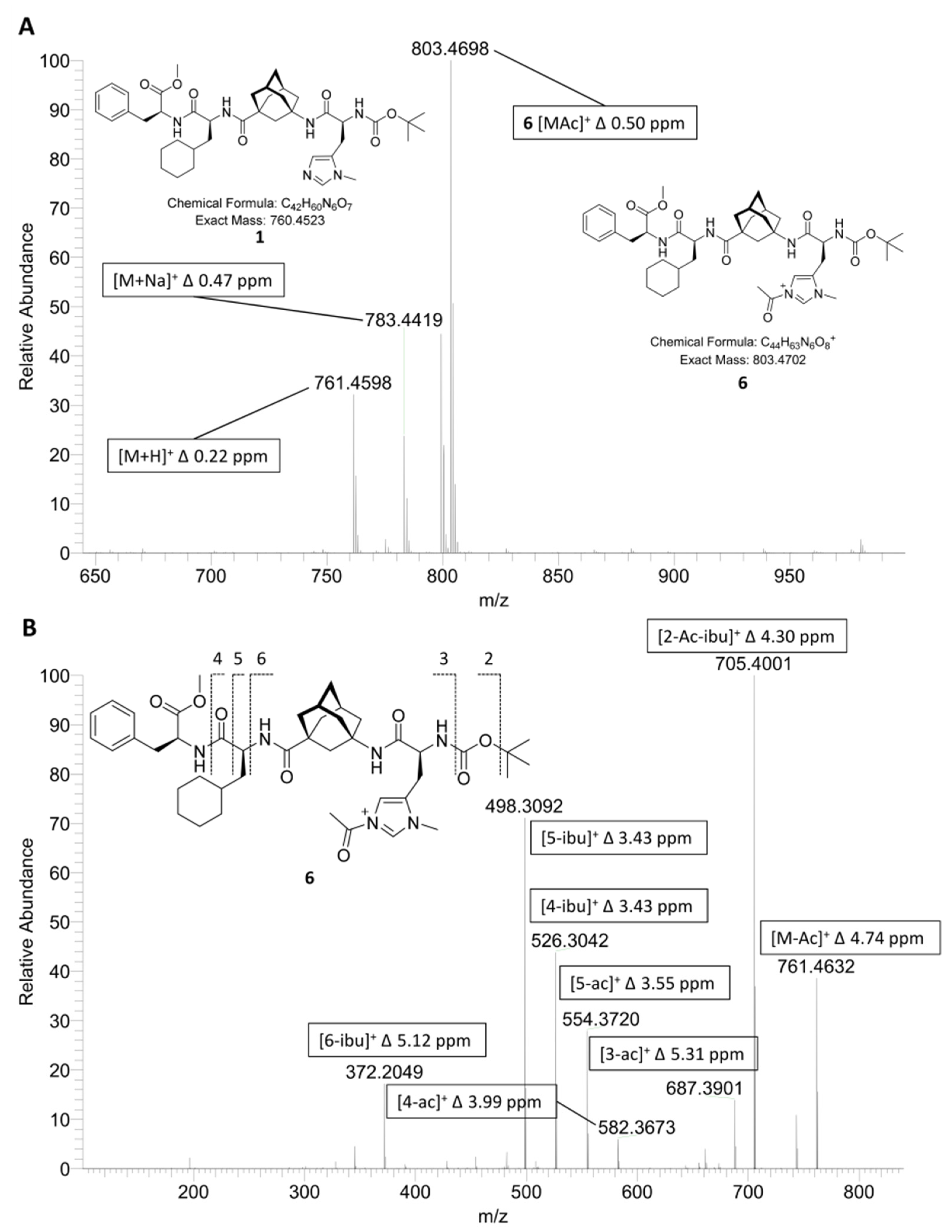
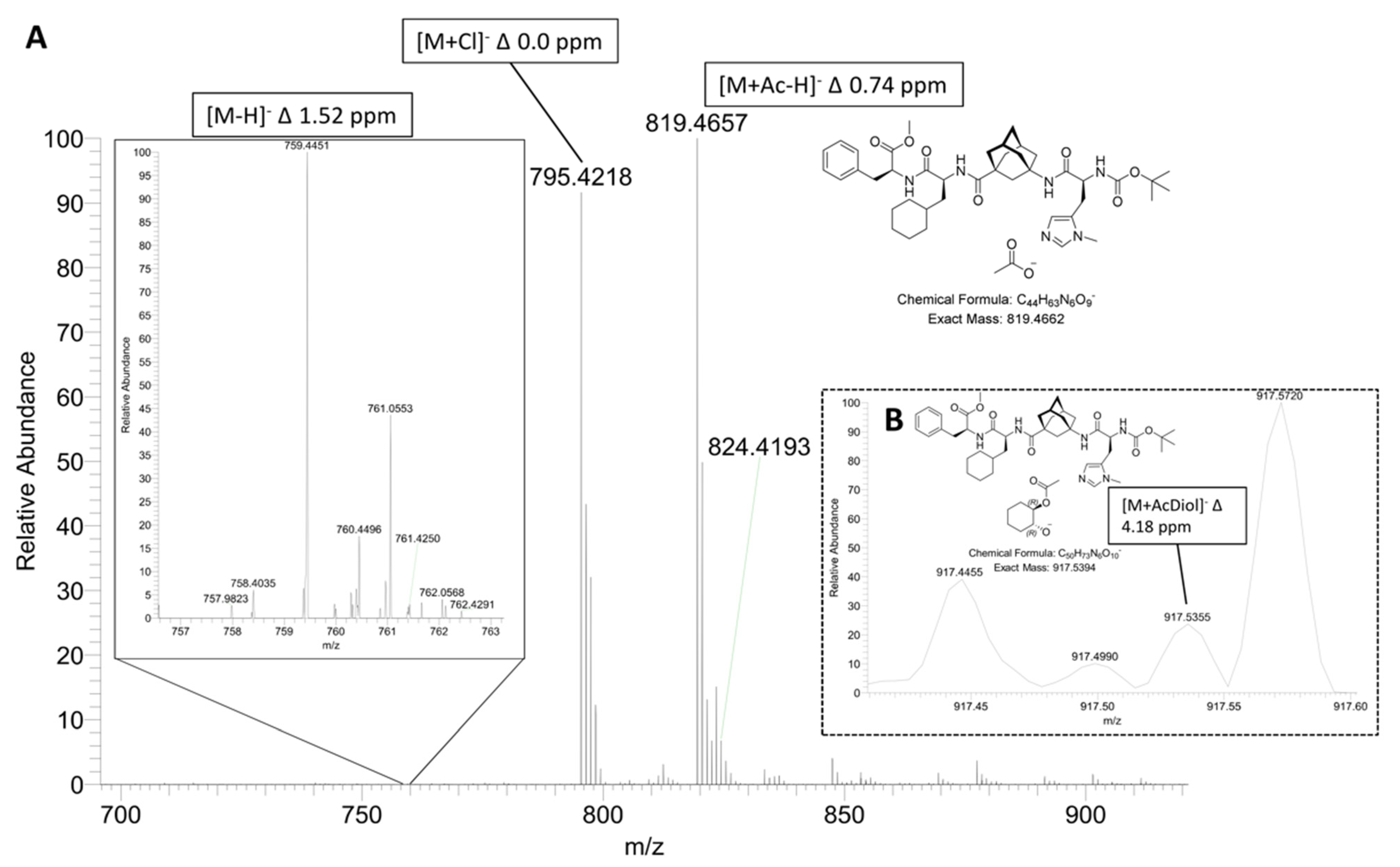
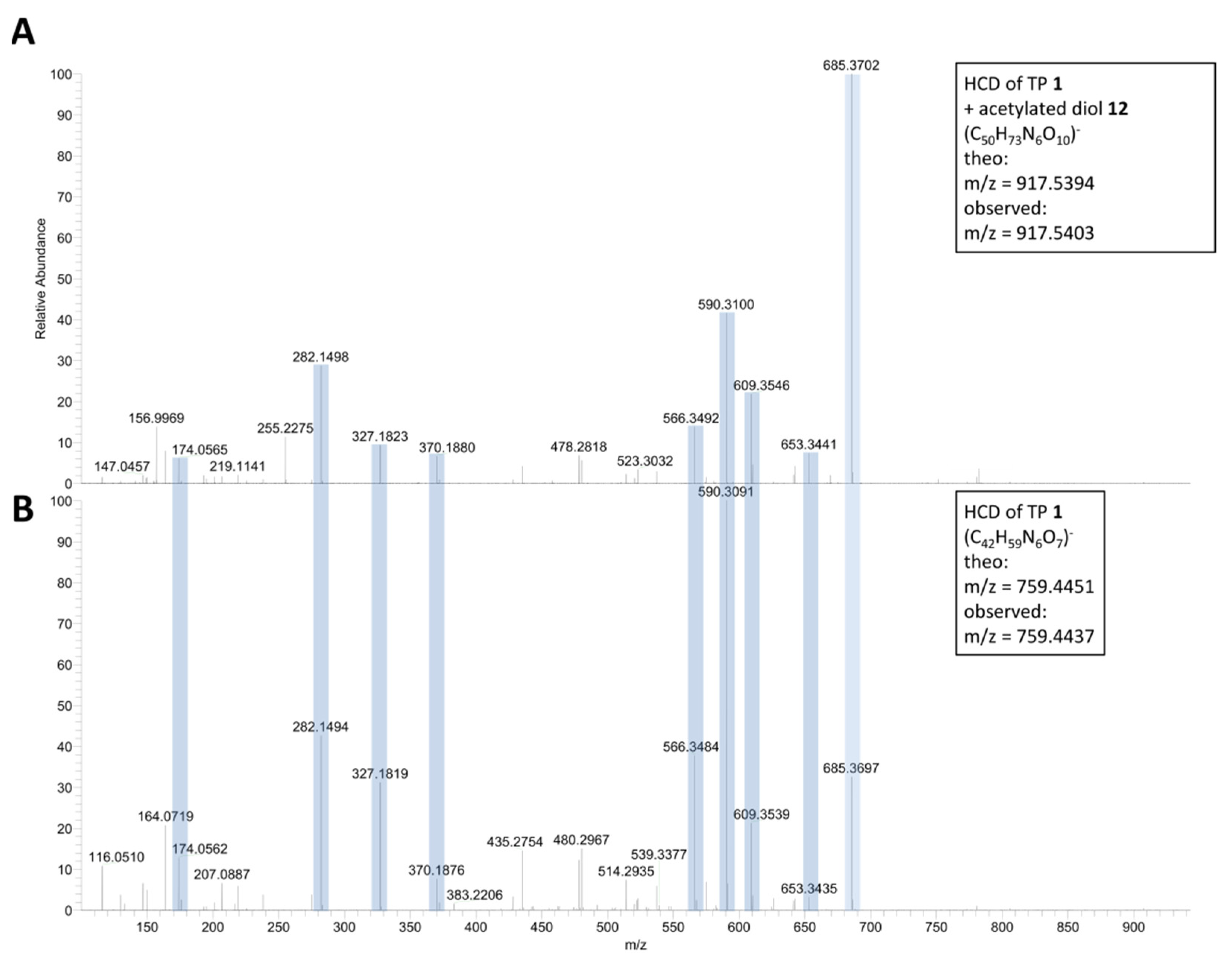
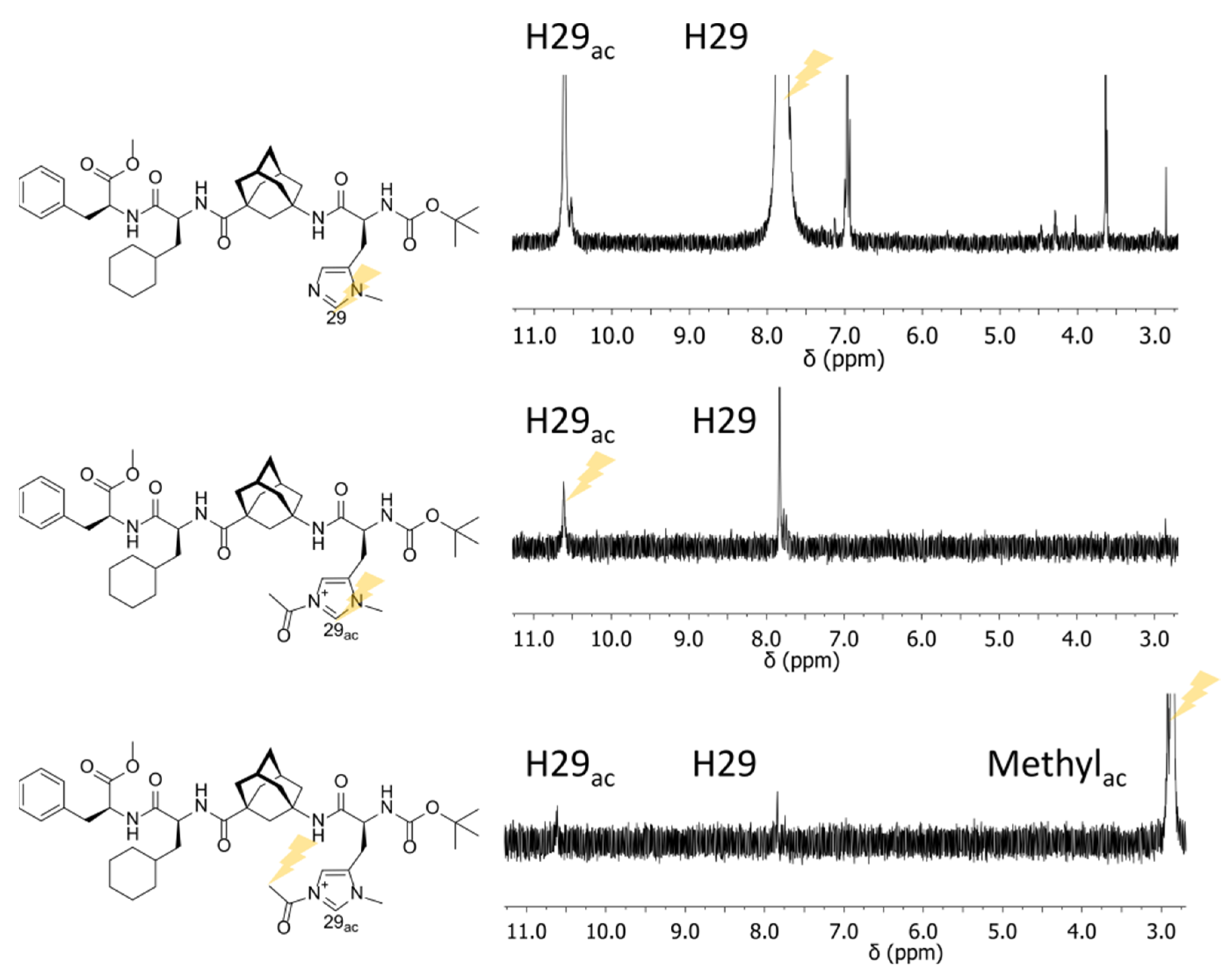
2.1. ESI-HRMS Measurements
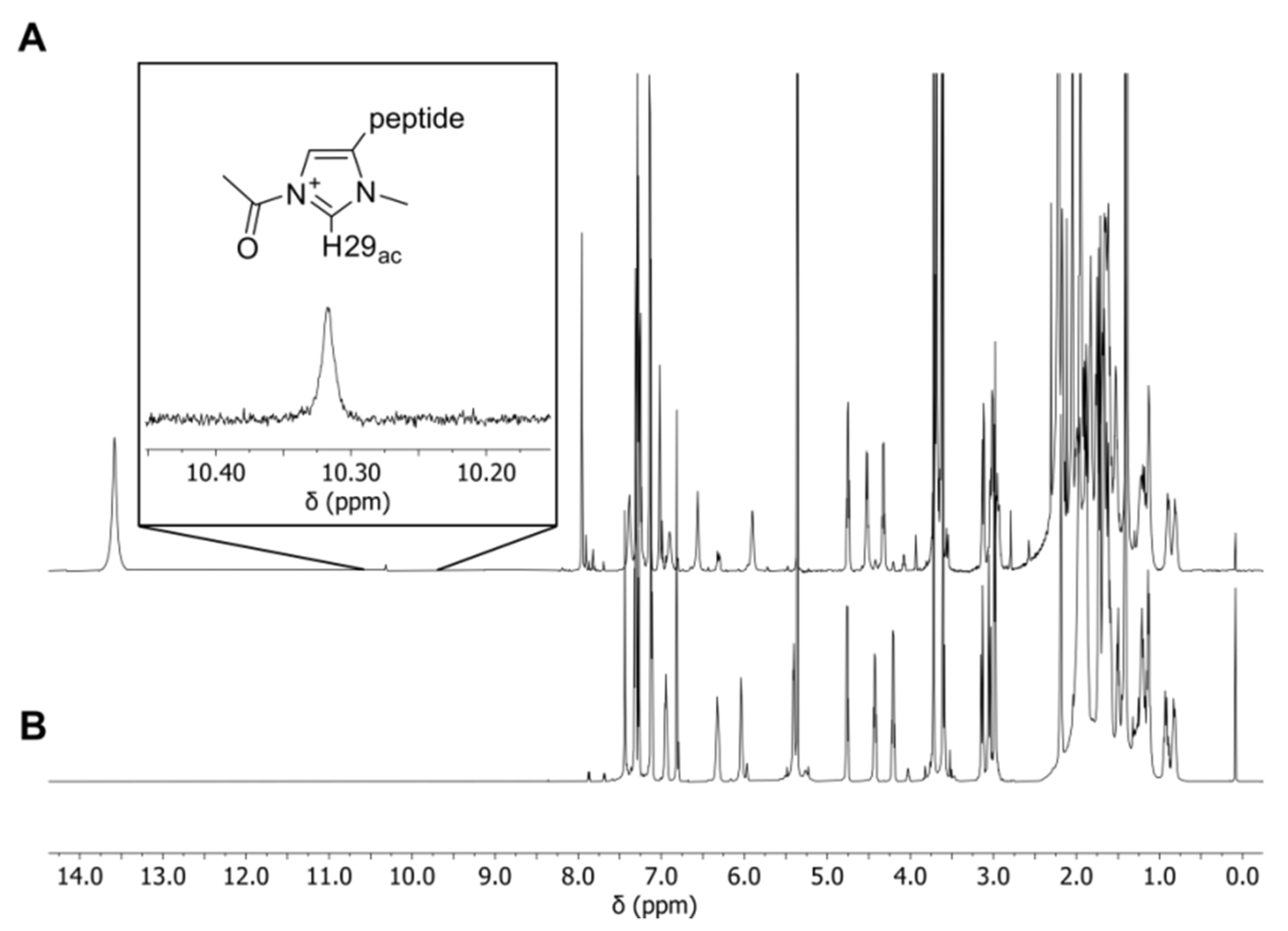
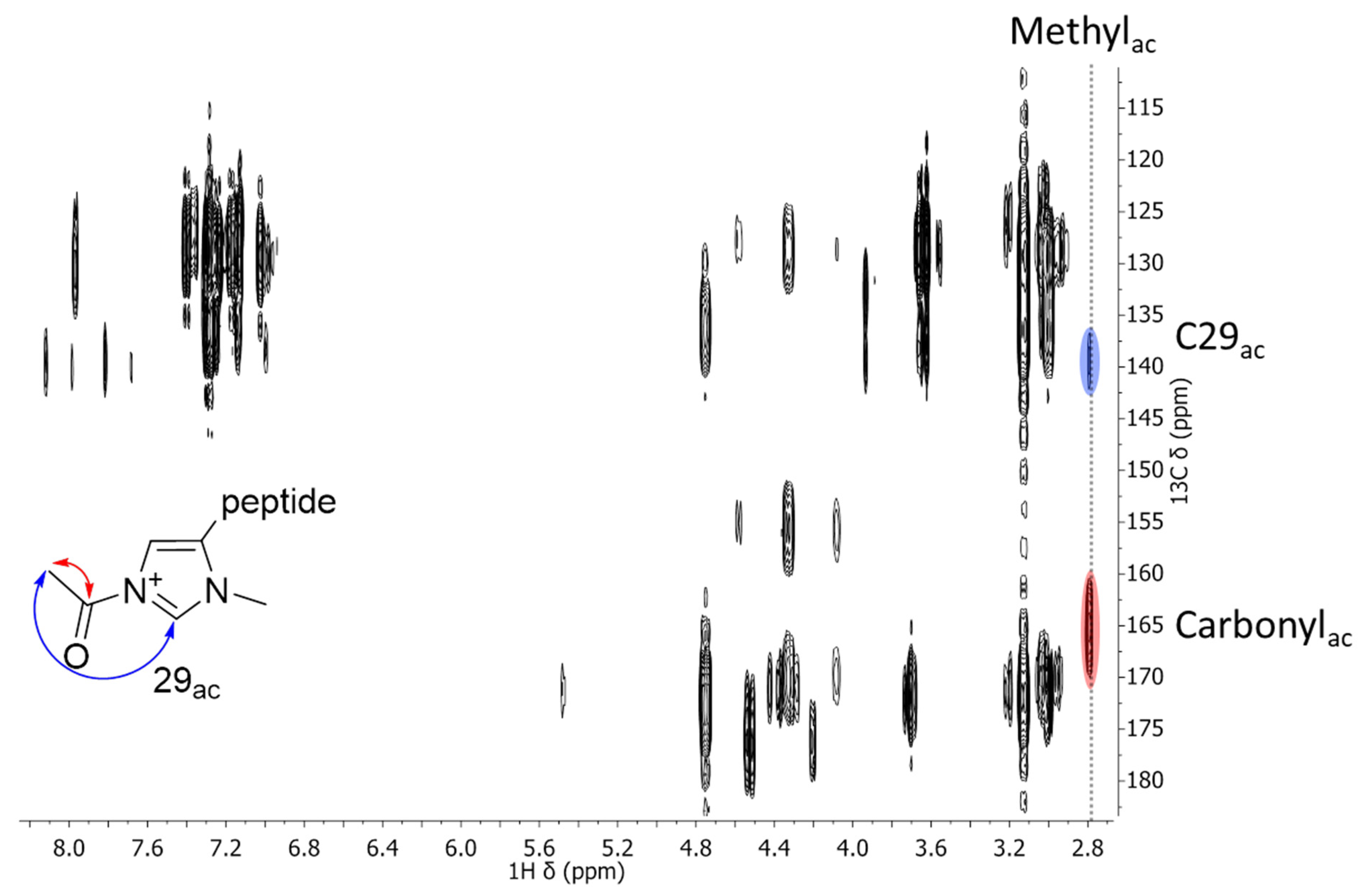
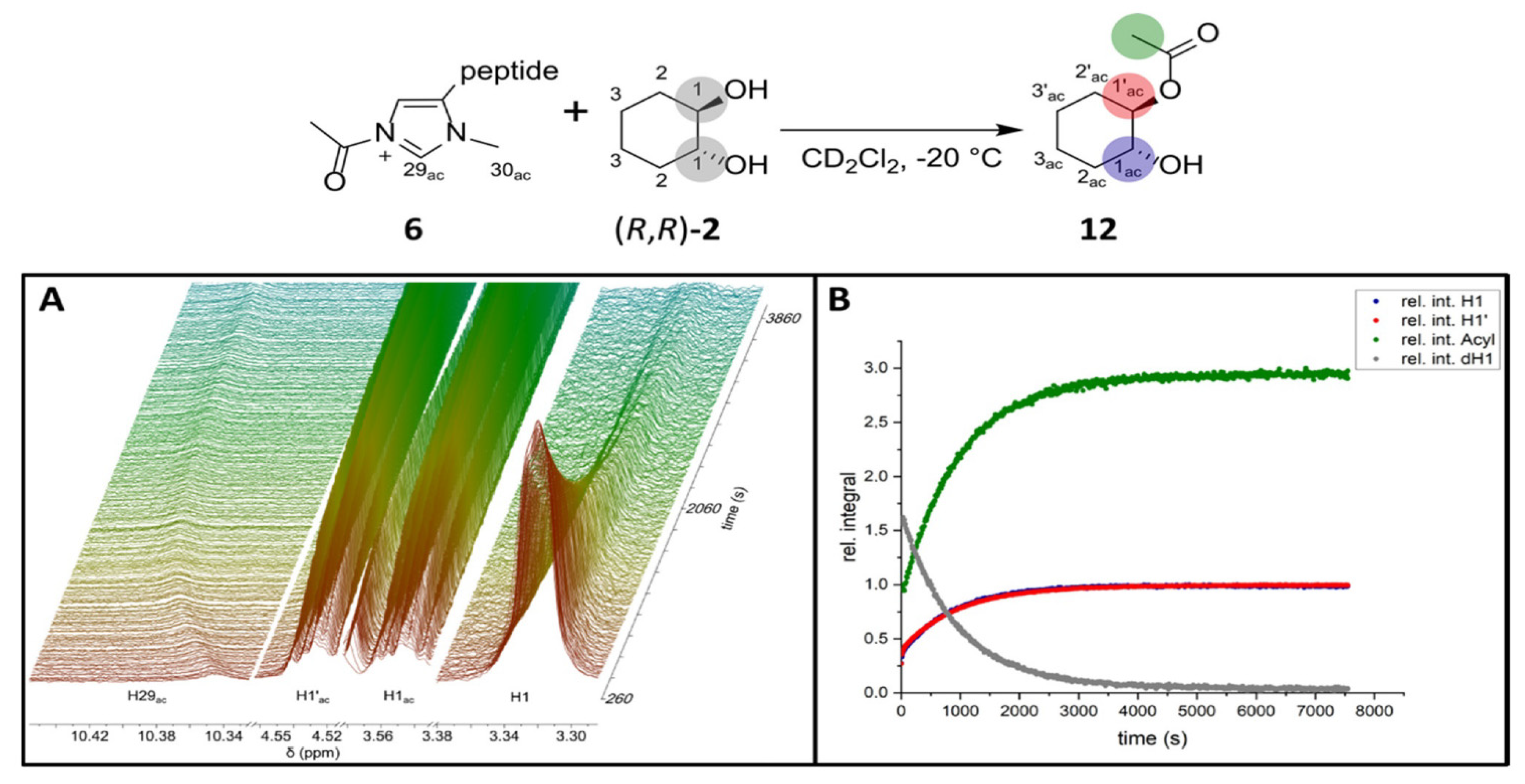
2.2. NMR Measurements
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yauk, Y.-K.; Souleyre, E.J.F.; Matich, A.J.; Chen, X.; Wang, M.Y.; Plunkett, B.; Dare, A.P.; Espley, R.V.; Tomes, S.; Chagné, D.; et al. Alcohol acyl transferase 1 links two distinct volatile pathways that produce esters and phenylpropenes in apple fruit. Plant J. 2017, 91, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Nachmansohn, D.; Machado, A.L. The formation of acetylcholine. A new enzyme: “Choline acetylase”. J. Neurophysiol. 1943, 6, 397–403. [Google Scholar] [CrossRef]
- Metrano, A.J.; Chinn, A.J.; Shugrue, C.R.; Stone, E.A.; Kim, B.; Miller, S.J. Asymmetric Catalysis Mediated by Synthetic Peptides, Version 2.0: Expansion of Scope and Mechanisms. Chem. Rev. 2020, 120, 11479–11615. [Google Scholar] [CrossRef]
- Lewandowski, B.; Wennemers, H. Asymmetric catalysis with short-chain peptides. Curr. Opin. Chem. Biol. 2014, 22, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Wennemers, H. Asymmetric catalysis with peptides. Chem. Comm. 2011, 47, 12036–12041. [Google Scholar] [CrossRef]
