

The Conversion of Superoxide to Hydroperoxide on Cobalt(III) Depends on the Structural and Electronic Properties of Azole-Based Chelating Ligands

 , and
, and
Abstract
:1. Introduction
2. Results
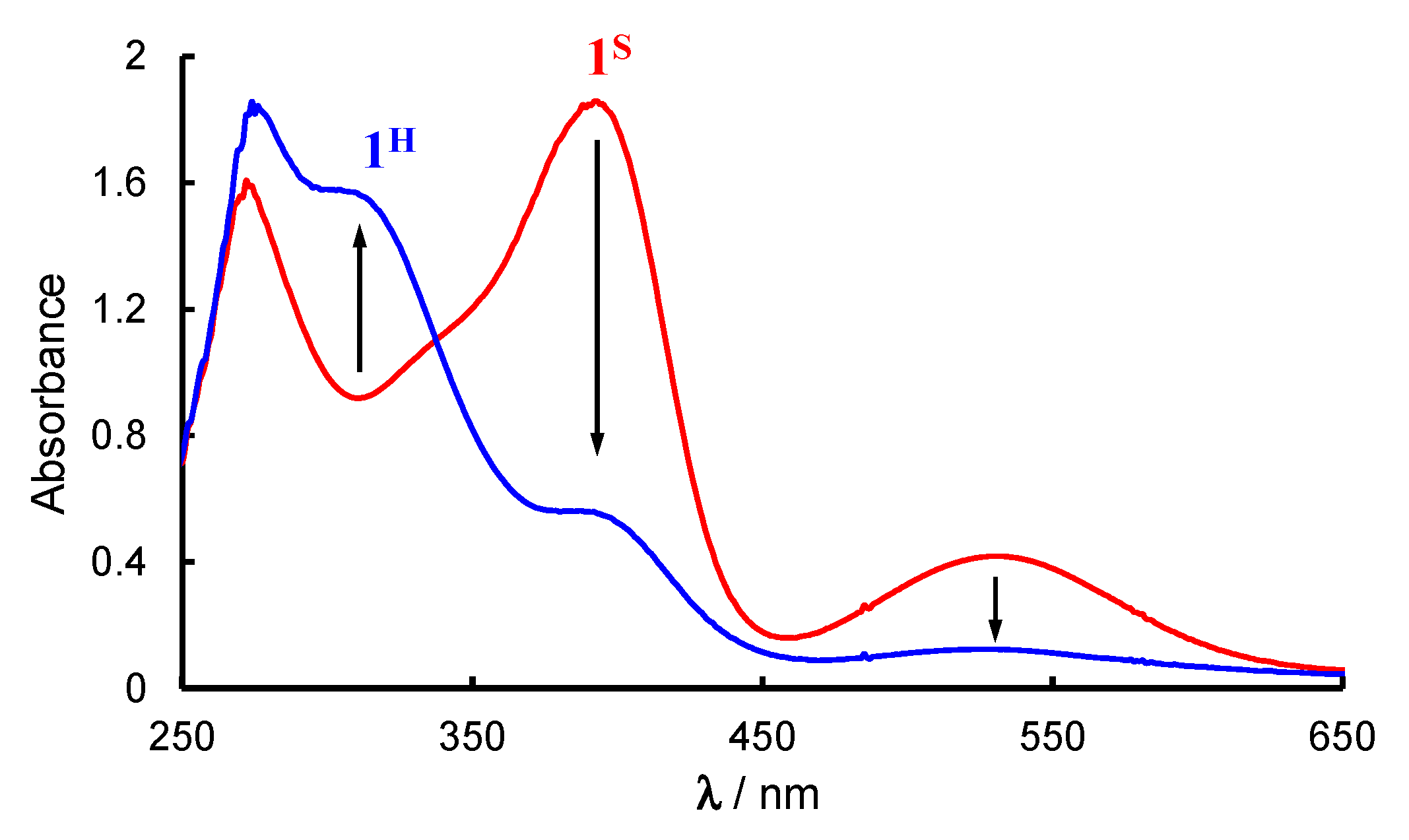
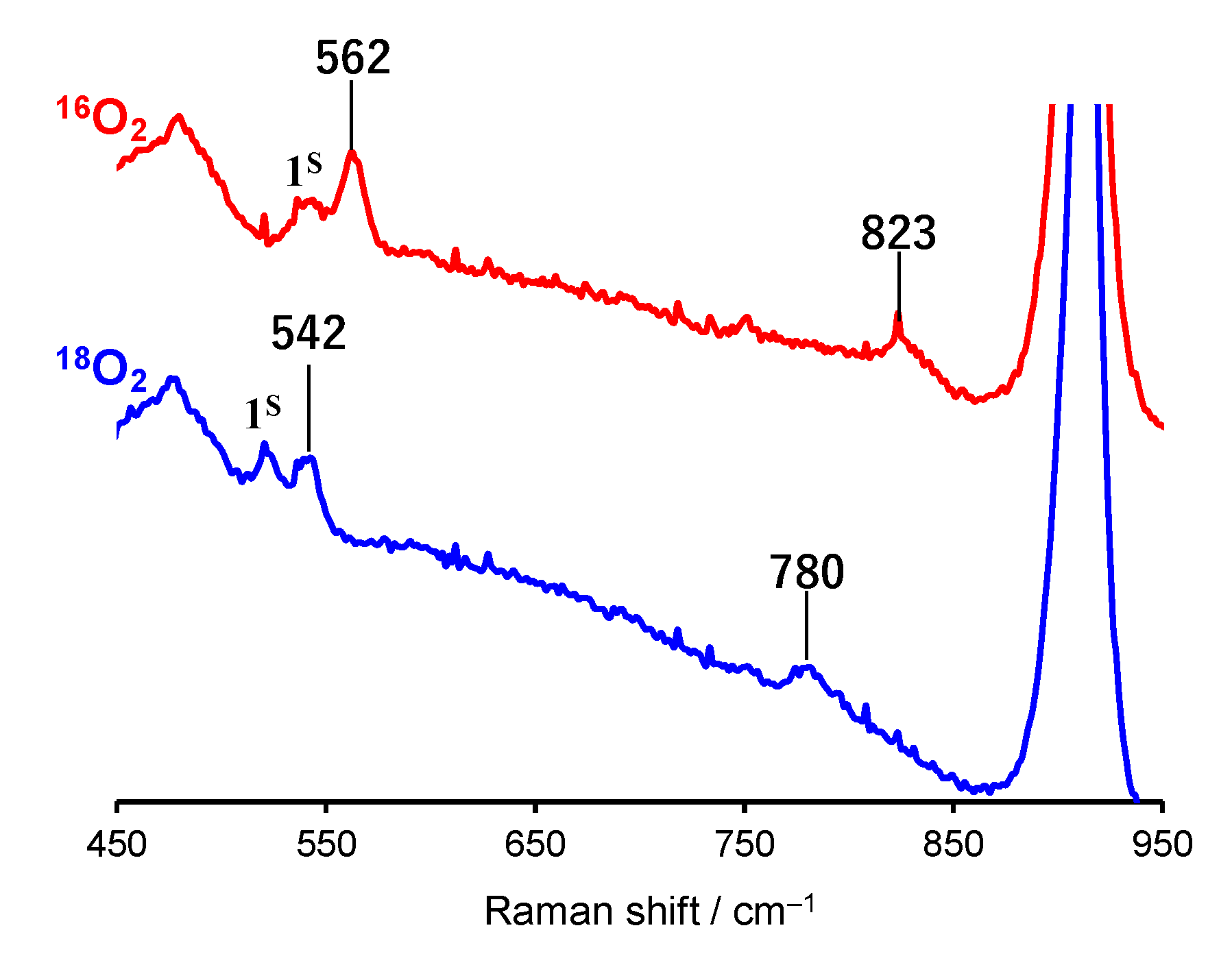
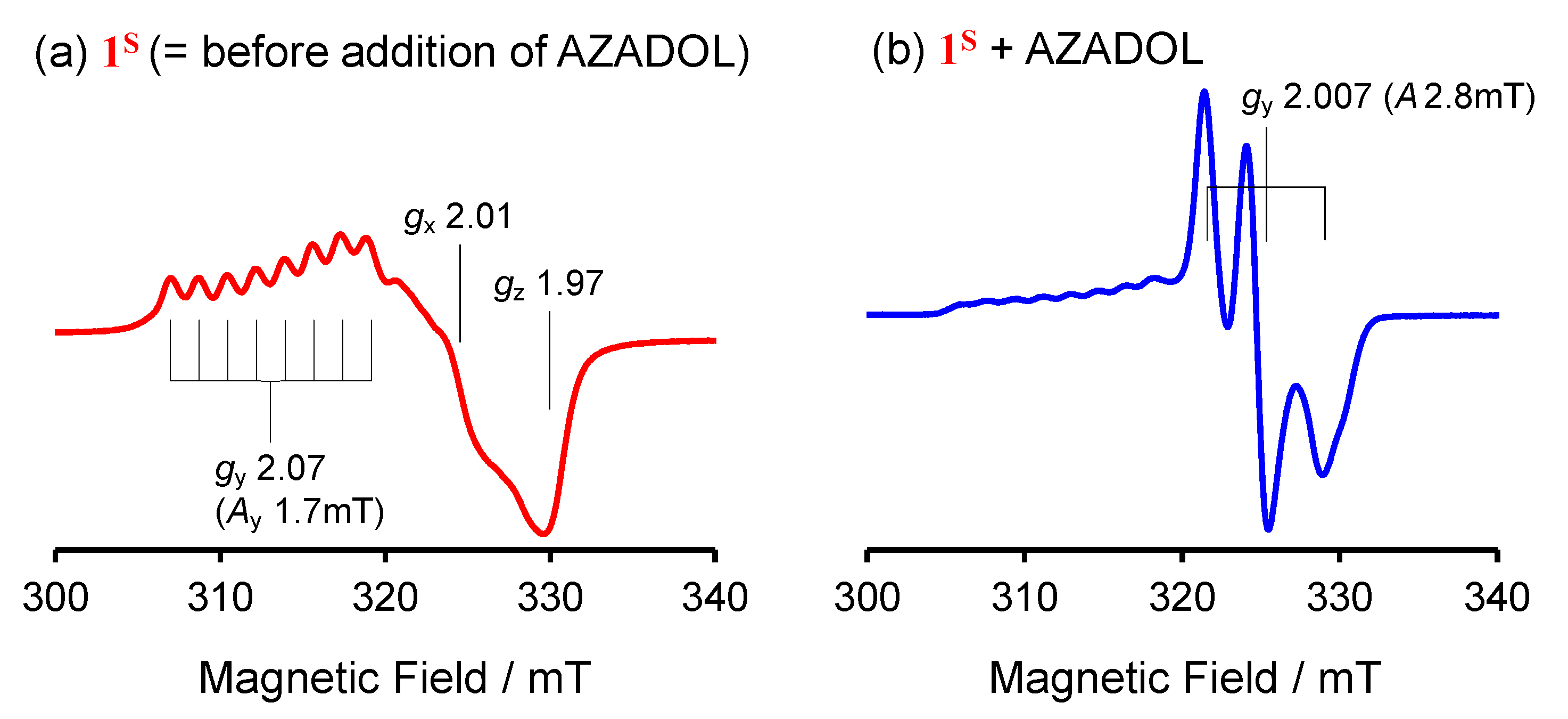
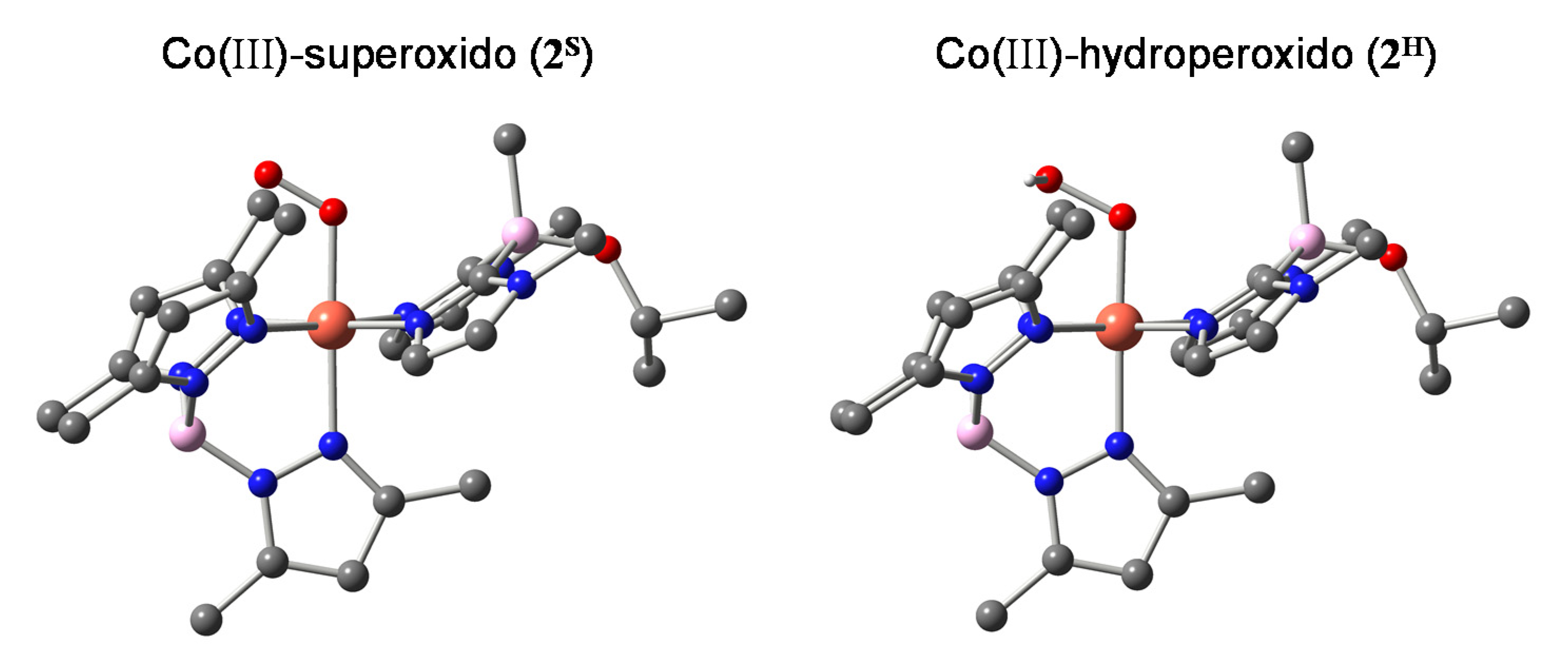
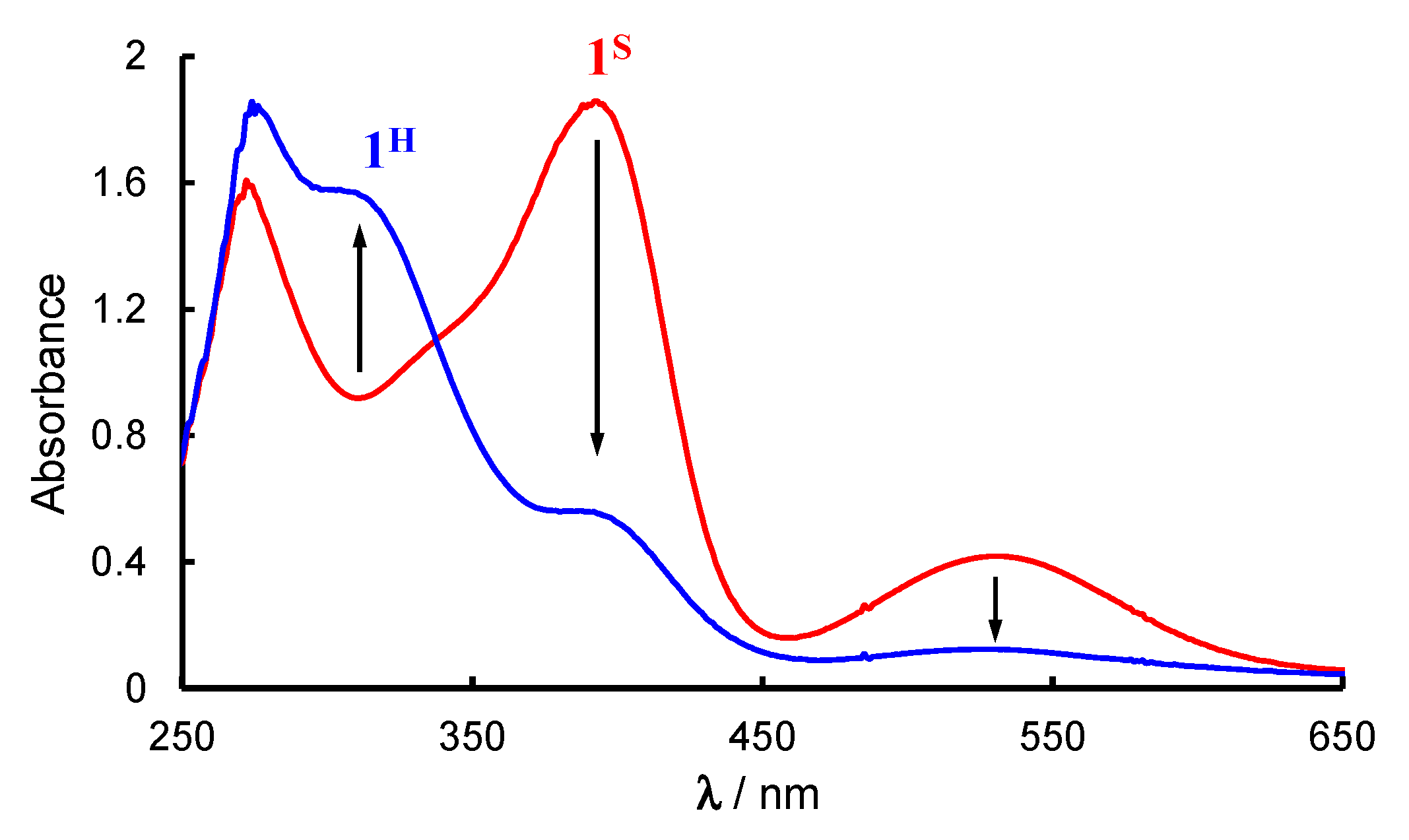
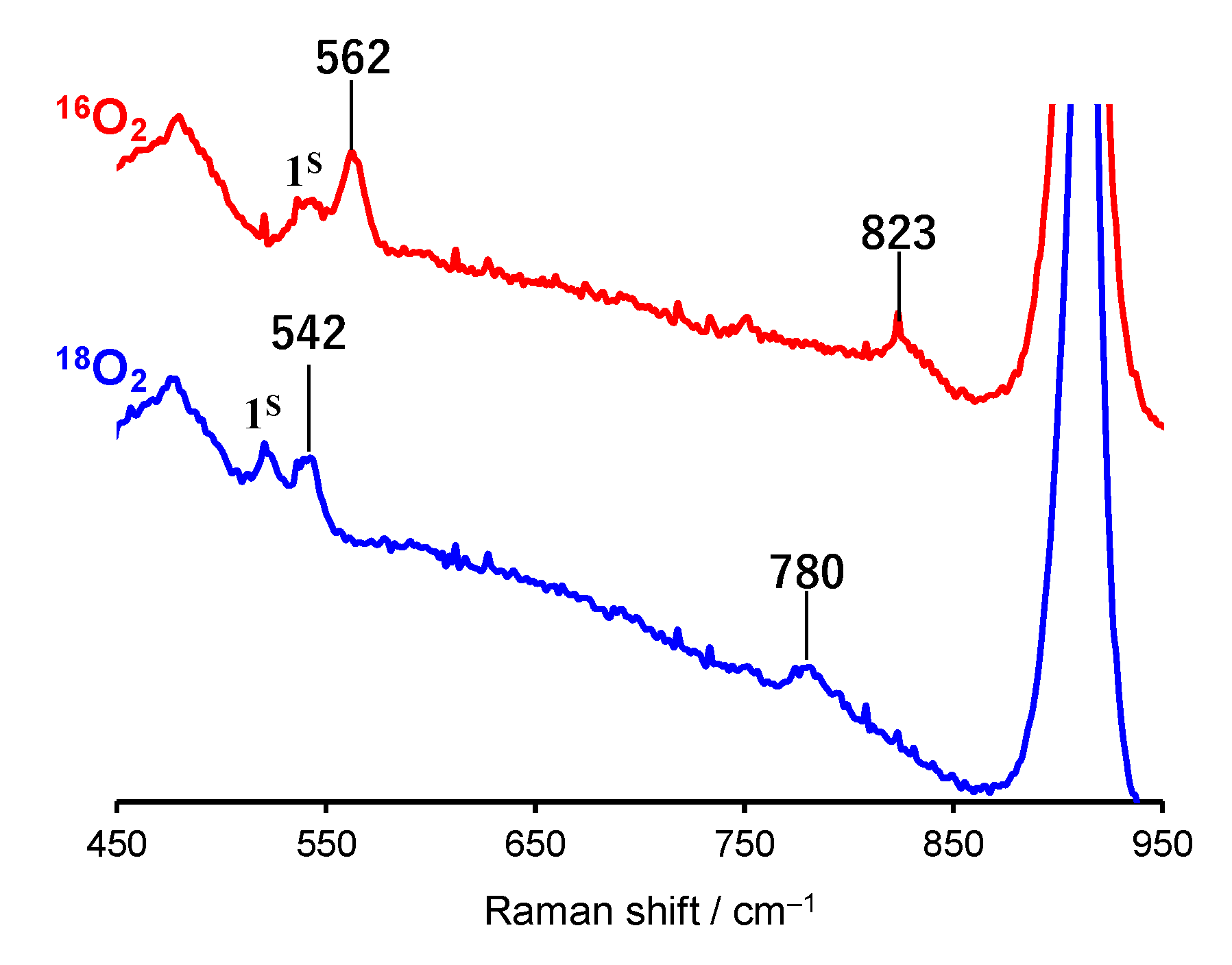
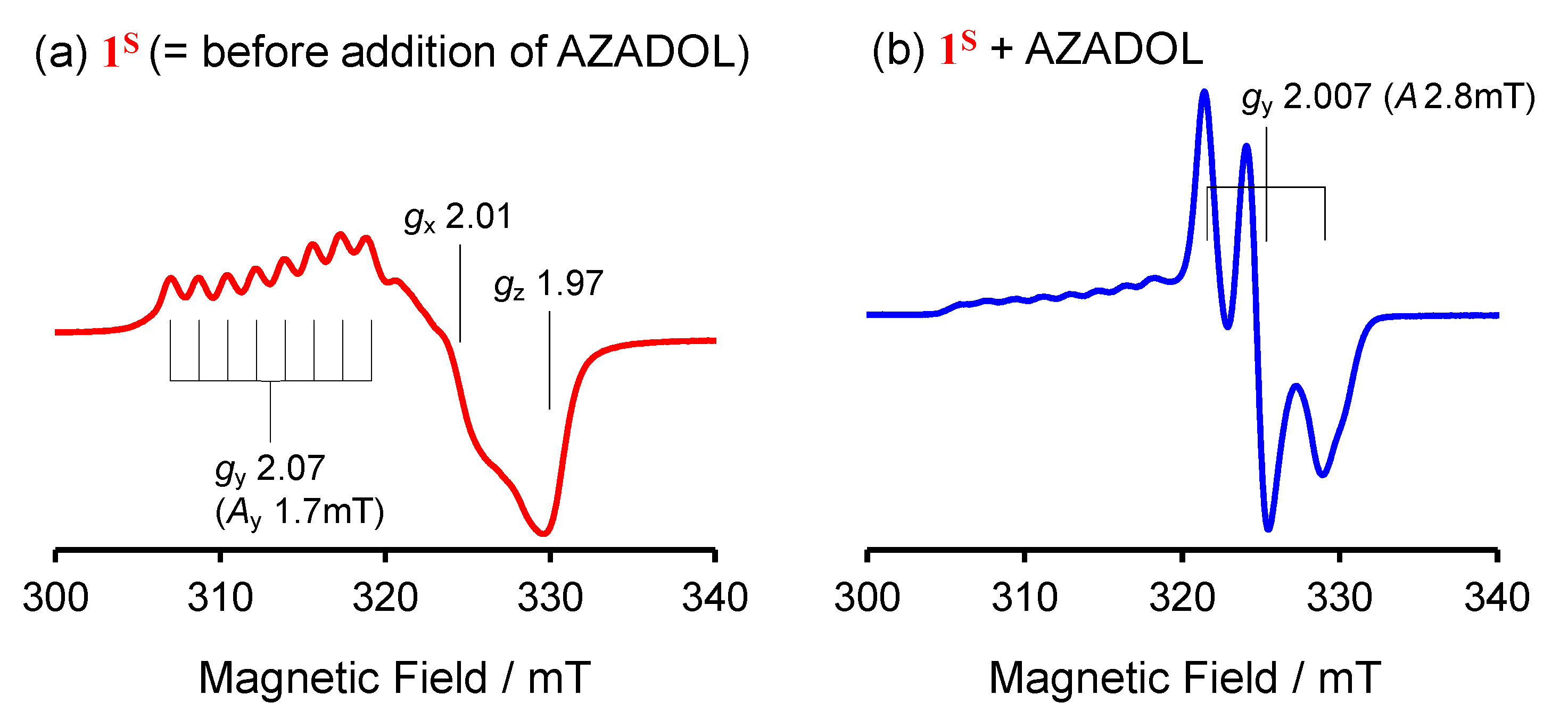
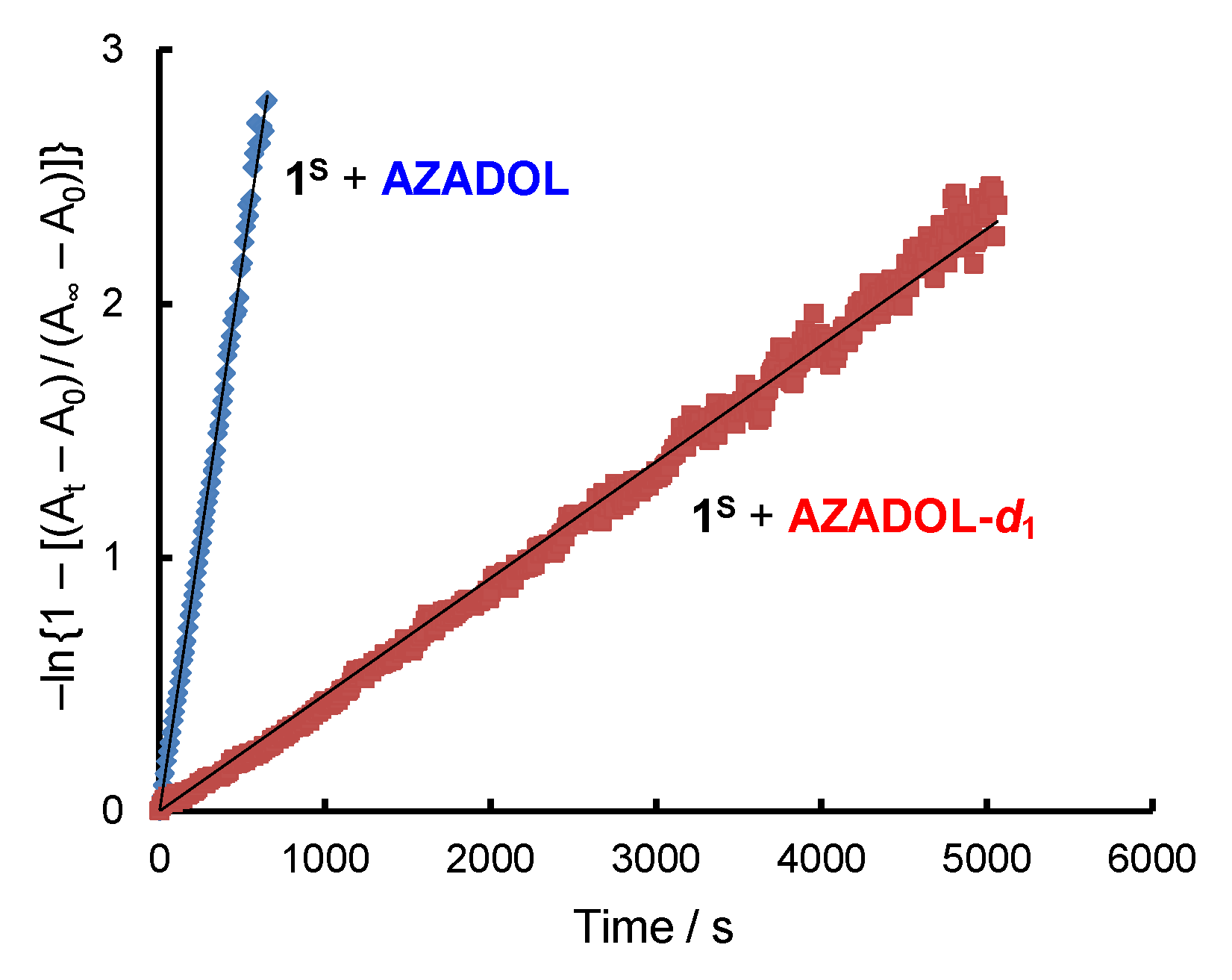
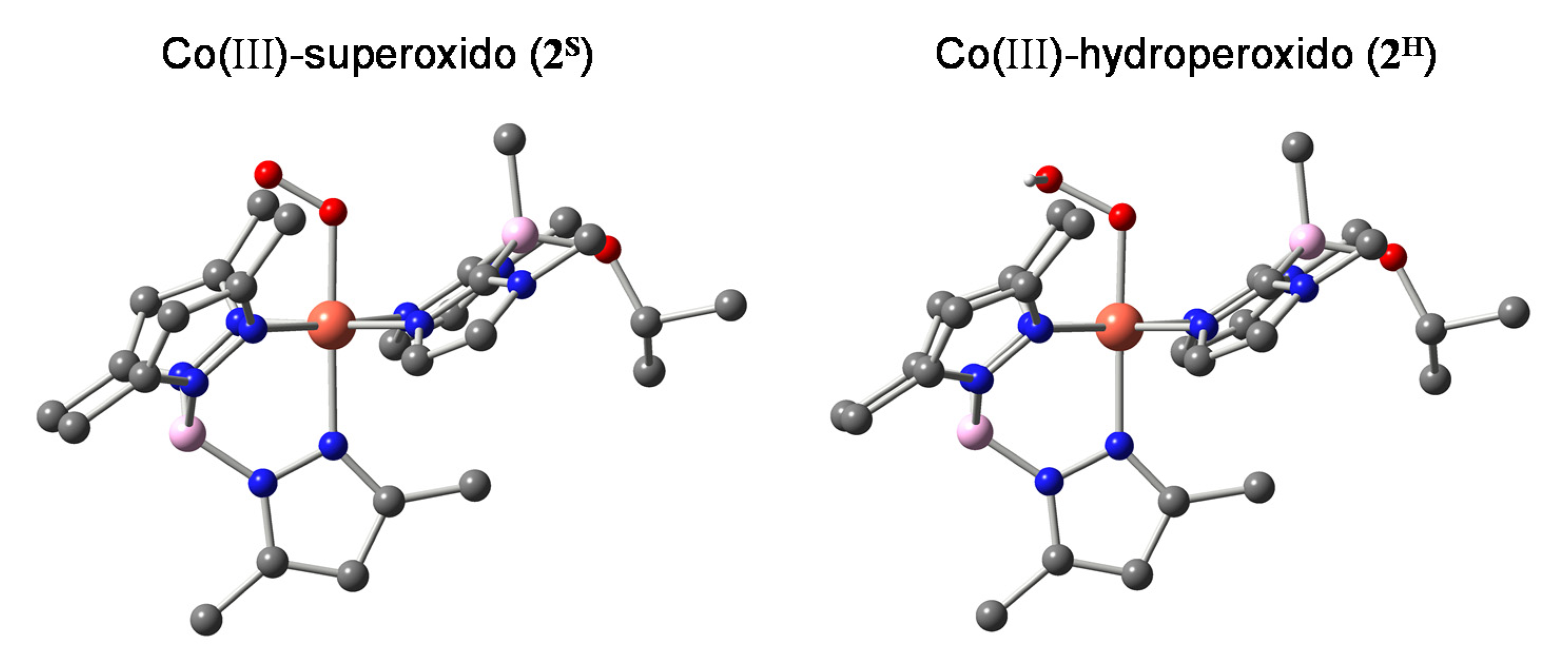
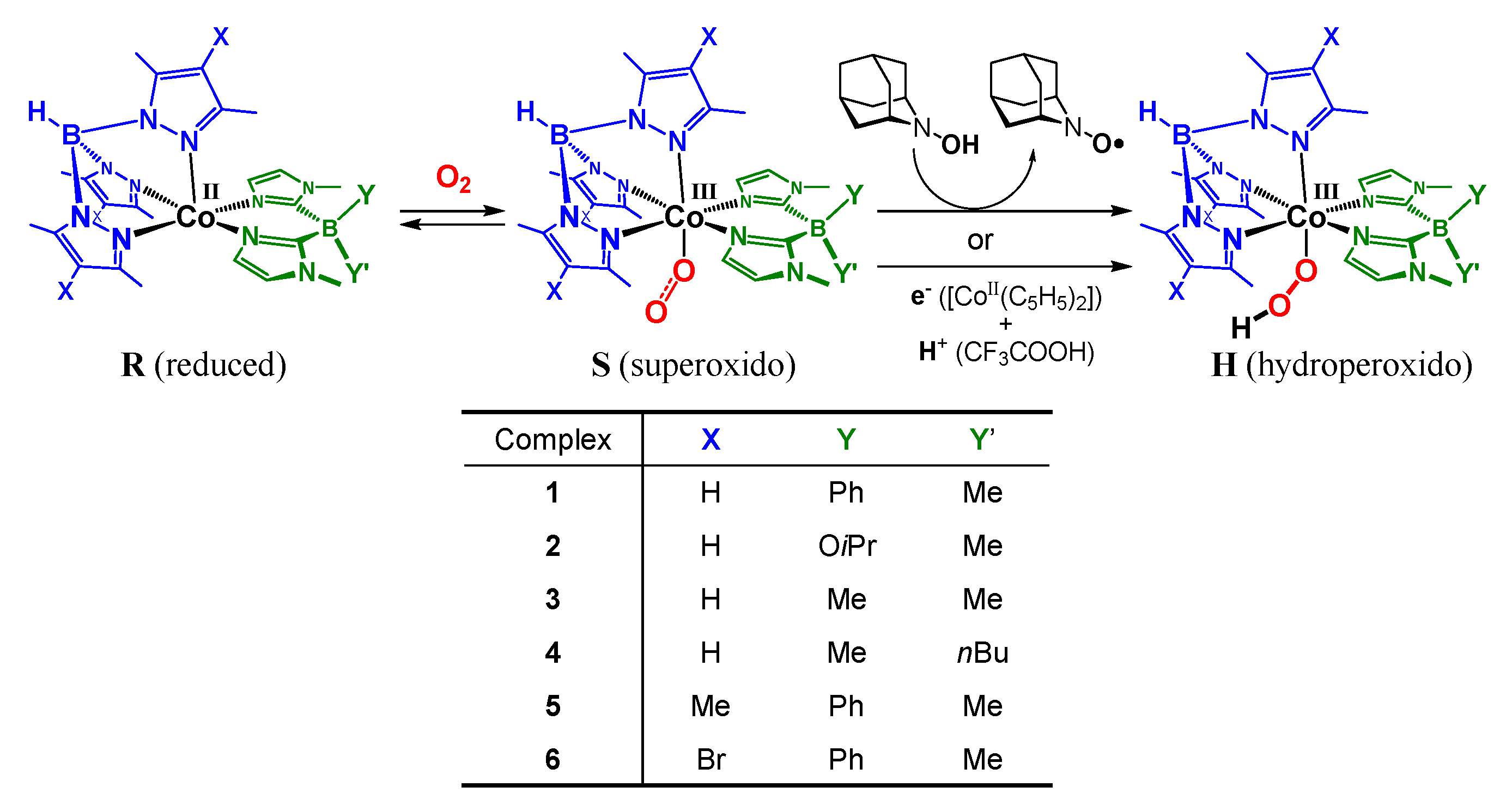
2.1. Formation and Characterization of Cobalt(III)-Hydroperoxo Species
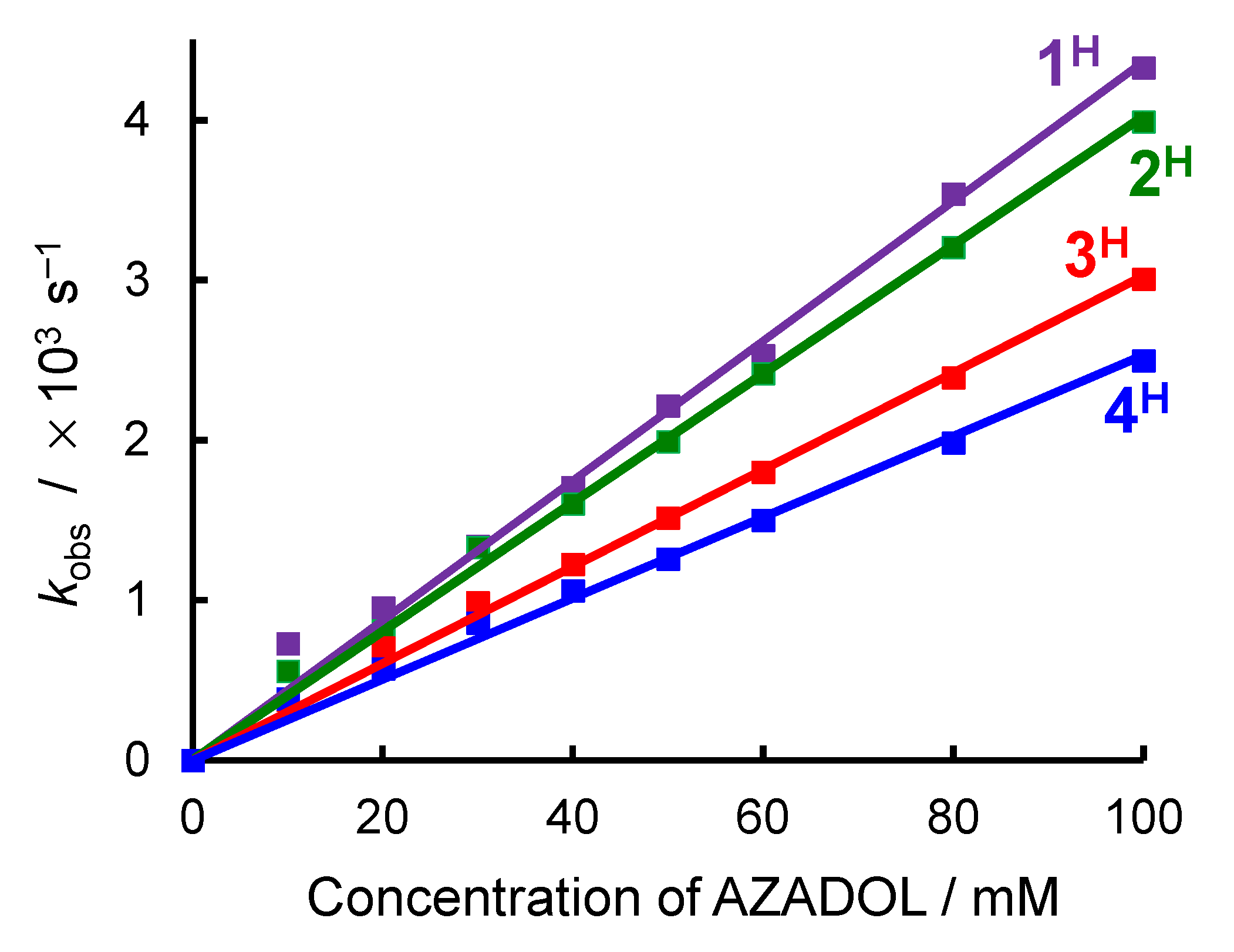
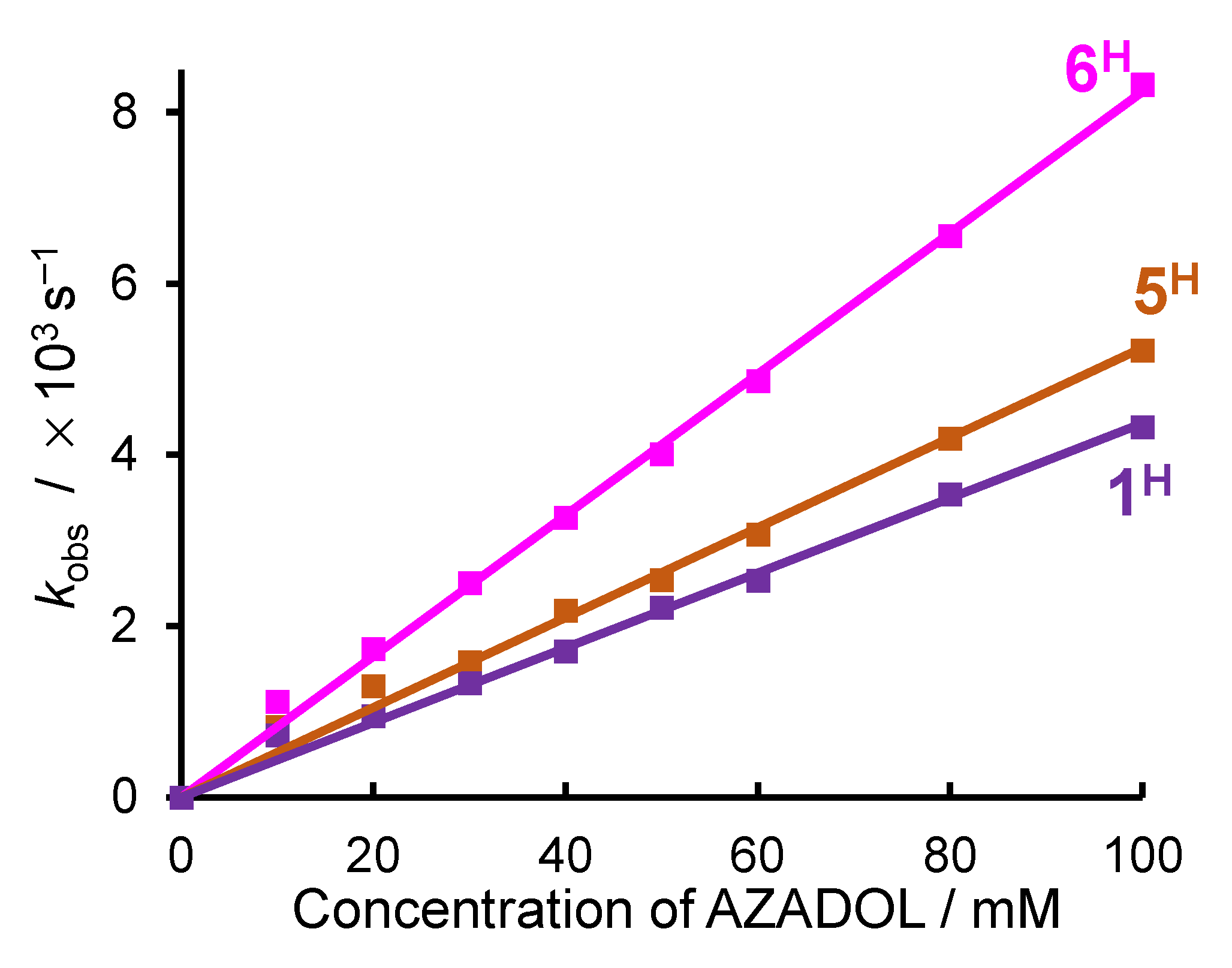
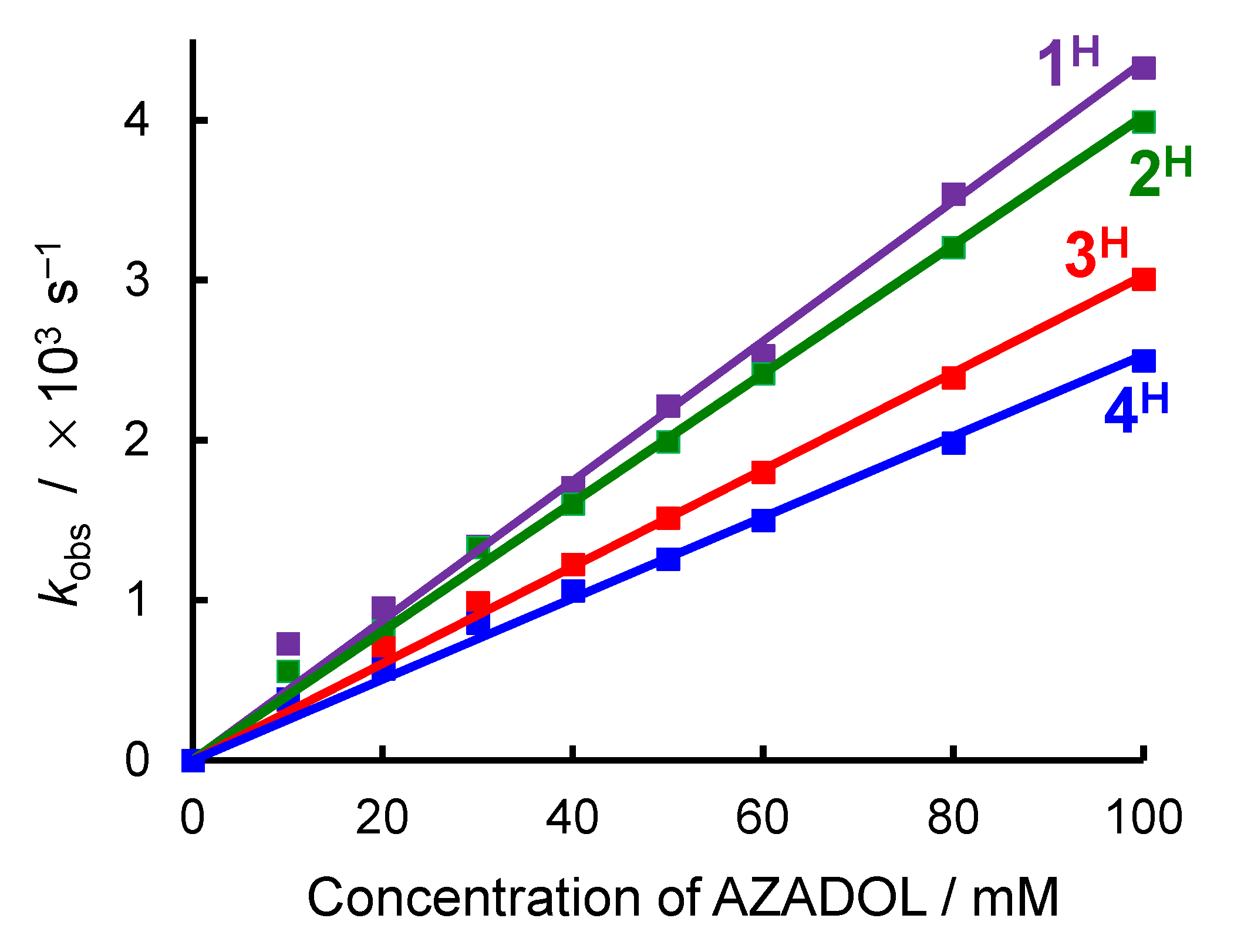
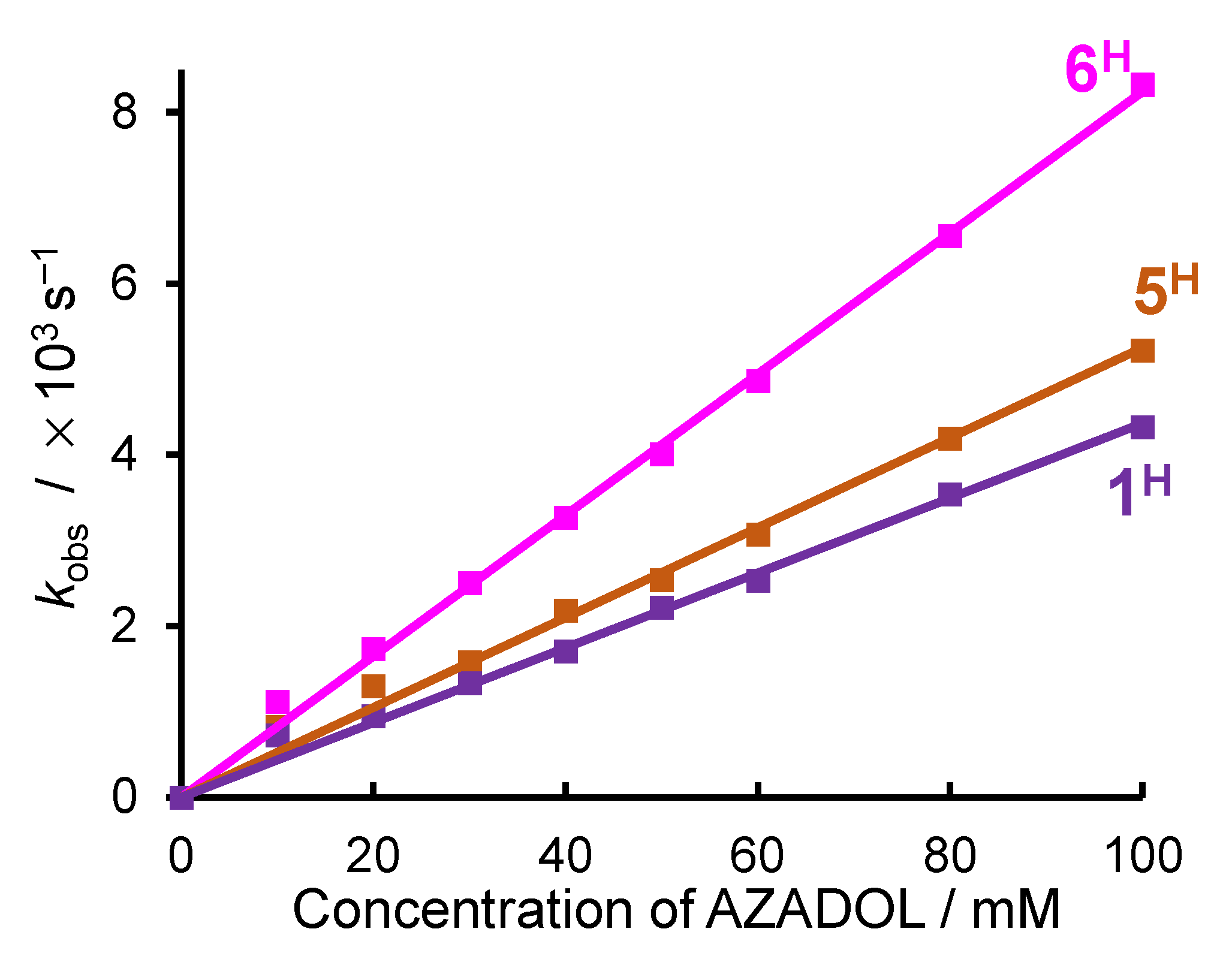
2.2. Ligand Effect on the Formation of the Cobalt(III)-Hydroperoxido Species
3. Discussion
4. Materials and Methods
4.1. General
4.2. Reaction of the Cobalt(III)-Superoxido Complex [CoIII(O2)(TpMe2,H)(LPh)] (1S) with AZADOL
4.3. Reaction of 1S with [CoII(Cp)2] and Trifluoroacetic Acid
