
The Steric Effect in Preparations of Vanadium(II)/(III) Dinitrogen Complexes of Triamidoamine Ligands Bearing Bulky Substituents


 and
and
Abstract
:1. Introduction
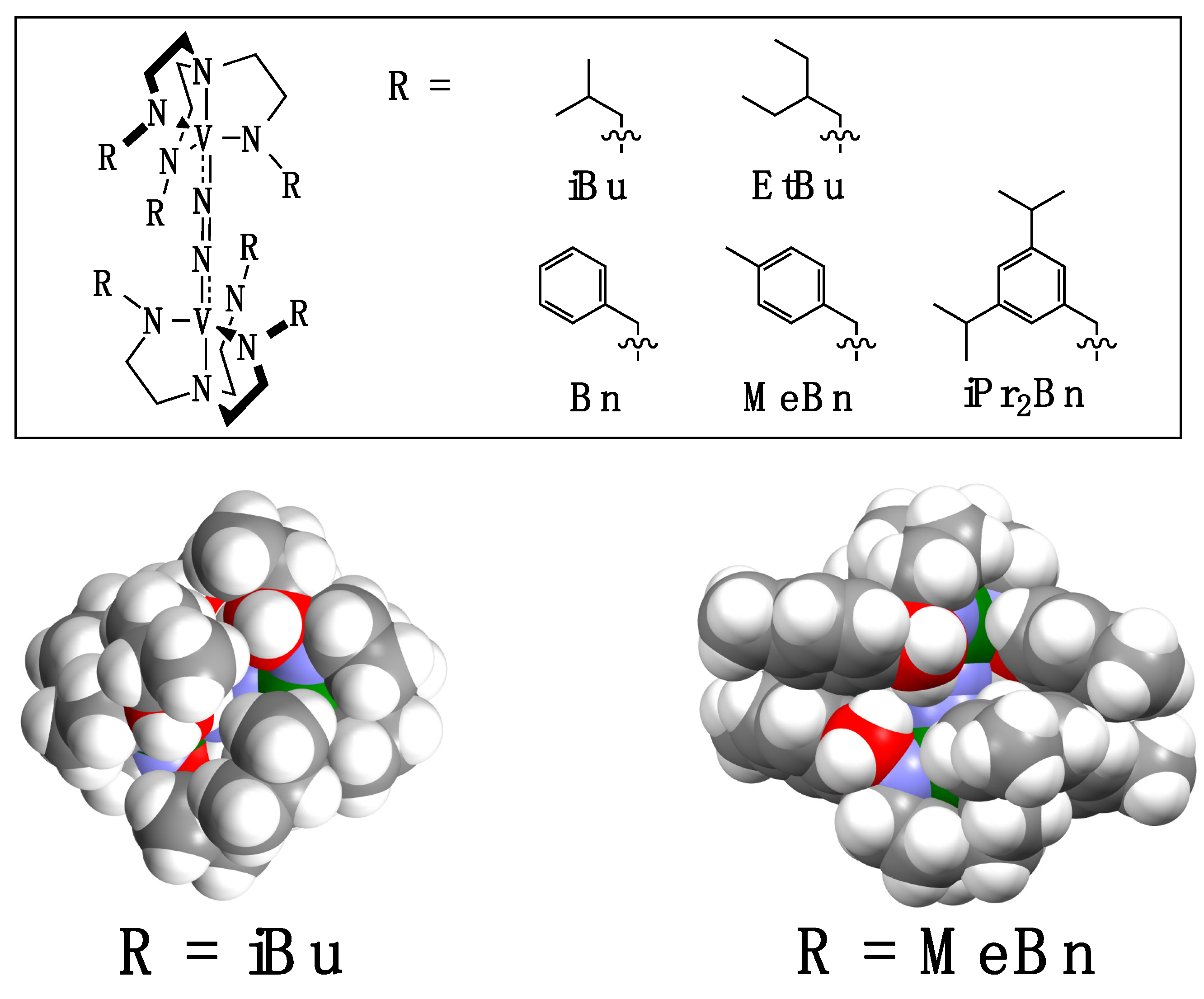
2. Results and Discussion
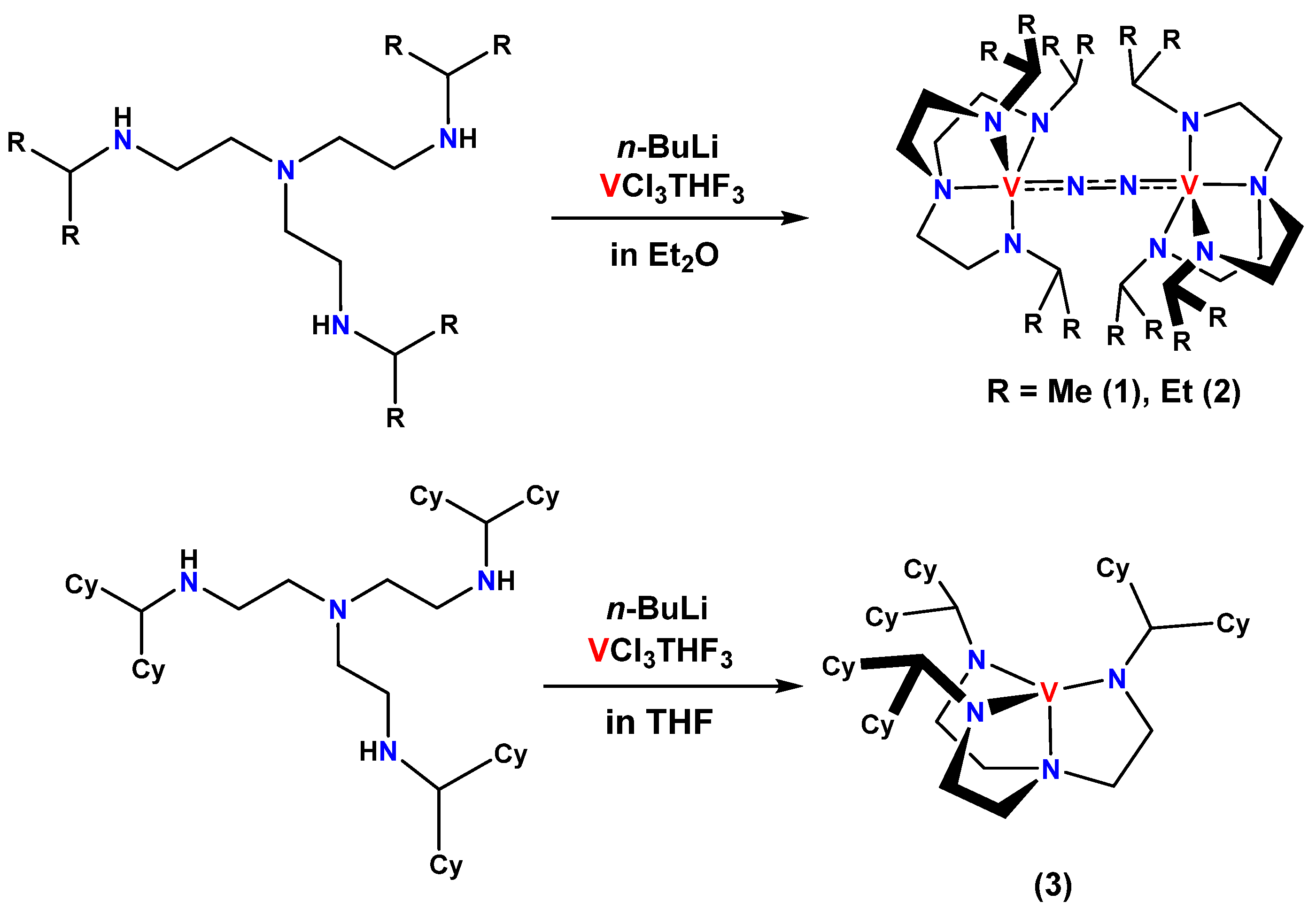
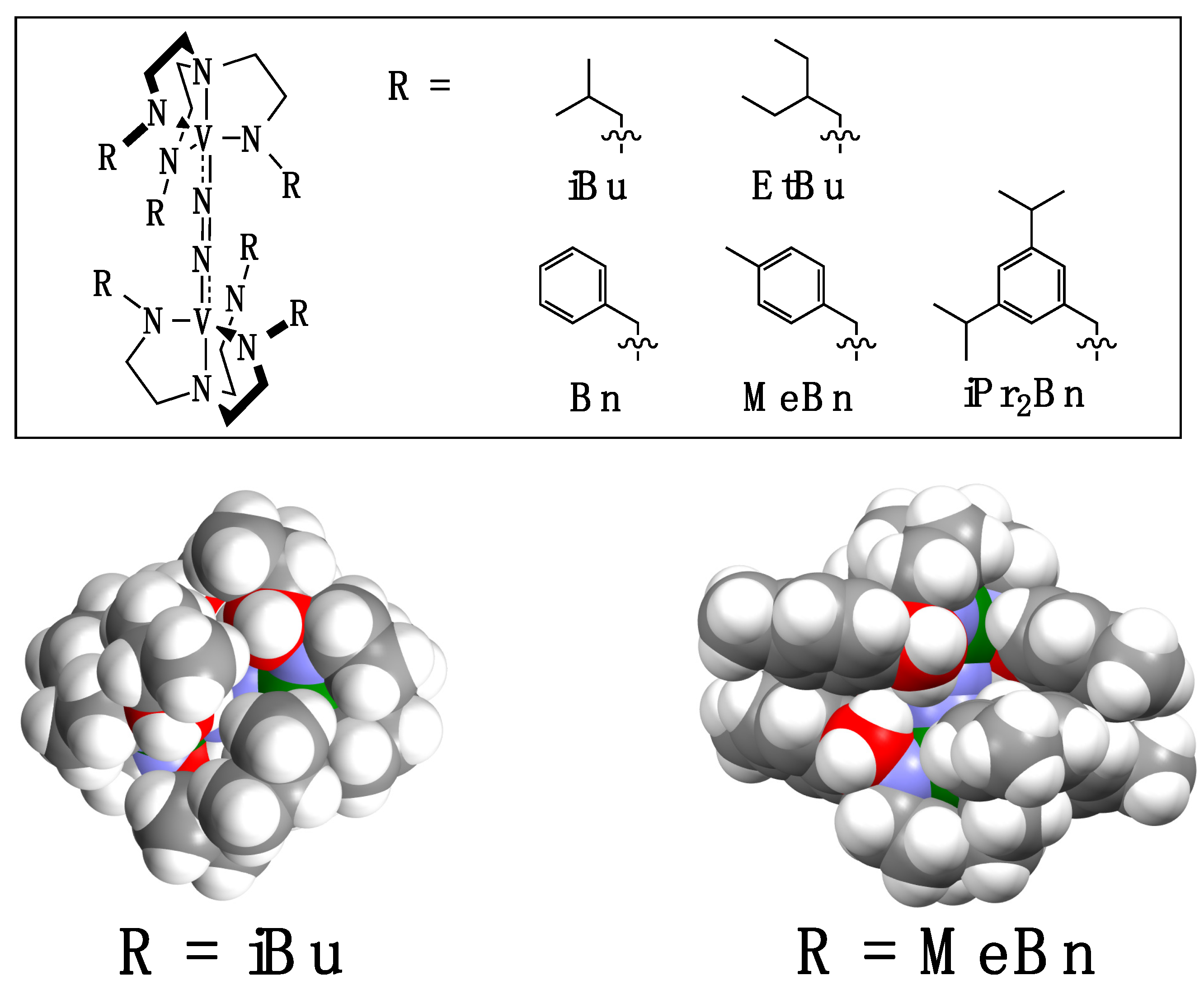
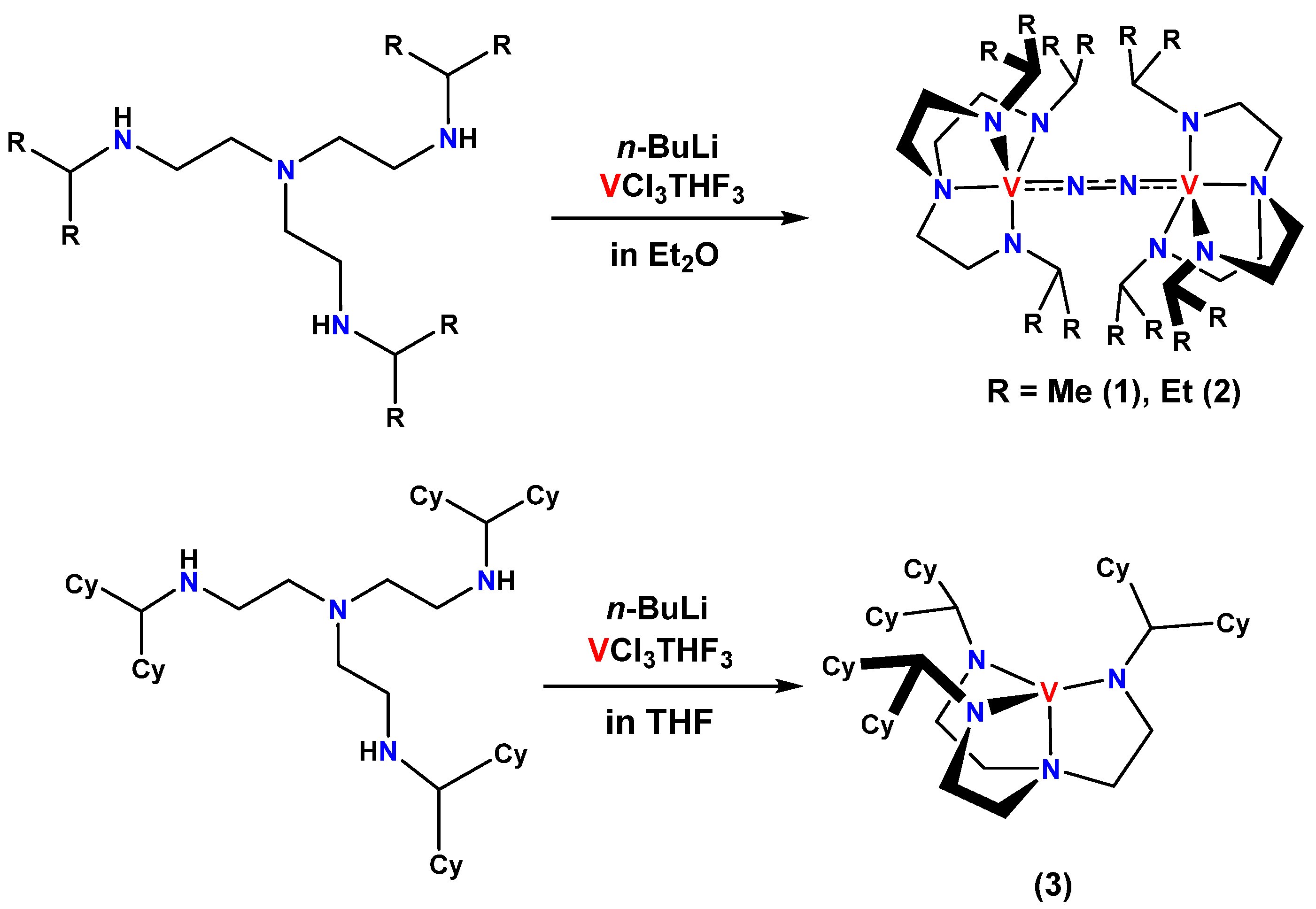
2.1. Syntheses of Ligands and Their Vanadium Complexes 1, 2, and 3
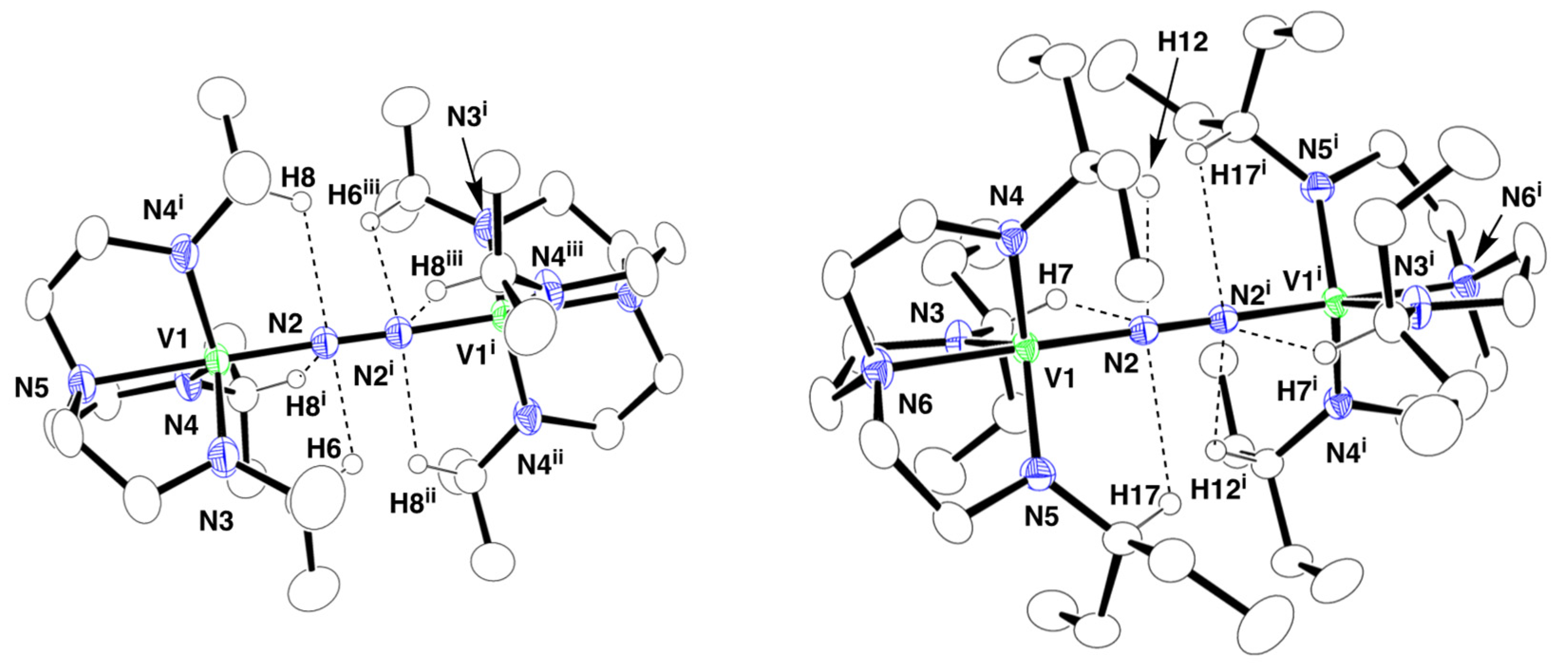
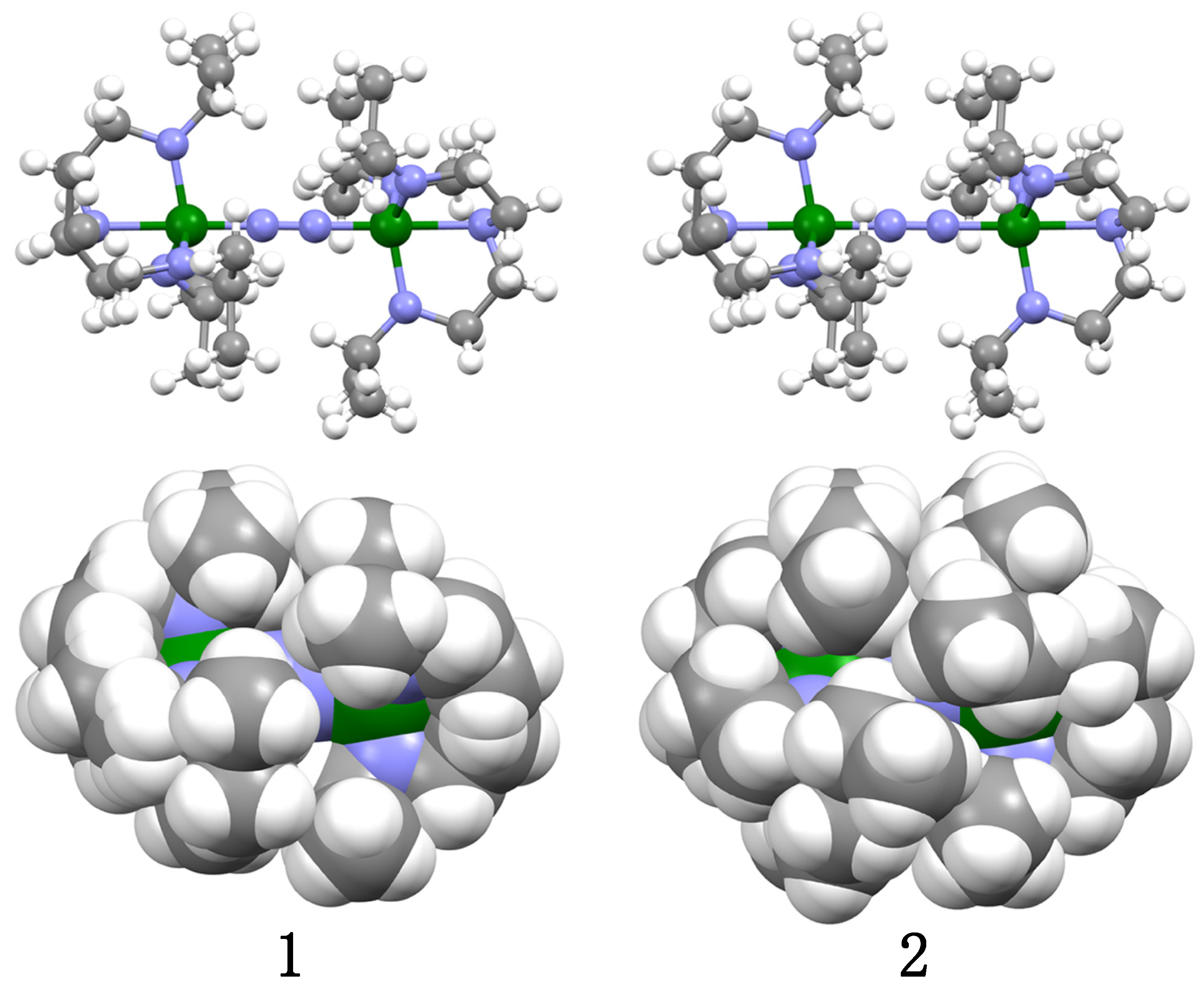
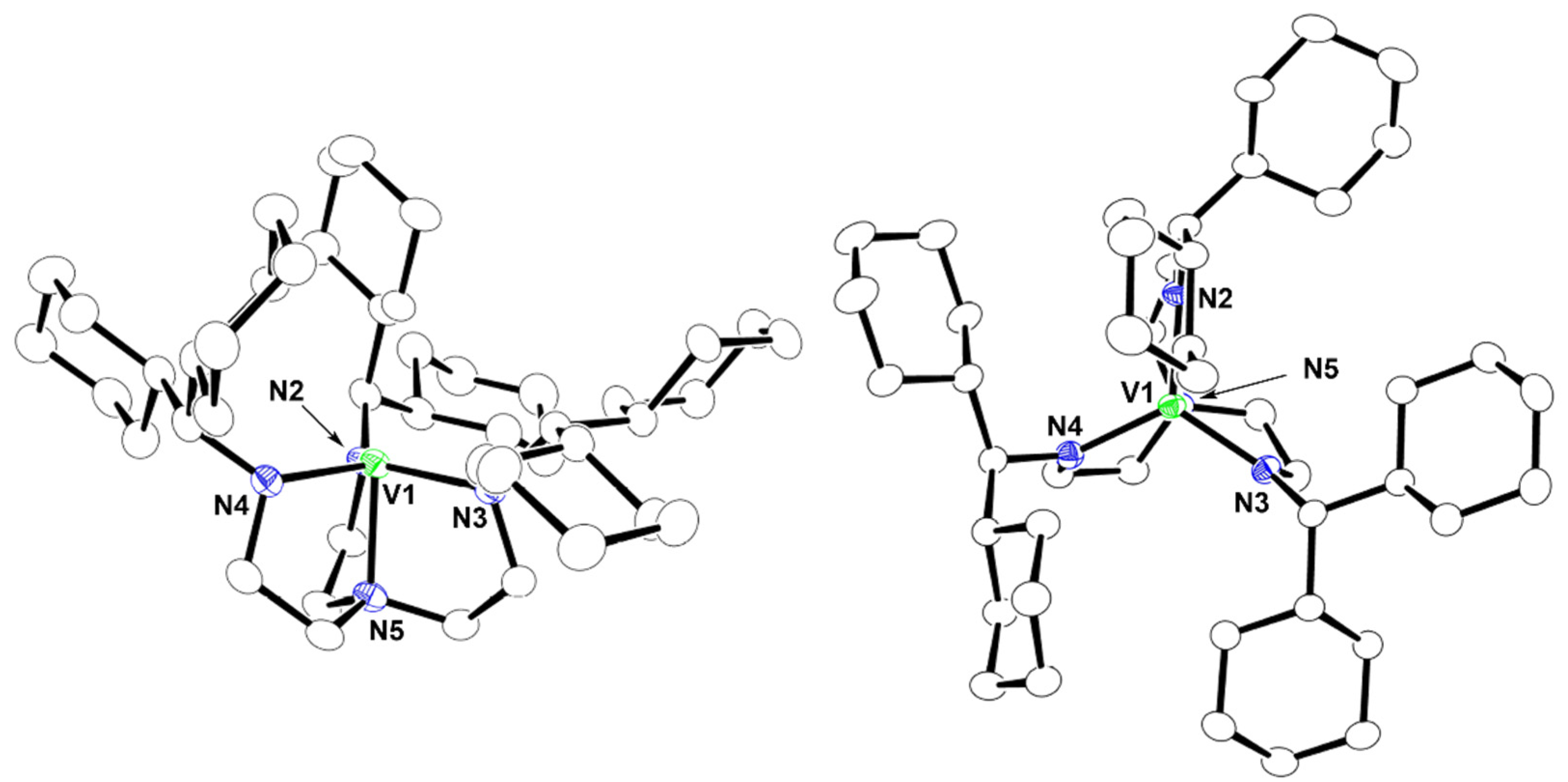
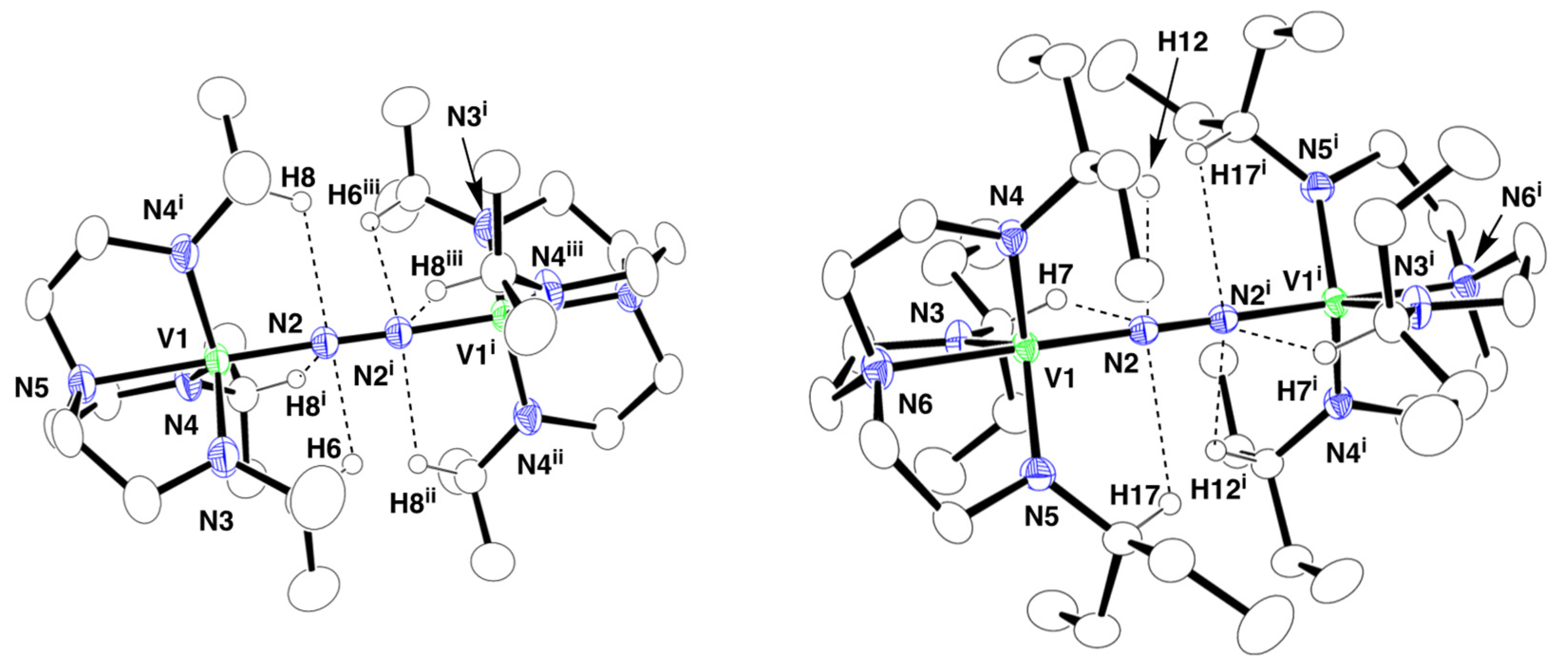
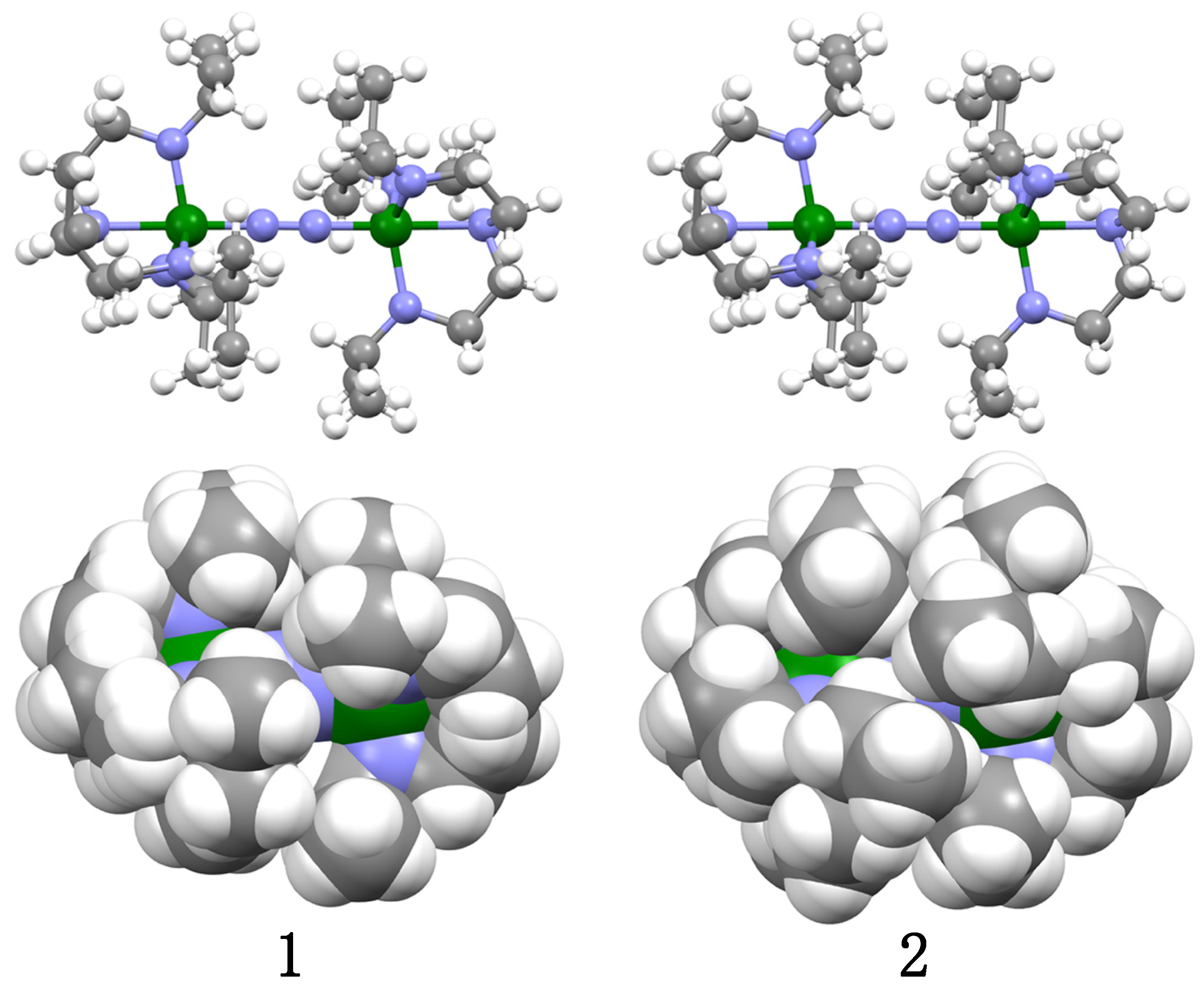
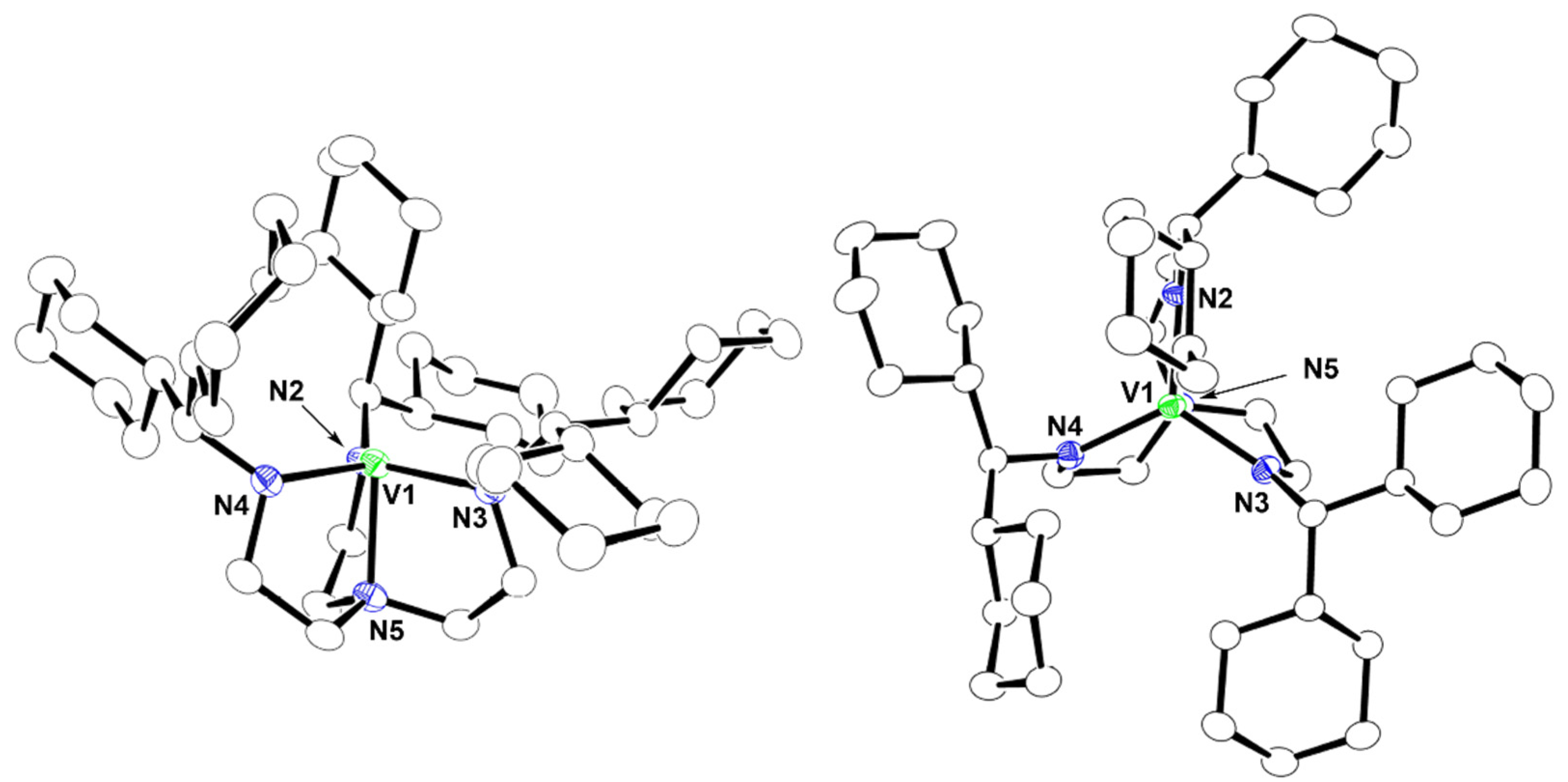
2.2. Crystal Structures of 1 and 2
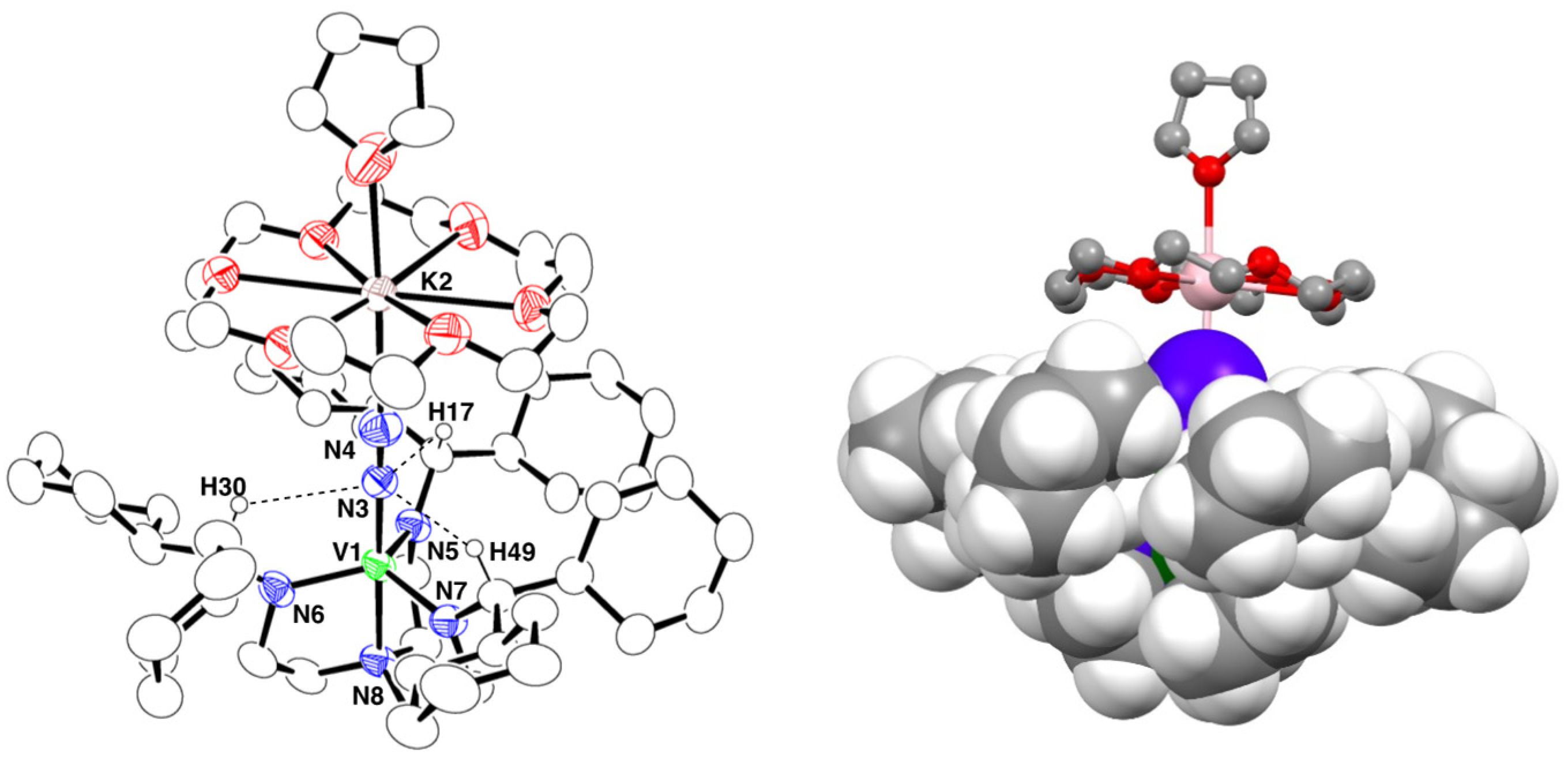
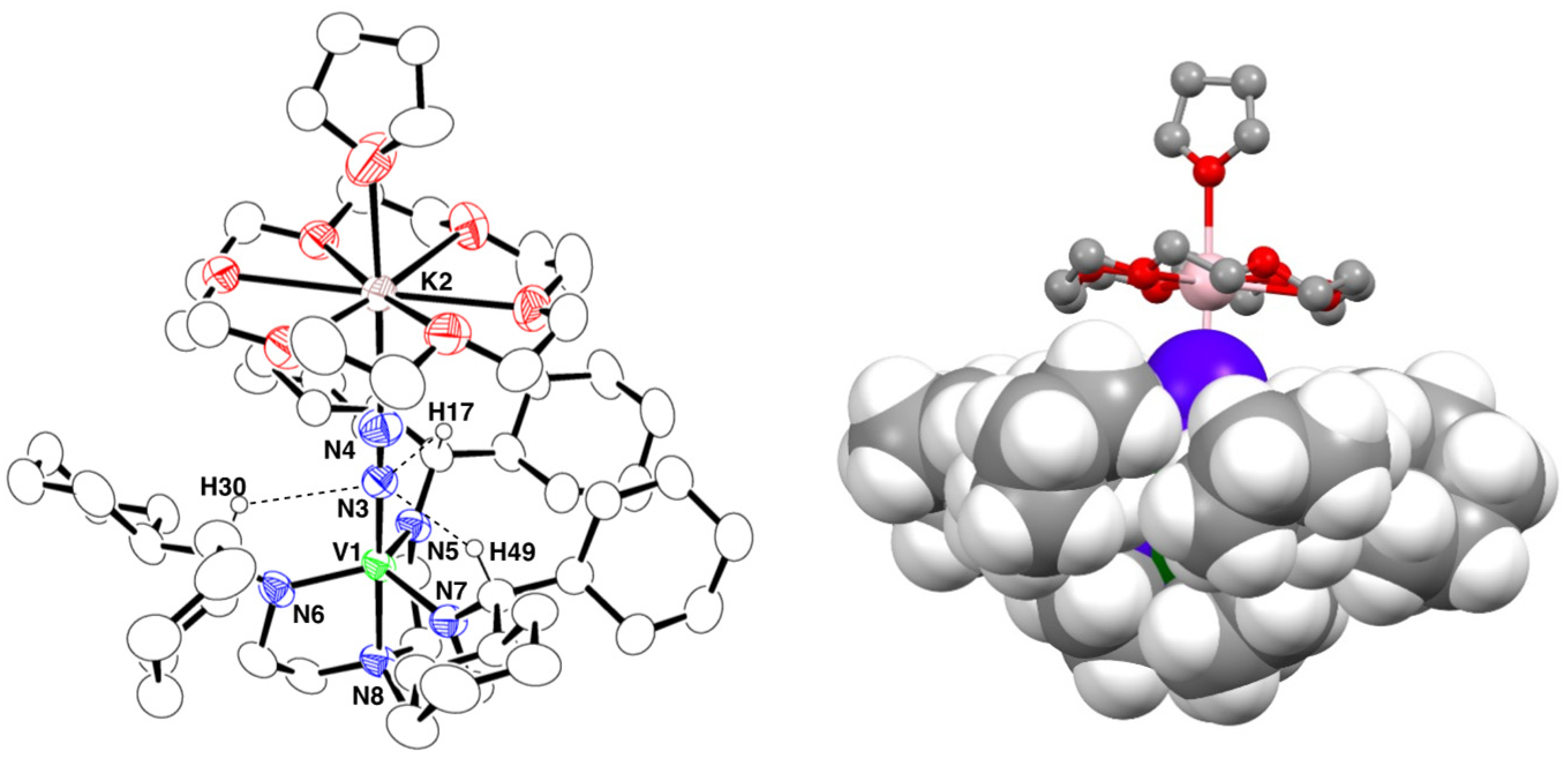
2.3. Crystal Structure of 3
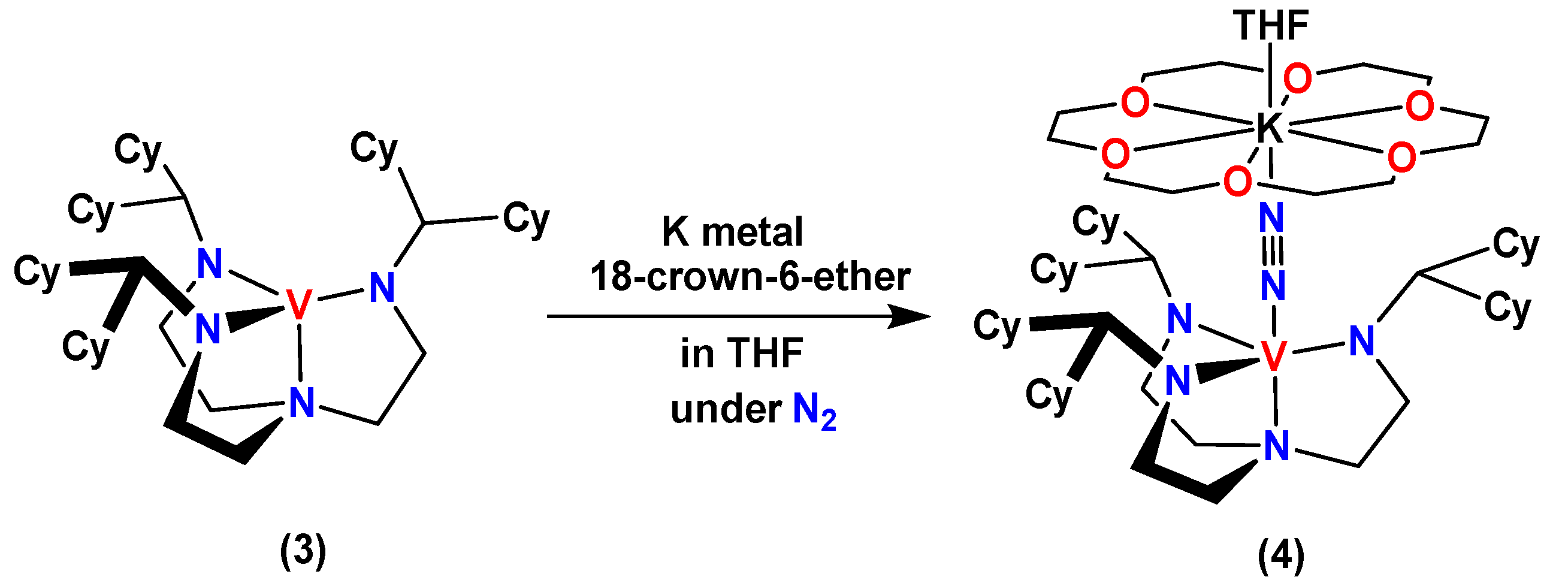
2.4. Synthesis and Crystal Structure of 4
2.5. Raman and Infrared Spectra of 1, 2, and 4
2.6. 1H-, 15N-, and 51V-NMR Spectra
2.7. Protonation of 1–4 in the Presence of Reductants
3. Materials and Methods
3.1. General Procedures
3.2. Physical Measurements
3.3. X-ray Crystallography Procedures
3.4. Synthesis of Tris(2-(3-pentylamino)ethyl)amine (H3LPen)
3.5. Synthesis of Tris(2-dicyclohexylmethylaminoethyl)amine (H3LCy2)
3.6. Synthesis of [{V(LiPr)}2(μ-14N2)] (1)
3.7. [{V(LiPr)}2(μ-15N2)] (1′)
3.8. Synthesis of [{V(LPen)}2(μ-14N2)] (2)
3.9. Synthesis of [{V(LPen)}2(μ-15N2)] (2′)
3.10. Synthesis of [V(LCy2)] (3)
3.11. Synthesis of [VK(LCy2)(μ-14N2)(18-crown-6)] (4)
3.12. Synthesis of [VK(LCy2)(μ-15N2)(18-crown-6)] (4′)
3.13. Protonation of 1, 2, 3, and 4 with Reductant and Proton Source
3.14. NH3 Quantification Procedure
3.15. N2H4 Quantification Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hales, B.J.; Case, E.E.; Morningstar, J.E.; Dzeda, M.F.; Mauterer, L.A. Isolation of a New Vanadium-Containing Nitrogenase from Azotobacter vinelandii. Biochemistry 1986, 25, 7251–7255. [Google Scholar] [CrossRef] [PubMed]