- Miller, S.J.; Copeland, G.T.; Papaioannou, N.; Horstmann, T.E.; Ruel, E.M. Kinetic Resolution of Alcohols Catalyzed by Tripeptides Containing the N-Alkylimidazole Substructure. J. Am. Chem. Soc. 1998, 120, 1629–1630. [Google Scholar] [CrossRef]
- Jarvo, E.R.; Copeland, G.T.; Papaioannou, N.; Bonitatebus, P.J.; Miller, S.J. A Biomimetic Approach to Asymmetric Acyl Transfer Catalysis. J. Am. Chem. Soc. 1999, 121, 11638–11643. [Google Scholar] [CrossRef]
- Jarvo, E.R.; Miller, S.J. Amino acids and peptides as asymmetric organocatalysts. Tetrahedron 2002, 58, 2481–2495. [Google Scholar] [CrossRef]
- Copeland, G.T.; Miller, S.J. Selection of Enantioselective Acyl Transfer Catalysts from a Pooled Peptide Library through a Fluorescence-Based Activity Assay: An Approach to Kinetic Resolution of Secondary Alcohols of Broad Structural Scope. J. Am. Chem. Soc. 2001, 123, 6496–6502. [Google Scholar] [CrossRef]
- Davie, E.A.C.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric Catalysis Mediated by Synthetic Peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef]
