
Glycosyl Formates: Glycosylations with Neighboring-Group Participation

Abstract
:1. Introduction
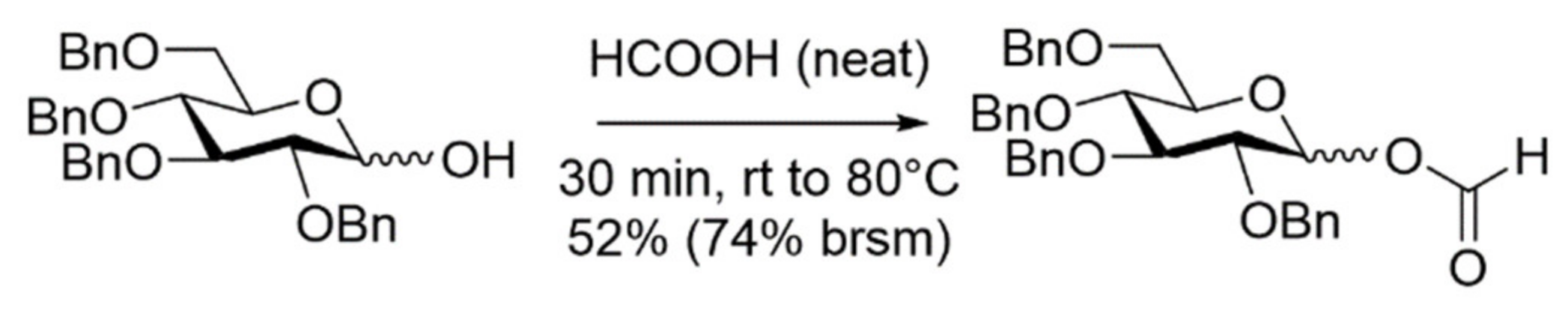
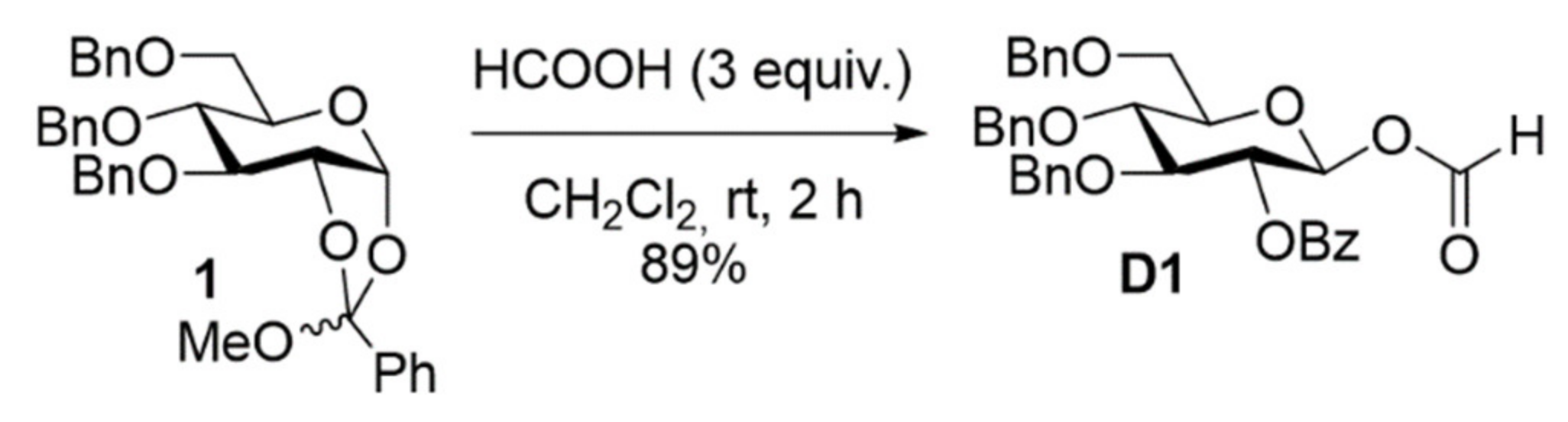
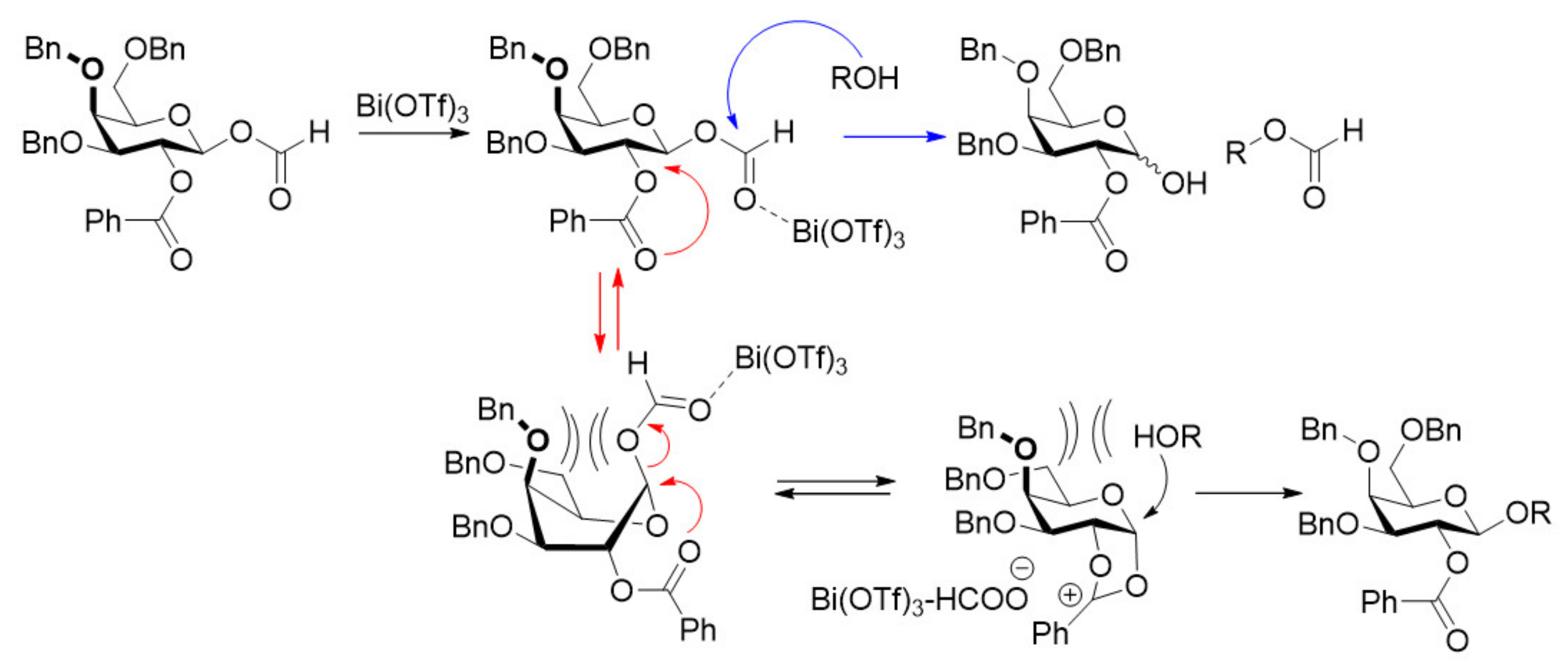
2. Results
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nielsen, M.M.; Pedersen, C.M. Catalytic Glycosylations in Oligosaccharide Synthesis. Chem. Rev. 2018, 118, 8285–8358. [Google Scholar] [CrossRef] [PubMed]
- Kochetkov, N.K.; Khorlin, A.J.; Bochkov, A.F. New synthesis of glycosides. Tetrahedron Lett. 1964, 5, 289–293. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Murai YShoda, S. An Efficient Method for Glucosylation of Hydroxy Compounds Using Glucopyranosyl Fluoride. Chem. Lett. 1981, 10, 431–432. [Google Scholar] [CrossRef]
- Schmidt, R.R.; Michel, J. Facile Synthesis of α- and β-O-Glycosyl Imidates; Preparation of Glycosides and Disaccharides. Angew. Chem. Int. Ed. Engl. 1980, 19, 731–732. [Google Scholar] [CrossRef]
- Yu, B.; Tao, H. Glycosyl trifluoroacetimidates. Part 1: Preparation and application as new glycosyl donors. Tetrahedron Lett. 2001, 42, 2405–2407. [Google Scholar] [CrossRef]
- Hotha, S.; Kashyap, S. Propargyl Glycosides as Stable Glycosyl Donors: Anomeric Activation and Glycoside Syntheses. J. Am. Chem. Soc. 2006, 128, 9620–9621. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Yu, B. An efficient glycosylation protocol with glycosyl ortho-alkynylbenzoates as donors under the catalysis of Ph3PAuOTf. Tetrahedron Lett. 2008, 49, 3604–3608. [Google Scholar] [CrossRef]
- Shaw, M.; Kumar, A. Additive-Free Gold(III)-Catalyzed Stereoselective Synthesis of 2-Deoxyglycosides Using Phenylpropiolate Glycosides as Donors. Chem.-Asian J. 2019, 14, 4651–4658. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, R.; Zhang, S.; Fang, J.; Liu, S.; Sun, G.; Xu, B.; Xiao, Y.; Fu, D.; Wan, Q.; et al. Hydrogen-Bonding-Assisted Exogenous Nucleophilic Reagent Effect for β-Selective Glycosylation of Rare 3-Amino Sugars. J. Am. Chem. Soc. 2019, 141, 8509–8515. [Google Scholar] [CrossRef]
