
An Improved Isotope Labelling Method for Quantifying Deamidated Cobratide Using High-Resolution Quadrupole-Orbitrap Mass Spectrometry
Abstract
:1. Introduction
2. Results
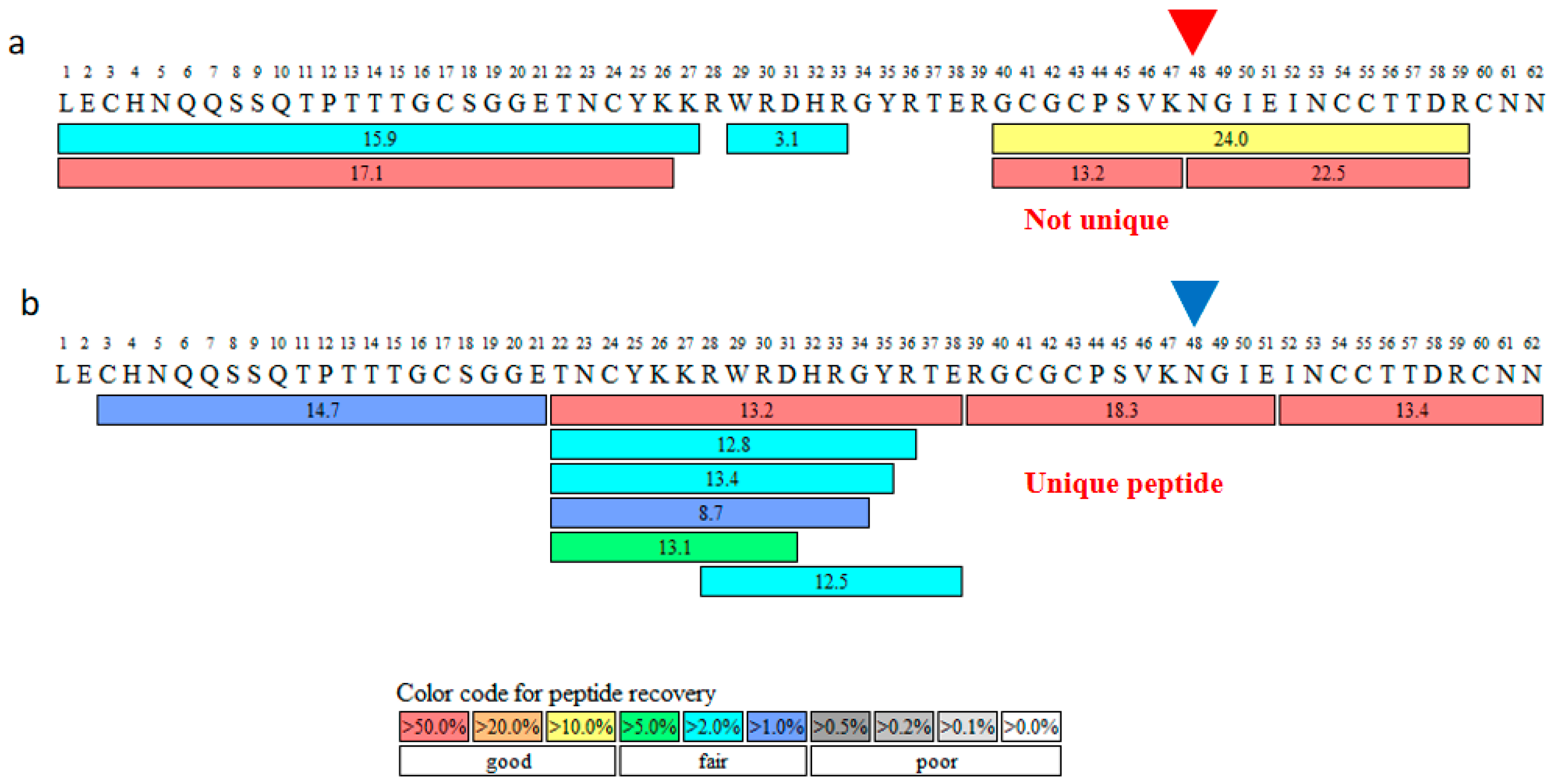
2.1. Glu-C for Enzymatic Digestion
2.2. Introduction to the Calculation
2.3. Calculation of the Percentages of Asp and isoAsp Contents
2.3.1. Calculation of Total Deamidation
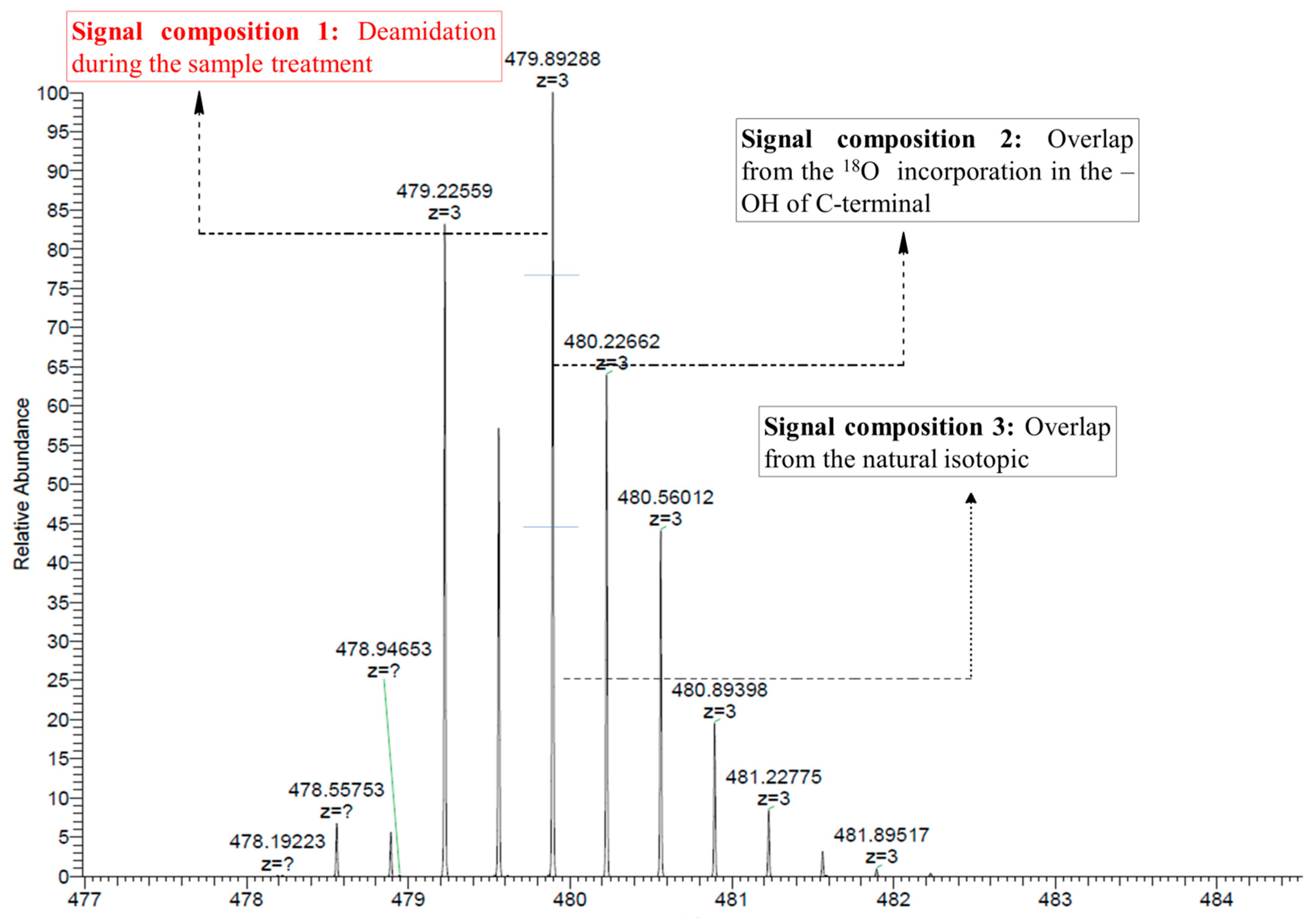
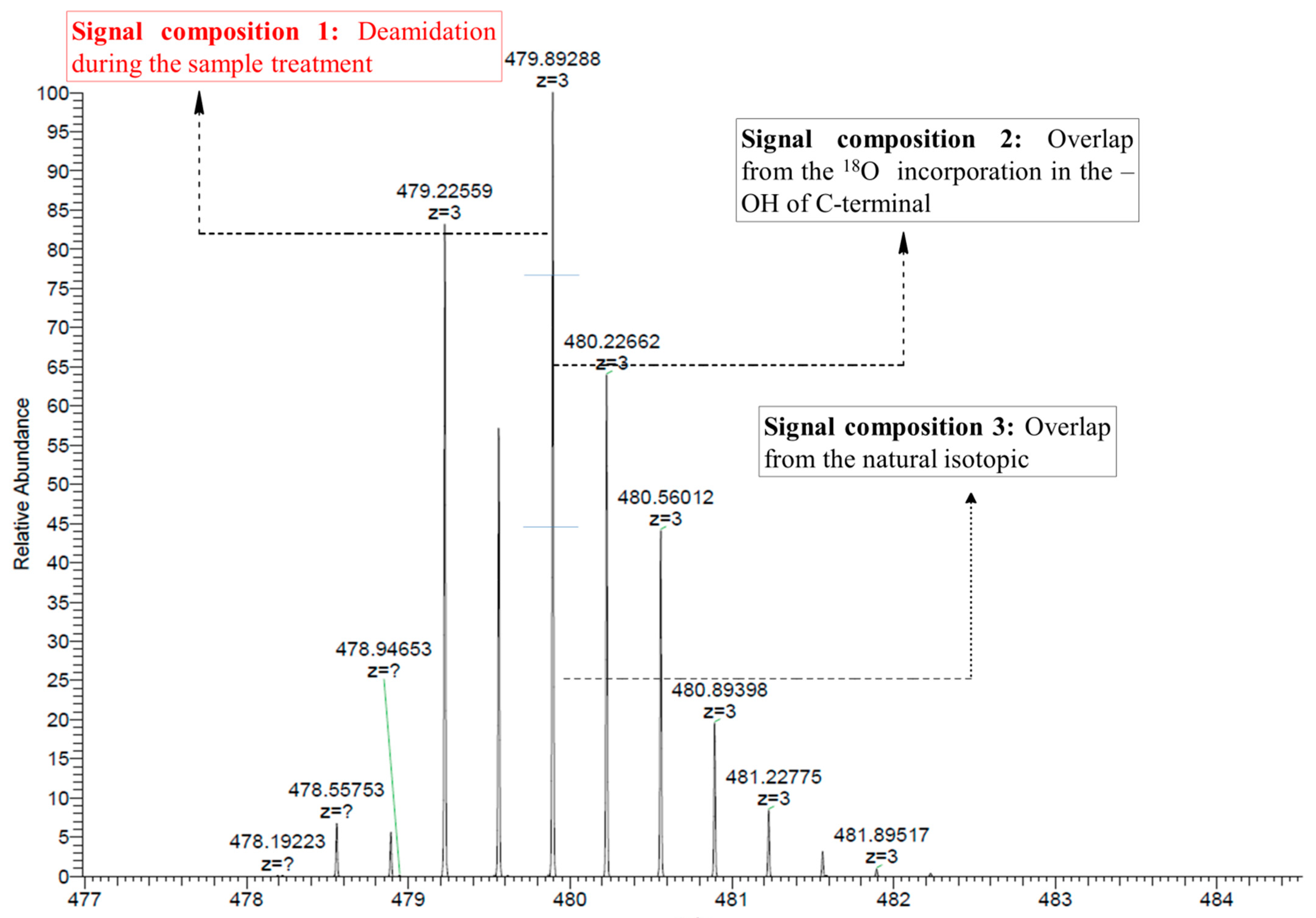
2.3.2. Calculation of Isotope Overlaps
2.3.2.1. Determination of the Overlap by the Natural Isotopic Abundance
2.3.2.2. Determination of the Overlap by Peptides with Two C-Terminal 18O Atoms
2.3.3. Calculation of the Deamidation Ratio Prior to Sample Preparation
2.3.4. Correction of the Deamidation Ratio
2.3.5. Calculation of the Relative Percentage of Asp/isoAsp Present in the Sample Prior to Sample Preparation
2.4. Validation
2.4.1. Specificity
2.4.2. Precision
2.4.3. Limit of Detection (LOD) and Limit of Quantification (LOQ)
2.4.4. Robustness
2.4.5. Recovery
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Preparation of the Test Solution
4.3. Liquid Chromatography/Mass Spectrometry
4.4. Validation Parameters of the Proposed Method
4.4.1. Specificity
4.4.2. Precision
4.4.3. Accuracy
4.4.4. Sensitivity
4.4.5. Robustness
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wang, J.; Lundström, S.L.; Seelow, S.; Rodin, S.; Meng, Z.; Astorga-Wells, J.; Jia, Q.; Zubarev, R.A. First Immunoassay for Measuring Isoaspartate in Human Serum Albumin. Molecules 2021, 26, 6709. [Google Scholar] [CrossRef] [PubMed]
- Kawasue, S.; Sakaguchi, Y.; Koga, R.; Yoshida, H.; Nohta, H. Assessment method for deamidation in proteins using carboxylic acid derivatization-liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2020, 181, 113095. [Google Scholar] [CrossRef] [PubMed]