- Arber, J.M.; Dobson, B.R.; Eady, R.R.; Stevens, P.; Hasnain, S.S.; Garner, C.D.; Smith, B.E. Vanadium K-edge X-ray absorption spectrum of the VFe protein of the vanadium nitrogenase of Azotobacter chroococcum. Nature 1986, 325, 372–374. [Google Scholar] [CrossRef]
- Eady, R.R. Structure-Function Relationships of Alternative Nitrogenases. Chem. Rev. 1996, 96, 3013–3030. [Google Scholar] [CrossRef]
- Rehder, D. The coordination chemistry of vanadium as related to its biological functions. Coord. Chem. Rev. 1999, 182, 297–322. [Google Scholar] [CrossRef]
- Smith, B.E. Structure, function, and biosynthesis of the metallosulfur clusters in nitrogenases. Adv. Inorg. Chem. 1999, 47, 159–218. [Google Scholar]
- Eady, R.R. Current status of structure function relationships of vanadium nitrogenase. Coord. Chem. Rev. 2003, 237, 23–30. [Google Scholar] [CrossRef]
- Janas, Z.; Sobota, P. Aryloxo and thiolato vanadium complexes as chemical models of the active site of vanadium nitrogenase. Coord. Chem. Rev. 2005, 249, 2144–2155. [Google Scholar] [CrossRef]
- Sippel, D.; Einsle, O. The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat. Chem. Biol. 2017, 13, 956–960. [Google Scholar] [CrossRef]
- Sippel, D.; Rohde, M.; Netzer, J.; Trncik, C.; Gies, J.; Grunau, K.; Djurdjevic, I.; Decamps, L.; Einsle, O. A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 2018, 359, 1484–1489. [Google Scholar] [CrossRef]
- Duman, L.M.; Sita, L.R. Group 5 Transition Metal-Dinitrogen Complexes. In Transition Metal-Dinitrogen Complexes, 1st ed.; Nishibayashi, Y., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; pp. 159–220. [Google Scholar]
- Rehder, D.; Woitha, C.; Priebsch, W.; Gailus, H. Trans-[Na(thf)][V(N2)2(Ph2PCH2CH2PPh2)2]: Structural characterization of a dinitrogenvanadium complex, a functional model for vanadiumnitrogenase. J. Chem. Soc. Chem. Commun. 1992, 28, 364–365. [Google Scholar] [CrossRef]