- Müller, C.E.; Wanka, L.; Jewell, K.; Schreiner, P.R. Enantioselective Kinetic Resolution of trans-Cycloalkane-1,2-diols. Angew. Chem. Int. Ed. 2008, 47, 6180–6183. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.E.; Zell, D.; Hrdina, R.; Wende, R.C.; Wanka, L.; Schuler, S.M.M.; Schreiner, P.R. Lipophilic Oligopeptides for Chemo- and Enantioselective Acyl Transfer Reactions onto Alcohols. J. Org. Chem. 2013, 78, 8465–8484. [Google Scholar] [CrossRef] [PubMed]
- Shinisha, C.; Sunoj, R.B. On the Origins of Kinetic Resolution of Cyclohexane-1, 2-diols through Stereoselective Acylation by Chiral Tetrapeptides. Org. Lett. 2009, 11, 3242–3245. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Wagner, J.P.; Schreiner, P.R. London Dispersion in Molecular Chemistry—Reconsidering Steric Effects. Angew. Chem. Int. Ed. 2015, 54, 12274–12296. [Google Scholar] [CrossRef]
- Williamson, M.P. The Nuclear Overhauser Effect. In Modern Magnetic Resonance; Webb, G.A., Ed.; Springer: Dordrecht, The Netherlands, 2006; pp. 409–412. [Google Scholar]
- Procházková, E.; Kolmer, A.; Ilgen, J.; Schwab, M.; Kaltschnee, L.; Fredersdorf, M.; Schmidts, V.; Wende, R.C.; Schreiner, P.R.; Thiele, C.M. Uncovering Key Structural Features of an Enantioselective Peptide-Catalyzed Acylation Utilizing Advanced NMR Techniques. Angew. Chem. Int. Ed. 2016, 55, 15754–15759. [Google Scholar] [CrossRef]