4.4. Kinetic Analyses for the Formation of the Cobalt(III)-Superoxido Complexes 1H–6H
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef]
- Bollinger, J.M., Jr.; Krebs, C. Enzymatic C−H activation by metal−superoxo intermediates. Curr. Opin. Chem. Biol. 2007, 11, 151–158. [Google Scholar] [CrossRef]
- Klinman, J.P. The Copper-Enzyme Family of Dopamine-Monooxygenase and Peptidylglycine—Hydroxylating Monooxygenase: Resolving the Chemical Pathway for Substrate Hydroxylation. J. Biol. Chem. 2006, 281, 3013–3016. [Google Scholar] [CrossRef]
- Whittaker, J.W. Free Radical Catalysis by Galactose Oxidase. Chem. Rev. 2003, 103, 2347–2363. [Google Scholar] [CrossRef]
- Hedegard, E.D.; Ryde, U. Molecular mechanism of lytic polysaccharide monooxygenases. Chem. Sci. 2018, 9, 3866–3880. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Lee, Y.-M.; Nam, W. Structure and reactivity of the first-row d-block metal-superoxo complexes. Dalton. Trans. 2019, 48, 9469–9489. [Google Scholar] [CrossRef]
- Noh, H.; Cho, J. Synthesis, characterization and reactivity of non-heme 1st row transition metal-superoxo intermediates. Coord. Chem. Rev. 2019, 382, 126–144. [Google Scholar] [CrossRef]