- Woitha, C.; Rehder, D. Vanadium(-I) Dinitrogen Complexes with N2 Coordinated End-on: Functional Models for the “Alternative Nitrogenase”. Angew. Chem. Int. Ed. Engl. 1990, 29, 1438–1440. [Google Scholar] [CrossRef]
- Gailus, H.; Woitha, C.; Rehder, D. Dinitrogenvanadates(-I): Synthesis, reactions and conditions for their stability. J. Chem. Soc. Dalton Trans. 1994, 23, 3471–3477. [Google Scholar] [CrossRef]
- Smythe, N.C.; Schrock, R.R.; Müller, P.; Weare, W.W. Synthesis of [(HIPTNCH2CH2)3N]V Compounds (HIPT) 3,5-(2,4,6-i-Pr3C6H2)2C6H3) and an Evaluation of Vanadium for the Reduction of Dinitrogen to Ammonia. Inorg. Chem. 2006, 45, 9197–9205. [Google Scholar] [CrossRef] [PubMed]
- Nishibayashi, Y.; Tanabe, Y. Recent advances in nitrogen fixation upon vanadium complexes. Coord. Chem. Rev. 2019, 381, 135–150. [Google Scholar]
- Ferguson, R.; Solari, E.; Floriani, C.; Chiesi-Villa, A.; Rizzoli, C. Fixation and Reduction of Dinitrogen by Vanadium(II) and Vanadium(III): Synthesis and Structure of Dinitrogenmesitylvanadium Complexes. Angew. Chem. Int. Ed. Engl. 1993, 32, 396–397. [Google Scholar] [CrossRef]
- Ferguson, R.; Solari, E.; Floriani, C.; Osella, D.; Ravera, M.; Re, N.; Chiesi-Villa, A.; Rizzoli, C. Stepwise reduction of dinitrogen occurring on a divanadium model compound: A synthetic, structural, magnetic, electrochemical, and theoretical investigation on the [V=N=N=N](n+) [n = 4-6] based complexes. J. Am. Chem. Soc. 1997, 119, 10104–10115. [Google Scholar] [CrossRef]
- Liu, G.; Liang, X.; Meetsma, A.; Hessen, B. Synthesis and structure of an aminoethyl-functionalized cyclopentadienyl vanadium(i) dinitrogen complex. Dalton Trans. 2010, 39, 7891–7893. [Google Scholar] [CrossRef]
- Vidyaratne, I.; Crewdson, P.; Lefebvre, E.; Gambarotta, S. Dinitrogen Coordination and Cleavage Promoted by a Vanadium Complex of a σ,π,σ-Donor Ligand. Inorg. Chem. 2007, 46, 8836–8842. [Google Scholar] [CrossRef]
- Sekiguchi, Y.; Arashiba, K.; Tanaka, H.; Eizawa, A.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic Reduction of Molecular Dinitrogen to Ammonia and Hydrazine Using Vanadium Complexes. Angew. Chem. Int. Ed. 2018, 57, 9064–9068. [Google Scholar] [CrossRef]