- Soulby, A.J.; Heal, J.W.; Barrow, M.P.; Roemer, R.A.; O’Connor, P.B. Does deamidation cause protein unfolding? A top-down tandem mass spectrometry study. Protein Sci. 2015, 24, 850. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Lin, S.; De Mel, N.; Parupudi, A.; Delmar, J.; Pandey, M.; Wang, X.; Wang, J. Deamidation in Moxetumomab Pasudotox Leading to Conformational Change and Immunotoxin Activity Loss. J. Pharm. Sci. 2020, 109, 2676. [Google Scholar] [CrossRef] [PubMed]
- Cacia, J.; Keck, R.; Presta, L.G.; Frenz, J. Isomerization of an aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE: Identification and effect on binding affinity. Biochemistry 1996, 35, 1897. [Google Scholar] [CrossRef] [PubMed]
- Bults, P.; Bischoff, R.; Bakker, H.; Gietema, J.A.; van de Merbel, N.C. LC-MS/MS-Based Monitoring of In Vivo Protein Biotransformation: Quantitative Determination of Trastuzumab and Its Deamidation Products in Human Plasma. Anal. Chem. 2016, 88, 1871. [Google Scholar] [CrossRef]
- Jin, Y.; Yi, Y.; Yeung, B. Mass spectrometric analysis of protein deamidation—A focus on top-down and middle-down mass spectrometry. Methods 2022, 200, 58. [Google Scholar] [CrossRef]
- Clarke, S. Propensity for spontaneous succinimide formation from aspartyl and asparaginyl residues in cellular proteins. Int. J. Pept. Protein Res. 1987, 30, 808. [Google Scholar] [CrossRef]
- Gazerani, P.; Cairns, B.E. Venom-based biotoxins as potential analgesics. Expert Rev. Neurother. 2014, 14, 1261. [Google Scholar] [CrossRef]
- Zhang, Y. Why do we study animal toxins? Dongwuxue Yanjiu 2015, 36, 183. [Google Scholar] [CrossRef]