- Helferich, B.; Schmitz-Hillebrecht, E. Eine neue Methode zur Synthese von Glykosiden der Phenole. Ber. Der Dtsch. Chem. Ges. (A B Ser.) 1933, 66, 378–383. [Google Scholar] [CrossRef]
- Kim, K.S.; Lee, Y.J.; Kim, H.Y.; Kang, S.S.; Kwon, S.Y. Glycosylation with glycosyl benzyl phthalates as a new type of glycosyl donor. Org. Biomol. Chem. 2004, 2, 2408–2410. [Google Scholar] [CrossRef]
- Christensen, H.; Christiansen, M.S.; Petersen, J.; Jensen, H.H. Direct formation of [small beta]-glycosides of N-acetyl glycosamines mediated by rare earth metal triflates. Org. Biomol. Chem. 2008, 6, 3276–3283. [Google Scholar] [CrossRef]
- Rasmussen, M.R.; Marqvorsen, M.H.S.; Kristensen, S.K.; Jensen, H.H. A protocol for metal triflate catalyzed direct glycosylations with galnac 1-opiv donors. J. Org. Chem. 2014, 79, 11011–11019. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, S.K.; Salamone, S.; Rasmussen, M.R.; Marqvorsen, M.H.S.; Jensen, H.H. Glycosyl ortho-Methoxybenzoates: Catalytically Activated Glycosyl Donors with an Easily Removable and Recyclable Leaving Group. Eur. J. Org. Chem. 2016, 2016, 5365–5376. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Hanamoto, T.; Suzuki, S.; Shimizu, K.; Furuno, H.; Inanaga, J. Tandem Catalysis Strategy for Direct Glycosylation of 1-Hydroxy Sugars. Methoxyacetic Acid as an Effective Catalytic Mediator. Heterocycles 2009, 79, 967–983. [Google Scholar] [CrossRef]
- Yang, L.; Hammelev, C.H.; Pedersen, C.M. Catalytic and Atom-Economic Glycosylation using Glycosyl Formates and Cheap Metal Salts. ChemSusChem 2020, 13, 3166–3171. [Google Scholar] [CrossRef]
- Steber, H.B.; Singh, Y.; Demchenko, A.V. Bismuth(iii) triflate as a novel and efficient activator for glycosyl halides. Org. Biomol. Chem. 2021, 19, 3220–3233. [Google Scholar] [CrossRef]
- Babu, J.L.; Khare, A.; Vankar, Y.D. Bi(OTf)3 and SiO2-Bi(OTf)3 as Effective Catalysts for the Ferrier Rearrangement. Molecules 2005, 10, 884–892. [Google Scholar] [CrossRef]