- Alachraf, M.W.; Wende, R.C.; Schuler, S.M.M.; Schreiner, P.R.; Schrader, W. Functionality, Effectiveness, and Mechanistic Evaluation of a Multicatalyst-Promoted Reaction Sequence by Electrospray Ionization Mass Spectrometry. Chem. Eur. J. 2015, 21, 16203–16208. [Google Scholar] [CrossRef]
- Ganem, B.; Li, Y.T.; Henion, J.D. Detection of Noncovalent Receptor-Ligand Complexes by Mass Spectrometry. J. Am. Chem. Soc. 1991, 113, 6294–6296. [Google Scholar] [CrossRef]
- Hofstadler, S.A.; Sannes-Lowery, K.A. Applications of ESI-MS in drug discovery: Interrogation of noncovalent complexes. Nat. Rev. Drug Discov. 2006, 5, 585–595. [Google Scholar] [CrossRef]
- Gadagkar, S.R.; Call, G.B. Computational tools for fitting the Hill equation to dose–response curves. J. Pharmacol. Toxicol. Methods 2015, 71, 68–76. [Google Scholar] [CrossRef]
- OriginLab Corporation. OriginPro 9.8.0; OriginLab: Northampton, MA, USA, 2021. [Google Scholar]
- Wan, K.X.; Gross, M.L.; Shibue, T. Gas-Phase Stability of Double-Stranded Oligodeoxynucleotides and Their Noncovalent Complexes With DNA-Binding Drugs As Revealed by Collisional Activation in an Ion Trap. J. Am. Soc. Mass Spectrom. 2000, 11, 450–457. [Google Scholar] [CrossRef]