- Lin, F.; Reid, P.F.; Qin, Z.H. Cobrotoxin could be an effective therapeutic for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1258. [Google Scholar] [CrossRef] [PubMed]
- Hao, P.; Adav, S.S.; Gallart-Palau, X.; Sze, S.K. Recent advances in mass spectrometric analysis of protein deamidation. Mass Spectrom. Rev. 2017, 36, 677. [Google Scholar] [CrossRef]
- Rolland, A.D.; Prell, J.S. Approaches to Heterogeneity in Native Mass Spectrometry. Chem. Rev. 2022, 27, 122–7909. [Google Scholar] [CrossRef]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’Auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747. [Google Scholar] [CrossRef]
- Abdelhamid, H.N.; Wu, H.F. Proteomics analysis of the mode of antibacterial action of nanoparticles and their interactions with proteins. TrAC Trends Anal. Chem. 2015, 65, 30. [Google Scholar] [CrossRef]
- Gao, P.; Xu, G. Mass-spectrometry-based microbial metabolomics: Recent developments and applications. Anal. Bioanal. Chem. 2015, 407, 669. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, W.; Gao, T.; Huang, L.; Fan, H.; Chen, H.X. Separation, identification and quantification of associated impurities in cobratide using sheathless CE-MS and CE-UV. Anal. Methods 2021, 13, 3845–3851. [Google Scholar] [CrossRef]
- Alfaro, J.F.; Gillies, L.A.; Sun, H.G.; Dai, S.; Zang, T.; Klaene, J.J.; Kim, B.J.; Lowenson, J.D.; Clarke, S.G.; Karger, B.L.; et al. Chemo-enzymatic detection of protein isoaspartate using protein isoaspartate methyltransferase and hydrazine trapping. Anal. Chem. 2008, 80, 3882. [Google Scholar] [CrossRef]
- Ni, W.; Dai, S.; Karger, B.L.; Zhou, Z.S. Analysis of isoaspartic Acid by selective proteolysis with Asp-N and electron transfer dissociation mass spectrometry. Anal. Chem. 2010, 82, 7485. [Google Scholar] [CrossRef]
