
New Chemicals Suppressing SARS-CoV-2 Replication in Cell Culture

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Computer-Aided Initial Search for Inhibitors
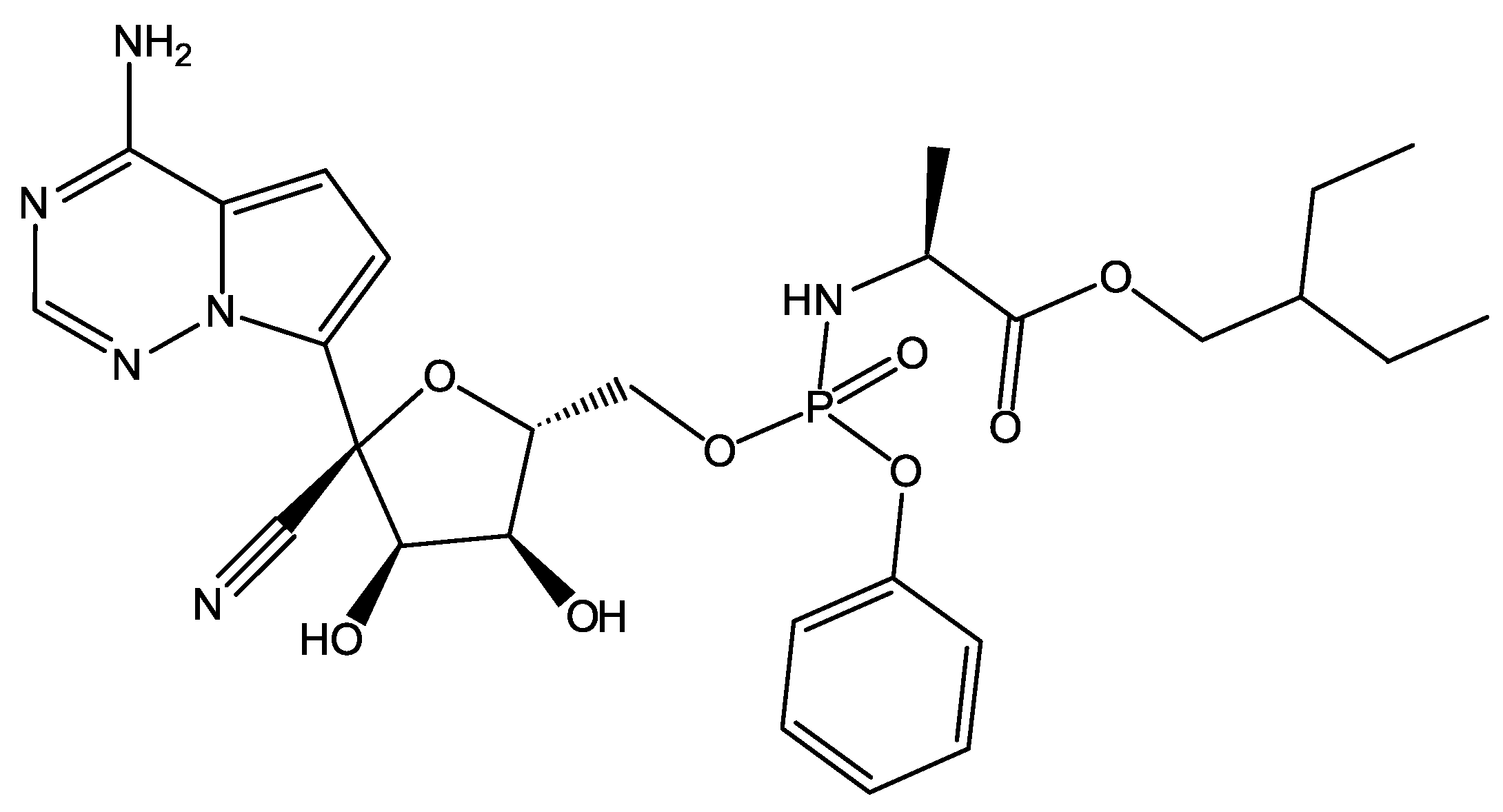
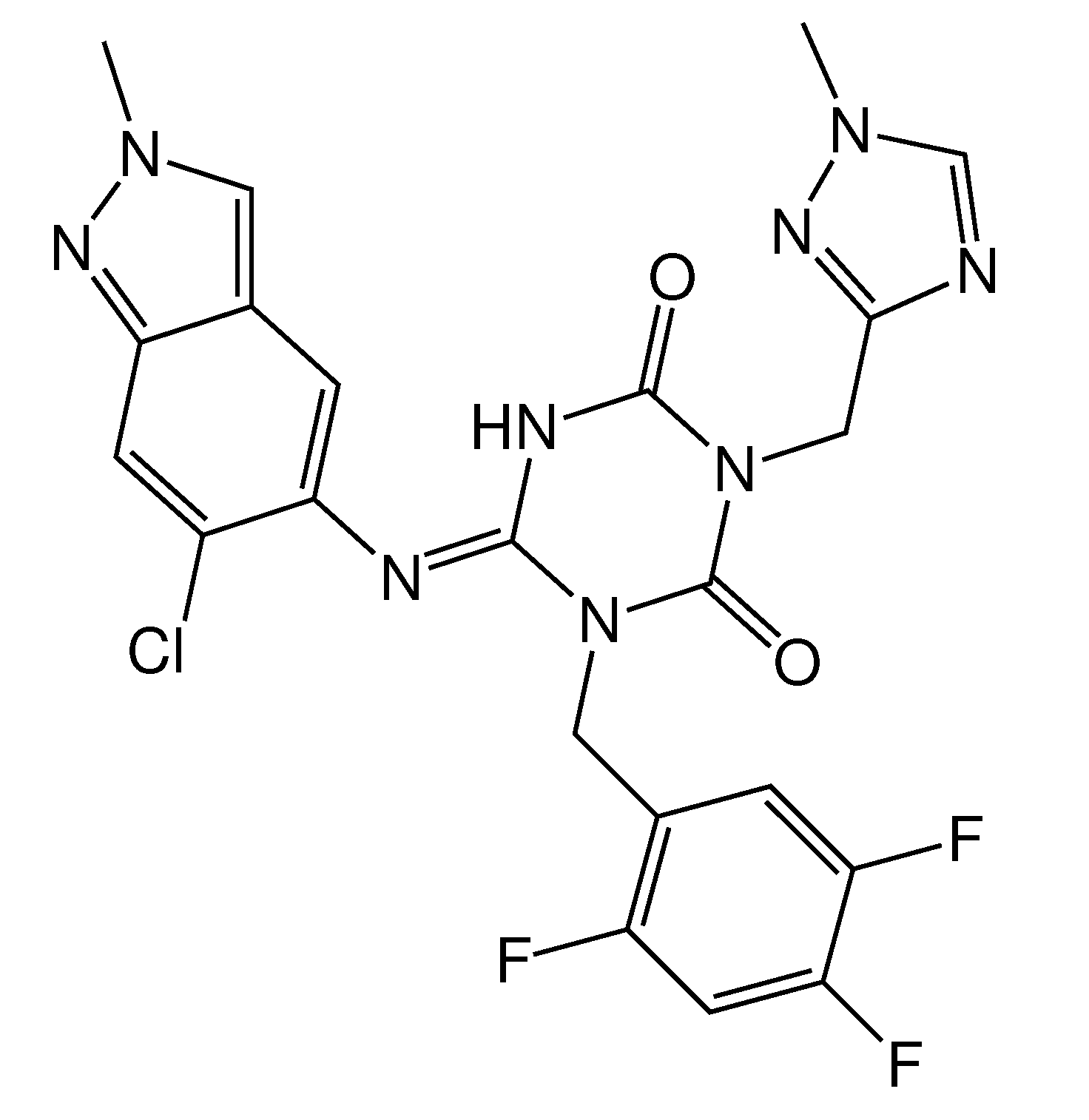