- Kokubo, Y.; Yamamoto, C.; Tsuzuki, K.; Nagai, T.; Katayama, A.; Ohta, T.; Ogura, T.; Wasada-Tsutsui, Y.; Kugimiya, S.; Masuda, H. Dinitrogen Fixation by Vanadium Complexes with a Triamidoamine Ligand. Inorg. Chem. 2018, 57, 11884–11894. [Google Scholar] [CrossRef]
- Kokubo, Y.; Wasada-Tsutsui, Y.; Yomura, S.; Yanagisawa, S.; Kubo, M.; Kugimiya, S.; Kajita, Y.; Ozawa, T.; Masuda, H. Syntheses, Characterizations, and Crystal Structures of Dinitrogen-Divanadium Complexes Bearing Triamidoamine Ligands. Eur. J. Inorg. Chem. 2020, 2020, 1456–1464. [Google Scholar] [CrossRef]
- Schrock, R.R. Transition Metal Complexes That Contain a Triamidoamine Ligand. Acc. Chem. Res. 1997, 30, 9–16. [Google Scholar] [CrossRef]
- Cummins, C.C.; Lee, J.; Schrock, R.R.; Davis, W.M. Trigonalmonopyramidal M(III) complexes of the type M(N3N) [M = titanium, vanadium, chromium, manganese, iron; N3N = [(tert-BuMe2Si)NCH2CH2]3N]. Angew. Chem. Int. Ed. Engl. 1992, 31, 1501–1503. [Google Scholar] [CrossRef]
- Cummins, C.C.; Lee, J.; Schrock, R.R. Phosphinidenetantalum(V) complexes [(N3N)Ta = PR] as phospha-Wittig reagents (R = Ph, Cy, tert-Bu; N3N = (Me3SiNCH2CH2)3N). Angew. Chem. Int. Ed. Engl. 1993, 32, 756–759. [Google Scholar] [CrossRef]
- Cummins, C.C.; Schrock, R.R. Synthesis of an iron(IV) cyanide complex that contains the triamidoamine ligand [(tert-BuMe2SiNCH2CH2)3N]3−. Inorg. Chem. 1994, 33, 395–396. [Google Scholar] [CrossRef]
- Cummins, C.C.; Schrock, R.R.; Davis, W.M. Synthesis of Terminal Vanadium(V) Imido, Oxo, Sulfido, Selenido, and Tellurido Complexes by Imido Group or Chalcogen Atom Transfer to Trigonal Monopyramidal V[N3N] (N3N = [(Me3SiNCH2CH2)3N]3-). Inorg. Chem. 1994, 33, 1448–1457. [Google Scholar] [CrossRef]
- Duan, Z.; Verkade, J.G. Synthesis and Characterization of a Novel Azatitanatrane. Inorg. Chem. 1995, 34, 4311–4316. [Google Scholar] [CrossRef]