- Krokhin, O.V.; Antonovici, M.; Ens, W.; Wilkins, J.A.; Standing, K.G. Deamidation of -Asn-Gly- sequences during sample preparation for proteomics: Consequences for MALDI and HPLC-MALDI analysis. Anal. Chem. 2006, 78, 6645. [Google Scholar] [CrossRef]
- Liu, S.; Moulton, K.R.; Auclair, J.R.; Zhou, Z.S. Mildly acidic conditions eliminate deamidation artifact during proteolysis: Digestion with endoprotease Glu-C at pH 4.5. Amino Acids 2016, 48, 1059. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liu, B.; Fan, H.H. Determination method of deamidation impurities in cobratide using mass spectrometry technique. Acta Pharm. Sin. 2021, 56, 2352–2359. [Google Scholar]
- Du, Y.; Wang, F.; May, K.; Xu, W.; Liu, H. Determination of deamidation artifacts introduced by sample preparation using 18O-labeling and tandem mass spectrometry analysis. Anal. Chem. 2012, 84, 6355. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, F.; Xu, W.; May, K.; Richardson, D. Quantitation of asparagine deamidation by isotope labeling and liquid chromatography coupled with mass spectrometry analysis. Anal. Biochem. 2013, 432, 16. [Google Scholar] [CrossRef] [PubMed]
- Validation of Analytical Procedures: Text and Methodology, ICH Q2 (R1). In Proceedings of the International Conference on Harmonization (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 6 November 2005. Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 29 July 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Deamidation Result (Proposed Method) | Deamidation Result (CE–UV) a | ||
|---|---|---|---|---|
| isoAsp | Asp | Total Deamidation | Total Deamidation | |
| S1 | 12.0% | 4.3% | 16.3% | 15.8% |
| S2 | 17.8% | 9.0% | 26.8% | 25.9% |
| S3 | 16.9% | 7.4% | 24.3% | 24.9% |
| S4 | 4.7% | 2.3% | 7.0% | 8.1% |