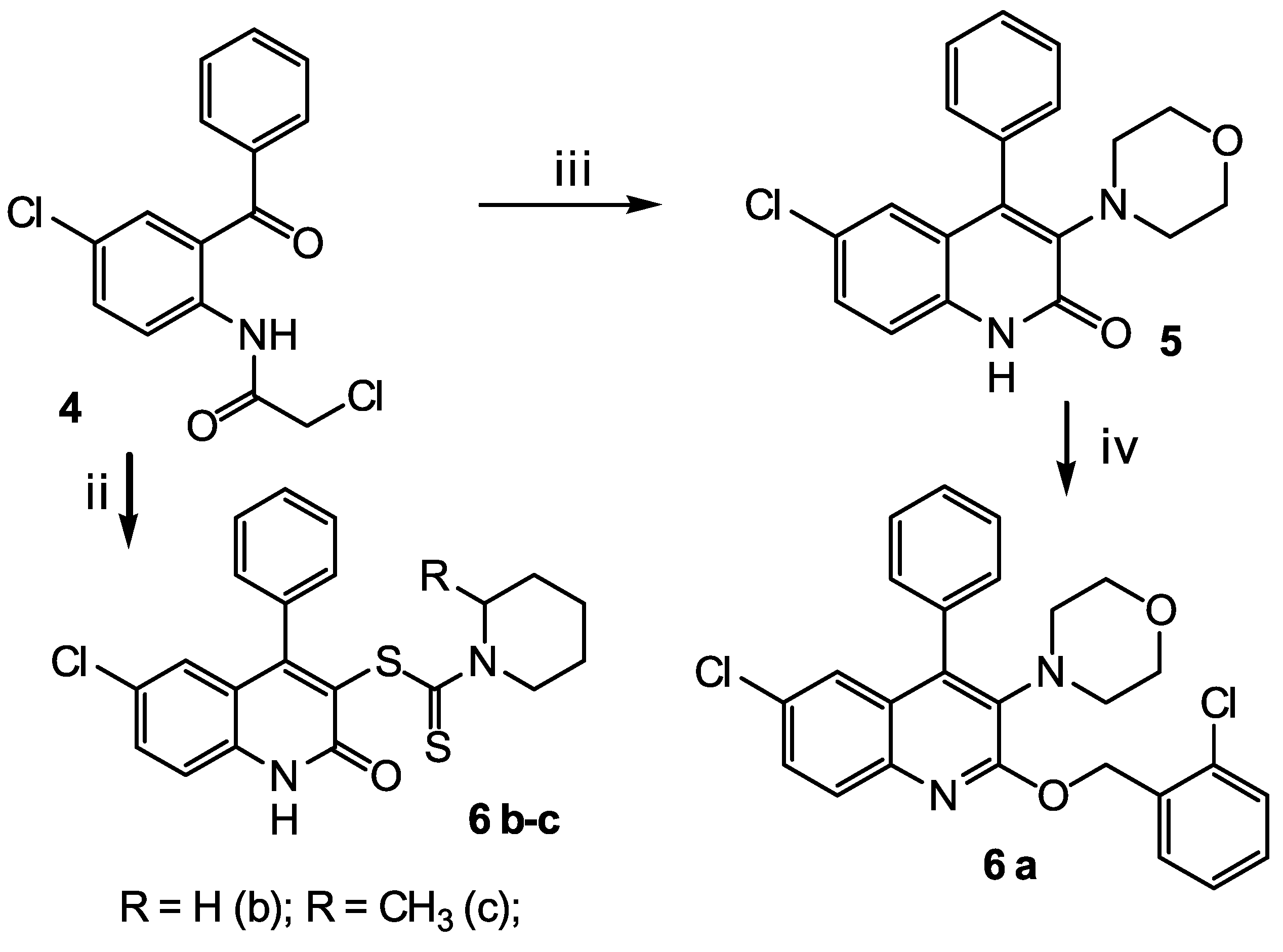
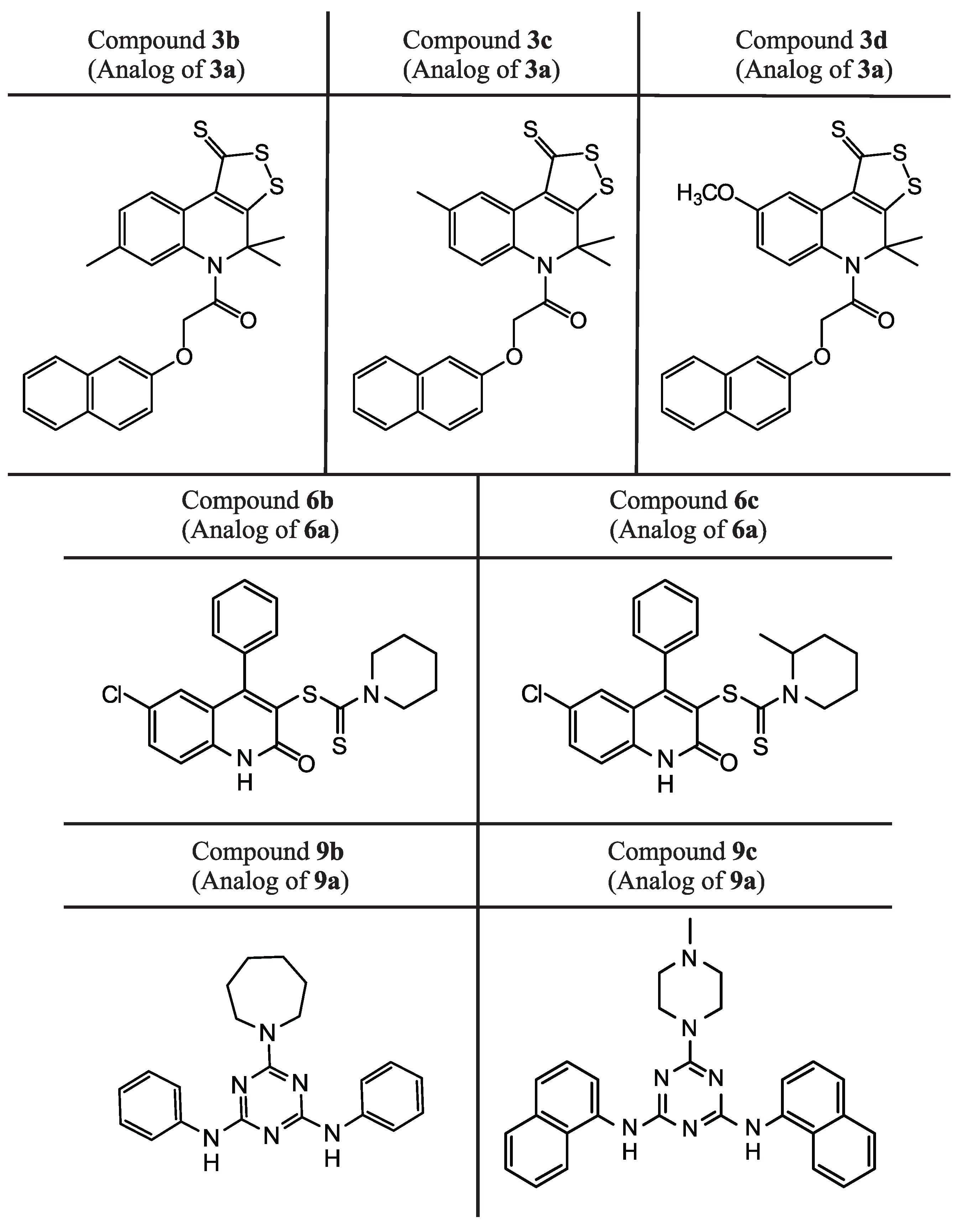