- Naiini, A.A.; Menge, W.M.P.B.; Verkade, J.G. Titanatranes and azatitanatranes: Nucleophilic substitution reactions on the axial position. Inorg. Chem. 1991, 30, 5009–5012. [Google Scholar] [CrossRef]
- Nomura, K.; Schrock, R.R.; Davis, W.M. Synthesis of Vanadium(III), -(IV), and -(V) Complexes That Contain the Pentafluorophenyl-Substituted Triamidoamine Ligand [(C6F5NCH2CH2)3N]3-. Inorg. Chem. 1996, 35, 3695–3701. [Google Scholar] [CrossRef]
- Freundlich, J.S.; Schrock, R.R. Synthesis of Triamidoamine Complexes of Niobium. Inorg. Chem. 1996, 35, 7459–7461. [Google Scholar] [CrossRef]
- Rosenberger, C.; Schrock, R.R.; Davis, W.M. Synthesis and Structure of a Trigonal Monopyramidal Vanadium(III) Complex, [(C6F5NCH2)3N]V, and the Vanadium(IV) Product of Its Oxidation, {[(C6F5NCH2CH2)2N(CH2CH2NHC6F5)]V(O)}2. Inorg. Chem. 1997, 36, 123–125. [Google Scholar] [CrossRef]
- Pinkas, J.; Gaul, B.; Verkade, J.G. Group 13 azatranes: Synthetic, conformational, and configurational features. J. Am. Chem. Soc. 1993, 115, 3925–3931. [Google Scholar] [CrossRef]
- Plass, W.; Verkade, J.G. A novel transmetalation reaction: A route to transition metallatranes. J. Am. Chem. Soc. 1992, 114, 2275–2276. [Google Scholar] [CrossRef]
- Cummins, C.C.; Schrock, R.R.; Davis, W.M. Synthesis of Vanadium and Titanium Complexes of the Type RM[(Me3SiNCH2CH2)3N] (R = Cl, Alkyl) and the Structure of ClV[(Me3SiNCH2CH2)3N]. Organometallics 1992, 11, 1452–1454. [Google Scholar] [CrossRef]
- Christou, V.; Arnold, J. Synthesis of monomeric terminal chalcogenides via template-induced disilylchalcogenide elimination: Structure of [ETa-{Me3SiNCH2CH2}3N]] (E = selenium, tellurium). Angew. Chem. Int. Ed. Engl. 1993, 32, 1450–1452. [Google Scholar] [CrossRef]
- Yandulov, D.V.; Schrock, R.R.; Rheingold, A.L.; Ceccarelli, C.; Davis, W.M. Synthesis and Reactions of Molybdenum Triamidoamine Complexes Containing Hexaisopropylterphenyl Substituents. Inorg. Chem. 2003, 42, 796–813. [Google Scholar] [CrossRef]