- Hough, L.; Lewis, A.W. 1-O-formyl-β-D-glucopyranose tetra-acetate. Carbohydr. Res. 1975, 42, 173–174. [Google Scholar] [CrossRef]
- Cecioni, S.; Vocadlo, D.J. Carbohydrate Bis-acetal-Based Substrates as Tunable Fluorescence-Quenched Probes for Monitoring exo-Glycosidase Activity. J. Am. Chem. Soc. 2017, 139, 8392–8395. [Google Scholar] [CrossRef]
- Yuan, J.; Lindner, K.; Frauenrath, H. 1-O-Vinyl Glycosides via Tebbe Olefination, Their Use as Chiral Auxiliaries and Monomers. J. Org. Chem. 2006, 71, 5457–5467. [Google Scholar] [CrossRef] [PubMed]
- Mydock, L.K.; Demchenko, A.V. Superarming the S-Benzoxazolyl Glycosyl Donors by Simple 2-O-Benzoyl-3,4,6-tri-O-benzyl Protection. Org. Lett. 2008, 10, 2103–2106. [Google Scholar] [CrossRef] [PubMed]
- Crich, D.; Hu, T.; Cai, F. Does Neighboring Group Participation by Non-Vicinal Esters Play a Role in Glycosylation Reactions? Effective Probes for the Detection of Bridging Intermediates. J. Org. Chem. 2008, 73, 8942–8953. [Google Scholar] [CrossRef] [PubMed]
- Mootoo, D.R.; Konradsson, P.; Udodong, U.; Fraser-Reid, B. Armed and disarmed n-pentenyl glycosides in saccharide couplings leading to oligosaccharides. J. Am. Chem. Soc. 1988, 110, 5583–5584. [Google Scholar] [CrossRef]
- Chang, C.-W.; Lin, M.H.; Chan, C.K.; Su, K.Y.; Wu, C.H.; Lo, W.C.; Lam, S.; Cheng, Y.-T.; Liao, P.-H.; Wong, C.-H.; et al. Automated Quantification of Hydroxyl Reactivities: Prediction of Glycosylation Reactions. Angew. Chem. Int. Ed. 2021, 60, 12413–12423. [Google Scholar] [CrossRef]
- Boullanger, P.; Banoub, J.; Descotes, G. N-Allyloxycarbonyl derivatives of D-glucosamine as promotors of 1,2-trans-glucosylation in Koenigs–Knorr reactions and in Lewis acid catalyzed condensations. Can. J. Chem. 1987, 65, 1343–1348. [Google Scholar] [CrossRef]
- Heinemann, F.; Hiegemann, M.; Welzel, P. Glycosylations with tetra-O-acetyl-N-allyloxycarbonylamino-2-deoxy-β-D-glucose in polar solvents. Tetrahedron 1992, 48, 3781–3788. [Google Scholar] [CrossRef]