- Daniel, J.M.; Friess, S.D.; Rajagopalan, S.; Wendt, S.; Zenobi, R. Quantitative determination of noncovalent binding interactions using soft ionization mass spectrometry. Int. J. Mass Spectrom. 2002, 216, 1–27. [Google Scholar] [CrossRef]
- Hesse-Ertelt, S.; Heinze, T.; Kosan, B.; Schwikal, K.; Meister, F. Solvent Effects on the NMR Chemical Shifts of Imidazolium-Based Ionic Liquids and Cellulose Therein. Macromol. Symp. 2010, 294, 75–89. [Google Scholar] [CrossRef]
- Bodenhausen, G.; Ruben, D.J. Natural Abundance Nitrogen-15 Nmr by Enhanced Heteronuclear Spectroscopy. Chem. Phys. Lett. 1980, 69, 185–189. [Google Scholar] [CrossRef]
- Bax, A.; Summers, M.F. Proton and carbon-13 assignments from sensitivity-enhanced detection of heteronuclear multiple-bond connectivity by 2D multiple quantum NMR. J. Am. Chem. Soc. 1986, 108, 2093–2094. [Google Scholar] [CrossRef]
- Kessler, H.; Oschkinat, H.; Griesinger, C.; Bermel, W. Transformation of Homonuclear Two-Dimensional NMR Techniques into One-Dimensional Techniques Using Gaussian Pulses. J. Magn. Reson. 1986, 70, 106–133. [Google Scholar] [CrossRef]
- Anderson, P.W. A Mathematical Model for the Narrowing of Spectral Lines by Exchange or Motion. J. Phys. Soc. Jpn. 1954, 9, 316–339. [Google Scholar] [CrossRef]