- Yandulov, D.V.; Schrock, R.R. Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Science 2003, 301, 76–78. [Google Scholar] [CrossRef]
- Yandulov, D.V.; Schrock, R.R. Studies Relevant to Catalytic Reduction of Dinitrogen to Ammonia by Molybdenum Triamidoamine Complexes. Inorg. Chem. 2005, 44, 1103–1117. [Google Scholar] [CrossRef]
- Smythe, N.C.; Schrock, R.R.; Müller, P.; Weare, W.W. Synthesis of [(HIPTNCH2CH2)3N]Cr Compounds (HIPT = 3,5-(2,4,6-i-Pr3C6H2)2C6H3 and an Evaluation of Chromium for the Reduction of Dinitrogen to Ammonia. Inorg. Chem. 2006, 45, 7111–7118. [Google Scholar] [CrossRef]
- Doyle, L.R.; Wooles, A.J.; Liddle, S.T. Liddle Bimetallic Cooperative Cleavage of Dinitrogen to Nitride and Tandem Frustrated Lewis Pair Hydrogenation to Ammonia. Angew. Chem. Int. Ed. 2019, 58, 6674–6677. [Google Scholar] [CrossRef]
- Ghana, P.; van Krüchten, F.D.; Spaniol, T.P.; van Leusen, J.; Kögerler, P.; Okuda, J. Conversion of dinitrogen to tris(trimethylsilyl)amine catalyzed by titanium triamido-amine complexes. Chem. Commun. 2019, 55, 3231–3234. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.R.; Wooles, A.J.; Jenkins, L.C.; Tuna, F.; McInnes, E.J.L.; Liddle, S.T. Catalytic Dinitrogen Reduction to Ammonia at a Triamidoamine–Titanium Complex. Angew. Chem. Int. Ed. 2018, 57, 6314–6318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, K.-Y.; Schrock, R.R.; Kempe, R. Synthesis of Molybdenum Complexes That Contain Silylated Triamidoamine Ligands. A p-Dinitrogen Complex, Methyl and Acetylide Complexes, and Coupling of Acetylides. J. Am. Chem. Soc. 1994, 116, 8804–8805. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua [1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar] [CrossRef]
- Singh, D.; Buratto, W.R.; Torres, J.F.; Murray, L.J. Activation of Dinitrogen by Polynuclear Metal Complexes. Chem. Rev. 2020, 120, 5517–5581. [Google Scholar] [CrossRef]