- Ekborg, G.; Glaudemans, C.P.J. p-(Trifluoroacetamido)phenyl 2-acetamido-2-deoxy-4-O-β-d-mannopyranosyl-β-d-glucopyranoside. Carbohydr. Res. 1984, 129, 287–292. [Google Scholar] [CrossRef]
- Hölemann, A.; Stocker, B.L.; Seeberger, P.H. Synthesis of a core arabinomannan oligosaccharide of Mycobacterium tuberculosis. J. Org. Chem. 2006, 71, 8071–8088. [Google Scholar] [CrossRef]
- Ma, T.; Li, C.; Zhang, Z.X.; Wang, Z.; Yu, L.; Xue, W. Indium(III) Iodide-Catalyzed Stereoselective Synthesis of β-Glucopyranosides by Using a Glucosyl Fluoride Donor with 2-O-Benzoyl-3,4,6-Tri-O-Benzyl Protection. Synlett 2017, 28, 2633–2636. [Google Scholar]
- Ravidà, A.; Liu, X.; Kovacs, L.; Seeberger, P.H. Synthesis of glycosyl phosphates from 1,2-orthoesters and application to in situ glycosylation reactions. Org. Lett. 2006, 8, 1815–1818. [Google Scholar] [CrossRef]
- Buhl, M.; Traboni, S.; Körsgen, M.; Lamping, S.; Arlinghaus, H.F.; Ravoo, B.J. On surface: O-glycosylation by catalytic microcontact printing. Chem. Commun. 2017, 53, 6203–6206. [Google Scholar] [CrossRef] [PubMed]
- Raposo, C.D.; Costa, R.; Petrova, K.T.; Brito, C.; Scotti, M.T.; Cardoso, M.M. Development of novel galactosylated PLGA nanoparticles for hepatocyte targeting using molecular modelling. Polymers 2020, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Kramer, S.; Langhanki, J.; Krumb, M.; Opatz, T.; Bros, M.; Zentel, R. HPMA-Based Nanocarriers for Effective Immune System Stimulation. Macromol. Biosci. 2019, 19, 1800481. [Google Scholar] [CrossRef]
- Wei, S.; Zhao, J.; Shao, H. A facile method for the preparation of sugar orthoesters promoted by anhydrous sodium bicarbonate. Can. J. Chem. 2009, 87, 1733–1737. [Google Scholar] [CrossRef]
- Bloemendal, V.R.; Moons, S.J.; Heming, J.J.; Chayoua, M.; Niesink, O.; van Hest, J.C.; Boltje, T.J.; Rutjes, F.P. Chemoenzymatic Synthesis of Sialic Acid Derivatives Using Immobilized N-Acetylneuraminate Lyase in a Continuous Flow Reactor. Adv. Synth. Catal. 2019, 361, 2443–2447. [Google Scholar] [CrossRef]