- Sack, R.A. A contribution to the theory of the exchange narrowing of spectral lines. Mol. Phys. 1958, 1, 163–167. [Google Scholar] [CrossRef]
- Jeener, J.; Meier, B.H.; Bachmann, P.; Ernst, R.R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar] [CrossRef]
- Nikitin, K.; O’Gara, R. Mechanisms and Beyond: Elucidation of Fluxional Dynamics by Exchange NMR Spectroscopy. Chem. Eur. J. 2019, 25, 4551–4589. [Google Scholar] [CrossRef]
- Maciejewski, M.W.; Liu, D.; Prasad, R.; Wilson, S.H.; Mullen, G.P. Backbone Dynamics and Refined Solution Structure of the N-terminal Domain of DNA Polymerase β. Correlation with DNA Binding and dRP Lyase Activity. J. Mol. Biol. 2000, 296, 229–253. [Google Scholar] [CrossRef] [PubMed]
- Otter, A.; Kotovych, G. The solution conformation of the synthetic tubulin fragment Ac-tubulin-α(430–441)-amide based on two-dimensional ROESY experiments. Can. J. Chem. 1988, 66, 1814–1820. [Google Scholar] [CrossRef]
- Mirau, P.A.; Bovey, F.A. Two-Dimensional NMR Studies of Molecular Weight and Concentration Effects on Polymer-Polymer Interactions. Macromolecules 1990, 23, 4548–4552. [Google Scholar] [CrossRef]