- Nishiura, T.; Chiba, Y.; Nakazawa, J.; Hikichi, S. Tuning the O2 Binding Affinity of Cobalt(II) Centers by Changing the Structural and Electronic Properties of the Distal Substituents on Azole-Based Chelating Ligands. Inorg. Chem. 2018, 57, 14218–14229. [Google Scholar] [CrossRef]
- Oddon, F.; Chiba, Y.; Nakazawa, J.; Ohta, T.; Ogura, T.; Hikichi, S. Characterization of Mononuclear Non-heme Iron(III)-Superoxo Complex with a Five-Azole Ligand Set. Angew. Chem. Int. 2015, 54, 7336–7339. [Google Scholar] [CrossRef]
- Rajani, C.; Kincaid, J.R.; Petering, D.H. Resonance Raman Studies of HOO-Co(III)Bleomycin and Co(III)Bleomycin: Identification of Two Important Vibrational Modes, (Co-OOH) and (O-OH). J. Am. Chem. Soc. 2004, 126, 3829–3836. [Google Scholar] [CrossRef]
- Wang, C.-C.; Chang, H.-C.; Lai, Y.-C.; Fang, H.; Li, C.-C.; Hsu, H.-K.; Li, Z.-Y.; Lin, T.-S.; Kuo, T.-S.; Neese, F.; et al. A Structurally Characterized Nonheme Cobalt−Hydroperoxo Complex Derived from Its Superoxo Intermediate via Hydrogen Atom Abstraction. J. Am. Chem. Soc. 2016, 138, 14186–14189. [Google Scholar] [CrossRef]