- Evans, D.F. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Sur, S.K. Measurement of magnetic susceptibility and magnetic moment of paramagnetic molecules in solution by high-field Fourier transform NMR spectroscopy. J. Magn. Reson. 1989, 82, 169–173. [Google Scholar] [CrossRef]
- Kokubo, Y.; Tsuzuki, K.; Sugiura, H.; Yomura, S.; Wasada-Tsutsui, Y.; Ozawa, T.; Yanagisawa, S.; Kubi, M.; Kugmiya, S.; Masuda, H.; et al. Syntheses, Characterizations, Crystal Structures, and Protonation Reactions of Dinitrogen Chromium Complexes Supported with Triamidoamine Ligands. Inorg. Chem. 2022; submitted for publication. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Hill, P.J.; Doyle, L.R.; Crawford, A.D.; Myers, W.K.; Ashley, A.E. Selective Catalytic Reduction of N2 to N2H4 by a Simple Fe Complex. J. Am. Chem. Soc. 2016, 138, 13521–13524. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 [a] | |||||
| V1–N2 | 1.7647(18) | V1–N3 | 1.896(2) | V1–N4 | 1.9123(14) |
| V1–N4i | 1.9123(14) | V1–N5 | 2.173(2) | N2–N2i | 1.219(4) |
| V•••Vi | 4.7482(8) | - | - | - | - |
| N2–V1–N5 | 178.98(9) | N2i–N2–V1 | 178.1(3) | N2–V1–N3 | 98.08(9) |
| N2–V1–N4 | 99.78(5) | N2–V1–N4i | 99.78(5) | N3–V1–N4 | 116.68(5) |
| N3–V1–N4i | 116.68(5) | N4–V1–N4i | 119.08(10) | - | - |
| 2 [b] | |||||
| V1–N2 | 1.7935(14) | V1–N3 | 1.9276(16) | V1–N4 | 1.9284(16) |
| V1–N5 | 1.9234(16) | V1–N6 | 2.1854(16) | N2–N2i | 1.226(3) |
| V•••Vi | 4.8128 (7) | - | - | - | - |
| N2–V1–N3 | 101.91(7) | N2–V1–N4 | 100.01(7) | N2–V1–N5 | 99.43(6) |
| N2–V1–N6 | 177.75(7) | N3–V1–N4 | 116.88(7) | N3–V1–N5 | 116.43(7) |
| N3–V1–N6 | 80.34(6) | N4–V1–N5 | 117.04(7) | N4–V1–N6 | 78.74(6) |
| N5–V1–N6 | 79.57(6) | N2i–N2–V1 | 177.80(19) | - | - |
| 3 | |||||
| V1–N2 | 1.9281 (13) | V1–N3 | 1.9433 (12) | V1–N4 | 1.9593 (12) |
| V1–N5 | 2.0687 (13) | - | - | - | - |
| N2–V1–N3 | 119.11 (6) | N2–V1–N4 | 118.23 (5) | N2–V1–N5 | 84.13 (5) |
| N3–V1–N4 | 119.29 (5) | N3–V1–N5 | 83.35 (5) | N4–V1–N5 | 84.17 (5) |
| 4 | |||||
| V1–N3 | 1.853 (3) | V1–N5 | 1.965 (2) | V1–N6 | 1.960 (2) |