- Bothner-By, A.A.; Stephens, R.L.; Lee, J.; Warren, C.D.; Jeanloz, R.W. Structure Determination of a Tetrasaccharide: Transient Nuclear Overhauser Effects in the Rotating Frame. J. Am. Chem. Soc. 1984, 106, 811–813. [Google Scholar] [CrossRef]
- Thiele, C.M.; Petzold, K.; Schleucher, J. EASY ROESY: Reliable Cross-Peak Integration in Adiabatic Symmetrized ROESY. Chem. Eur. J. 2009, 15, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Kuzmič, P. Chapter 10—DynaFit—A Software Package for Enzymology. In Methods in Enzymology; Johnson, M.L., Brand, L., Eds.; Academic Press: Cambridge, MA, USA, 2009; Volume 467, pp. 247–280. [Google Scholar]
- Nehls, T.; Heymann, T.; Meyners, C.; Hausch, F.; Lermyte, F. Fenton-Chemistry-Based Oxidative Modification of Proteins Reflects Their Conformation. Int. J. Mol. Sci. 2021, 22, 9927. [Google Scholar] [CrossRef] [PubMed]
- Thrippleton, M.J.; Keeler, J. Elimination of Zero-Quantum Interference in Two-Dimensional NMR Spectra. Angew. Chem. Int. Ed. 2003, 42, 3938–3941. [Google Scholar] [CrossRef]
- Kupce, E.; Boyd, J.; Campbell, I.D. Short Selective Pulses for Biochemical Applications. J. Magn. Reson. Ser. B 1995, 106, 300–303. [Google Scholar] [CrossRef]