- Shin, B.; Sutherlin, K.D.; Ohta, T.; Ogura, T.; Solomon, E.I.; Cho, J. Reactivity of a Cobalt(III)−Hydroperoxo Complex in Electrophilic Reactions. Inorg. Chem. 2016, 55, 12391–12399. [Google Scholar] [CrossRef]
- Anandababu, K.; Muthuramalingam, S.; Velusamy, M.; Mayilmurugan, R. Single-step benzene hydroxylation by cobalt(II) catalysts via a cobalt(III)-hydroperoxo intermediate. Catal. Sci. Technol. 2020, 10, 2540–2548. [Google Scholar] [CrossRef]
- Gordon, J.B.; Vilbert, A.C.; Siegler, M.A.; Lancaster, K.M.; Moënne-Loccoz, P.; Goldberg, D.P. A Nonheme Thiolate-Ligated Cobalt Superoxo Complex: Synthesis and Spectroscopic Characterization, Computational Studies, and Hydrogen Atom Abstraction Reactivity. J. Am. Chem. Soc. 2019, 141, 3641–3653. [Google Scholar] [CrossRef]
- Tano, T.; Okubo, Y.; Kunishita, A.; Kubo, M.; Sugimoto, H.; Fujieda, N.; Ogura, T.; Itoh, S. Redox Properties of a Mononuclear Copper(II)-Superoxide Complex. Inorg. Chem. 2013, 52, 10431–10437. [Google Scholar] [CrossRef]
- Kindermann, N.; Günes, C.-J.; Dechert, S.; Meyer, F. Hydrogen Atom Abstraction Thermodynamics of a μ-1,2-Superoxo Dicopper(II) Complex. J. Am. Chem. Soc. 2017, 139, 9831–9834. [Google Scholar] [CrossRef]