- Lv, Y.M.; Laborda, P.; Huang, K.; Cai, Z.P.; Wang, M.; Lu, A.M.; Doherty, C.; Liu, L.; Flitsch, S.L.; Voglmeir, J. Highly efficient and selective biocatalytic production of glucosamine from chitin. Green Chem. 2017, 19, 527–535. [Google Scholar] [CrossRef]
- Späte, A.K.; Schart, V.F.; Schöllkopf, S.; Niederwieser, A.; Wittmann, V. Terminal Alkenes as Versatile Chemical Reporter Groups for Metabolic Oligosaccharide Engineering. Chem.—A Eur. J. 2014, 20, 16502–16508. [Google Scholar] [CrossRef]
- De Nisco, M.; Pedatella, S.; Bektaş, S.; Nucci, A.; Caputo, R. d-Glucosamine in a chimeric prolinamide organocatalyst for direct asymmetric aldol addition. Carbohydr. Res. 2012, 356, 273–277. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Glycosyl donor | Glycosyl acceptor | Glycoside | Yield a |
| 1 | D1 | A1 | G1 | 65 |
| 2 | D1 | A2 | G2 | 67 |
| 3 | D1 | A3 | G3 | 72 |
| 4 | D1 | A4 | G4 | 81 |
| 5 | D2 | A1 | G5 | 71 |
| 6 | D2 | A2 | G6 | 72 |
| 7 | D2 | A3 | G7 | 73 |
| 8 | D2 | A4 | G8 | 38 |
| 9 | D3 | A1 | G9 | 62 |
| 10 | D3 | A2 | G10 | 68 |
| 11 | D3 | A3 | G11 | 71 |
| 12 | D3 | A4 | G12 | 76 |
| 13 | D4 | A1 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Pedersen, C.M. Glycosyl Formates: Glycosylations with Neighboring-Group Participation. Molecules 2022, 27, 6244. https://doi.org/10.3390/molecules27196244
Yang L, Pedersen CM. Glycosyl Formates: Glycosylations with Neighboring-Group Participation. Molecules. 2022; 27(19):6244. https://doi.org/10.3390/molecules27196244
Chicago/Turabian StyleYang, Liang, and Christian Marcus Pedersen. 2022. "Glycosyl Formates: Glycosylations with Neighboring-Group Participation" Molecules 27, no. 19: 6244. https://doi.org/10.3390/molecules27196244
APA StyleYang, L., & Pedersen, C. M. (2022). Glycosyl Formates: Glycosylations with Neighboring-Group Participation. Molecules, 27(19), 6244. https://doi.org/10.3390/molecules27196244