- Fredi, A.; Nolis, P.; Cobas, C.; Parella, T. Access to experimentally infeasible spectra by pure-shift NMR covariance. J. Magn. Reson. 2016, 270, 161–168. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | 6 (C44H63N6O8)+ | 7 (C46H67N6O8)+ | 8 (C47H69N6O8)+ |
|---|---|---|---|
| exact mass | 803.4702 | 831.5015 | 845.5171 |
| Δ (DCM)/ppm | 0.50 | 0.60 | −4.97 |
| Δ (toluene)/ppm | 0.87 | −0.60 | 1.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brauser, M.; Heymann, T.; Thiele, C.M. Detection and Verification of a Key Intermediate in an Enantioselective Peptide Catalyzed Acylation Reaction. Molecules 2022, 27, 6351. https://doi.org/10.3390/molecules27196351
Brauser M, Heymann T, Thiele CM. Detection and Verification of a Key Intermediate in an Enantioselective Peptide Catalyzed Acylation Reaction. Molecules. 2022; 27(19):6351. https://doi.org/10.3390/molecules27196351
Chicago/Turabian StyleBrauser, Matthias, Tim Heymann, and Christina Marie Thiele. 2022. "Detection and Verification of a Key Intermediate in an Enantioselective Peptide Catalyzed Acylation Reaction" Molecules 27, no. 19: 6351. https://doi.org/10.3390/molecules27196351
APA StyleBrauser, M., Heymann, T., & Thiele, C. M. (2022). Detection and Verification of a Key Intermediate in an Enantioselective Peptide Catalyzed Acylation Reaction. Molecules, 27(19), 6351. https://doi.org/10.3390/molecules27196351