- Bailey, W.D.; Dhar, D.; Cramblitt, A.C.; Tolman, W.B. Mechanistic Dichotomy in Proton-Coupled Electron-Transfer Reactions of Phenols with a Copper Superoxide Complex. J. Am. Chem. Soc. 2019, 141, 5470–5480. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Cramer, H.H.; van Gastel, M.; Tsai, Y.-H.; Chu, C.-Y.; Kuo, T.-S.; Lee, I.-R.; Ye, S.; Bill, E.; Lee, W.-Z. Mononuclear Manganese(III) Superoxo Complexes: Synthesis, Characterization, and Reactivity. Inorg. Chem. 2019, 58, 9756–9765. [Google Scholar] [CrossRef]
- Goo, Y.R.; Maity, A.C.; Cho, K.-B.; Lee, Y.-M.; Seo, M.S.; Park, Y.J.; Cho, J.; Nam, W. Tuning the Reactivity of Chromium(III)-Superoxo Species by Coordinating Axial Ligands. Inorg. Chem. 2015, 54, 10513–10520. [Google Scholar] [CrossRef]
- Kim, H.; Rogler, P.J.; Sharma, S.K.; Schaefer, A.W.; Solomon, E.I.; Karlin, K.D. Heme-FeIII Superoxide, Peroxide and Hydroperoxide Thermodynamic Relationships: FeIII-O2•− Complex H-Atom Abstraction Reactivity. J. Am. Chem. Soc. 2020, 142, 3104–3116. [Google Scholar] [CrossRef]