| V1–N7 | 1.955 (2) | V1–N8 | 2.172 (2) | N3–N4 | 1.152 (3) |
| N4–K2 | 2.648 (3) | - | - | - | - |
| N8–V1–N3 | 179.07 (10) | V1–N3–N4 | 179.08 (3) | N3–N4–K2 | 173.3 (2) |
| N5–V1–N6 | 118.34 (10) | N6–V1–N7 | 118.80 (10) | N7–V1–N5 | 116.46 (10) |
| N5–V1–N8 | 81.59 (9) | N6–V1–N8 | 81.40 (10) | N7–V1–N8 | 81.59 (9) |
| Entry | Complex | Reductant | Proton Source | Yield [b] /% | |
|---|---|---|---|---|---|
| NH3 [c] | N2H4 [c] | ||||
| 1 | 1 | Na[C10H8] | HOTf | 8 | 0.4 |
| 2 | K[C10H8] | 47 | 11 | ||
| 3 | 2 | Na[C10H8] | HOTf | 5 | 11 |
| 4 | K[C10H8] | 38 | 16 | ||
| 5 | 3 | Na[C10H8] | HOTf | 7 | n.d. |
| 6 | K[C10H8] | 77 | 7 | ||
| 7 | 4 | K[C10H8] | HOTf | 80 | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokubo, Y.; Igarashi, I.; Nakao, K.; Hachiya, W.; Kugimiya, S.; Ozawa, T.; Masuda, H.; Kajita, Y. The Steric Effect in Preparations of Vanadium(II)/(III) Dinitrogen Complexes of Triamidoamine Ligands Bearing Bulky Substituents. Molecules 2022, 27, 5864. https://doi.org/10.3390/molecules27185864
Kokubo Y, Igarashi I, Nakao K, Hachiya W, Kugimiya S, Ozawa T, Masuda H, Kajita Y. The Steric Effect in Preparations of Vanadium(II)/(III) Dinitrogen Complexes of Triamidoamine Ligands Bearing Bulky Substituents. Molecules. 2022; 27(18):5864. https://doi.org/10.3390/molecules27185864
Chicago/Turabian StyleKokubo, Yoshiaki, Itsuki Igarashi, Kenichi Nakao, Wataru Hachiya, Shinichi Kugimiya, Tomohiro Ozawa, Hideki Masuda, and Yuji Kajita. 2022. "The Steric Effect in Preparations of Vanadium(II)/(III) Dinitrogen Complexes of Triamidoamine Ligands Bearing Bulky Substituents" Molecules 27, no. 18: 5864. https://doi.org/10.3390/molecules27185864
APA StyleKokubo, Y., Igarashi, I., Nakao, K., Hachiya, W., Kugimiya, S., Ozawa, T., Masuda, H., & Kajita, Y. (2022). The Steric Effect in Preparations of Vanadium(II)/(III) Dinitrogen Complexes of Triamidoamine Ligands Bearing Bulky Substituents. Molecules, 27(18), 5864. https://doi.org/10.3390/molecules27185864