| S5 | 13.1% | 6.0% | 19.1% | 18.3% |
| RSD (%) | ||
|---|---|---|
| Impurity | Intra-Day a | Inter-Day b |
| IsoAsp | 1.6% | 2.8% |
| Asp | 2.0% | 2.2% |
| Total Deamidation | 1.7% | 2.1% |
| NO | Parameter | Level a | Area Ratio | |
|---|---|---|---|---|
| Mean (%) a | RSD (%) | |||
| 1 | Flow rate | A | 1.54 | 0.0 |
| B | 1.54 | 0.0 | ||
| 2 | Mobile phase | A | 1.57 | 1.3 |
| B | 1.55 | 0.4 | ||
| 3 | Column oven temperature | A | 1.56 | 0.0 |
| B | 1.55 | 0.9 | ||
| 4 | Injection volume | A | 1.55 | 0.9 |
| B | 1.54 | 0.0 | ||
| Impurity | Recovery (%) | RSD (%) |
|---|---|---|
| IsoAsp | 101.52 ± 1.17 | 1.15 |
| Asp | 102.42 ± 1.82 | 1.78 |
| Total Deamidation | 103.55 ± 1.07 | 1.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Huang, L.; Xu, R.; Fan, H.; Wang, Y. An Improved Isotope Labelling Method for Quantifying Deamidated Cobratide Using High-Resolution Quadrupole-Orbitrap Mass Spectrometry. Molecules 2022, 27, 6154. https://doi.org/10.3390/molecules27196154
Liu B, Huang L, Xu R, Fan H, Wang Y. An Improved Isotope Labelling Method for Quantifying Deamidated Cobratide Using High-Resolution Quadrupole-Orbitrap Mass Spectrometry. Molecules. 2022; 27(19):6154. https://doi.org/10.3390/molecules27196154
Chicago/Turabian StyleLiu, Bo, Lu Huang, Rongrong Xu, Huihong Fan, and Yue Wang. 2022. "An Improved Isotope Labelling Method for Quantifying Deamidated Cobratide Using High-Resolution Quadrupole-Orbitrap Mass Spectrometry" Molecules 27, no. 19: 6154. https://doi.org/10.3390/molecules27196154
APA StyleLiu, B., Huang, L., Xu, R., Fan, H., & Wang, Y. (2022). An Improved Isotope Labelling Method for Quantifying Deamidated Cobratide Using High-Resolution Quadrupole-Orbitrap Mass Spectrometry. Molecules, 27(19), 6154. https://doi.org/10.3390/molecules27196154