- Chiang, C.-W.; Kleespies, S.T.; Stout, H.D.; Meier, K.K.; Li, P.-Y.; Bominaar, E.L.; Que, L., Jr.; Münck, E.; Lee, W.-Z. Characterization of a Paramagnetic Mononuclear Nonheme Iron-Superoxo Complex. J. Am. Chem. Soc. 2014, 136, 10846–10849. [Google Scholar] [CrossRef] [PubMed]
- Blakely, M.N.; Dedushko, M.A.; Poon, P.C.Y.; Villar-Acevedo, G.; Kovacs, J.A. Formation of a Reactive, Alkyl Thiolate-Ligated FeIII-Superoxo Intermediate Derived from Dioxygen. J. Am. Chem. Soc. 2019, 141, 1867–1870. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Weigend, F.; Ahlrichs., A. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.1; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V.J. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. Chem. Phys. 2002, 117, 43. [Google Scholar] [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R.J. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. Chem. Phys. 1998, 108, 4439. [Google Scholar] [CrossRef]
- Statmann, R.E.; Scuseria, G.E.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. Chem. Phys. 1998, 109, 8218. [Google Scholar]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | (Co-O)/cm−1 | ν(O-O)/cm−1 | Reference | ||
|---|---|---|---|---|---|
| 16O2 | 18O2 | 16O2 | 18O2 | ||
| 1S | 544 | 523 | 1150 | 1090 | [8] |
| 1H | 562 | 542 | 823 | 780 | This work |
| (bleomycin)CoIII(OOH) | 545 | 518 | 828 | 784 | [10] |
| [CoIII(BDPP)(OOH)] 1 | - | - | 795 | 748 | [11] |
| [CoIII(OOH)(Me3-TPADP)(MeCN)]2+ 2 | 571 | 551 | 851 | 803 | [12] |
| [CoIII(OOH)(L2)] 3 | 564 | 544 | 832 | 781 | [13] |
| Superoxido | Hydroperoxido | ||
|---|---|---|---|
| Experimental/Å 1 | Optimized/Å | Optimized/Å | |
| O–O | 1.301 (5) | 1.276 | 1.425 |
| Co–O | 1.901 (3) | 1.923 | 1.894 |
| Co–NPz,ax | 2.052 (4) | 2.056 | 2.050 |
| Co–NPz,eq | 1.978 (3) | 2.006 | 2.019 |
| Co–NPz,eq | 2.005 (4) | 2.003 | 2.002 |
| Co–NIm,eq | 1.962 (4) | 1.959 | 1.958 |
| Co–NIm,eq | 1.952 (3) | 1.957 | 1.954 |
| Complex | X of TpMe2,X | Y of LY | k2 for H /M−1 s−1 | KO2 of R /atm−1 1 | Co(II)/(III) Oxidation Potential of R/V 1 |
|---|---|---|---|---|---|
| 1 | H | Ph | 4.4 × 10−2 | 18.88 | 0.18 |
| 2 | H | OiPr | 4.0 × 10−2 | 7.40 | 0.21 |
| 3 | H | Me | 3.0 × 10−2 | 2.62 | 0.07 |
| 4 | H | Bu | 2.5 × 10−2 | 0.77 | 0.12 |
| 5 | Me | Ph | 5.3 × 10−2 | 61.41 | 0.11 |
| 6 | Br | Ph | 8.2 × 10−2 | 2.99 | 0.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishiura, T.; Ohta, T.; Ogura, T.; Nakazawa, J.; Okamura, M.; Hikichi, S. The Conversion of Superoxide to Hydroperoxide on Cobalt(III) Depends on the Structural and Electronic Properties of Azole-Based Chelating Ligands. Molecules 2022, 27, 6416. https://doi.org/10.3390/molecules27196416
Nishiura T, Ohta T, Ogura T, Nakazawa J, Okamura M, Hikichi S. The Conversion of Superoxide to Hydroperoxide on Cobalt(III) Depends on the Structural and Electronic Properties of Azole-Based Chelating Ligands. Molecules. 2022; 27(19):6416. https://doi.org/10.3390/molecules27196416
Chicago/Turabian StyleNishiura, Toshiki, Takehiro Ohta, Takashi Ogura, Jun Nakazawa, Masaya Okamura, and Shiro Hikichi. 2022. "The Conversion of Superoxide to Hydroperoxide on Cobalt(III) Depends on the Structural and Electronic Properties of Azole-Based Chelating Ligands" Molecules 27, no. 19: 6416. https://doi.org/10.3390/molecules27196416
APA StyleNishiura, T., Ohta, T., Ogura, T., Nakazawa, J., Okamura, M., & Hikichi, S. (2022). The Conversion of Superoxide to Hydroperoxide on Cobalt(III) Depends on the Structural and Electronic Properties of Azole-Based Chelating Ligands. Molecules, 27(19), 6416. https://doi.org/10.3390/molecules27196416