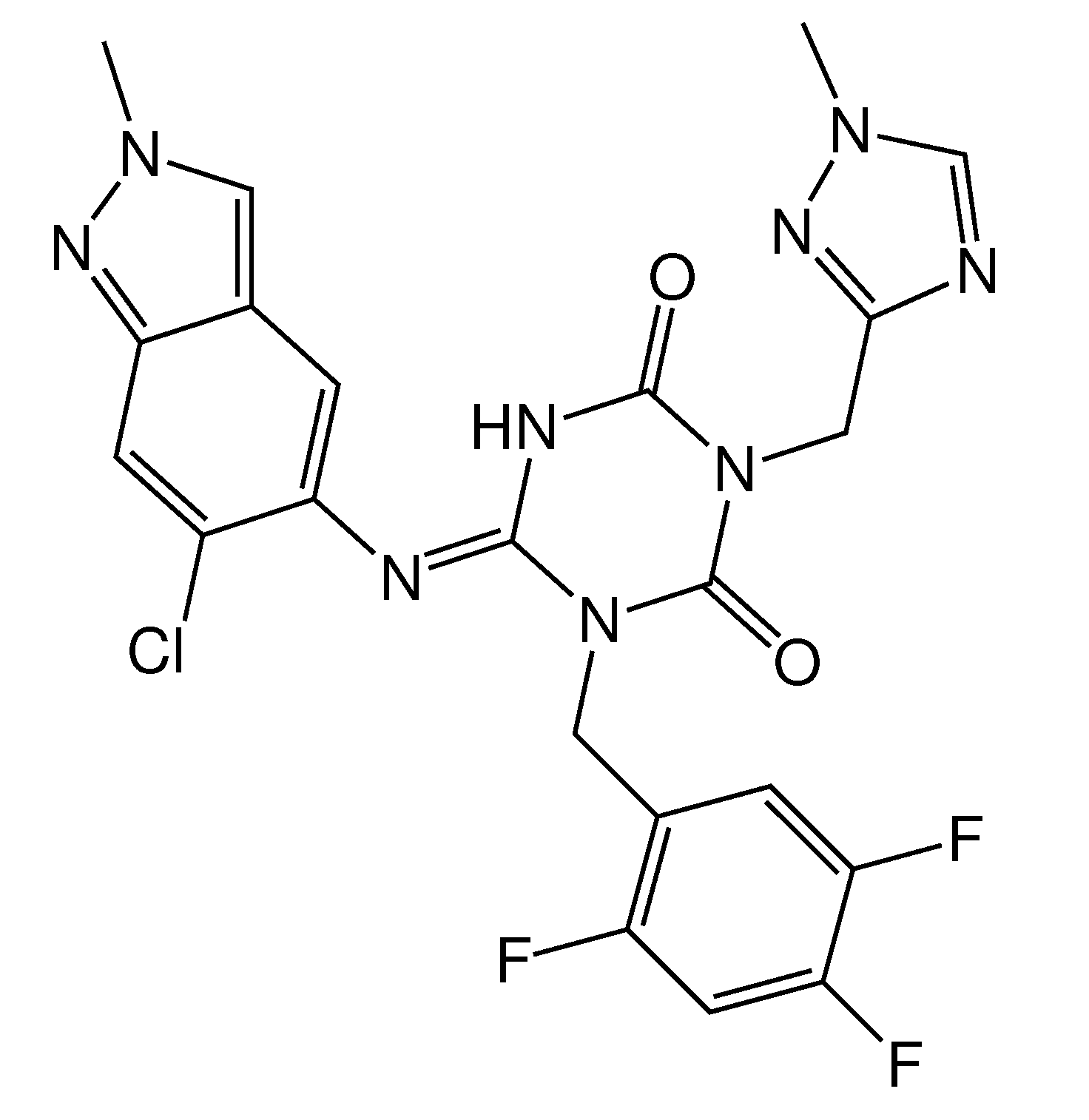
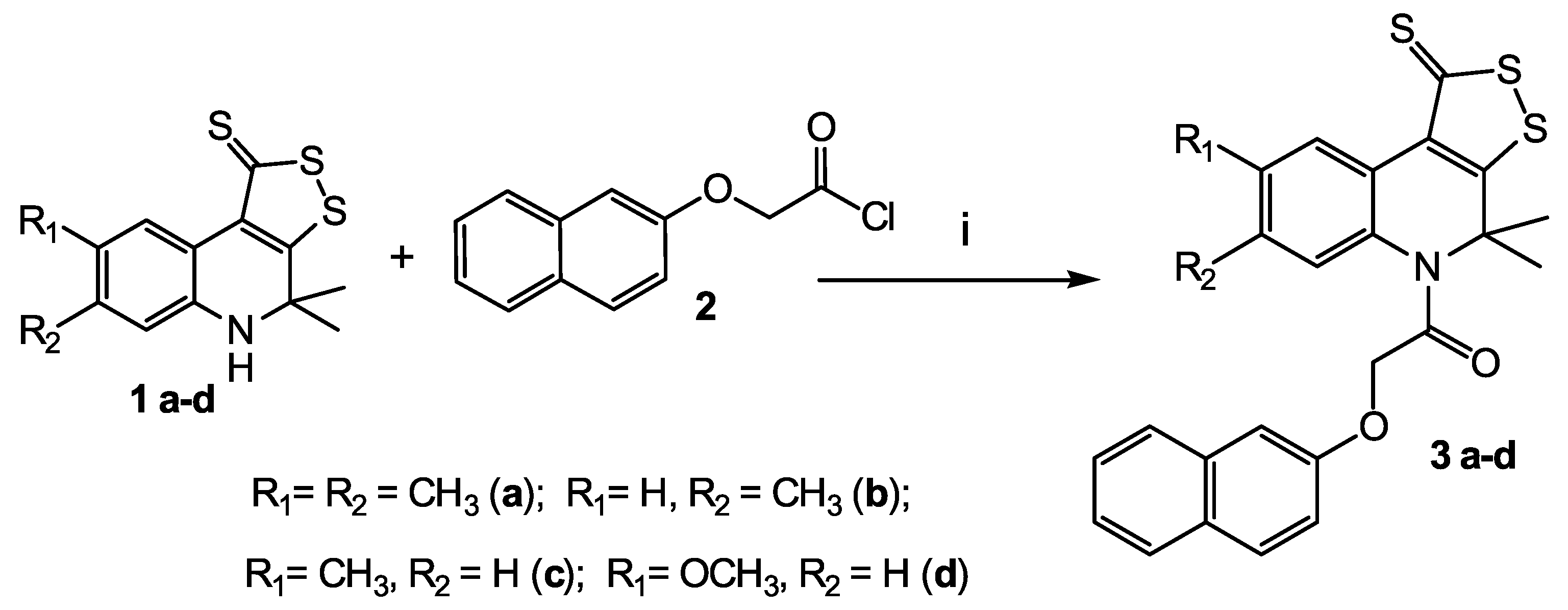
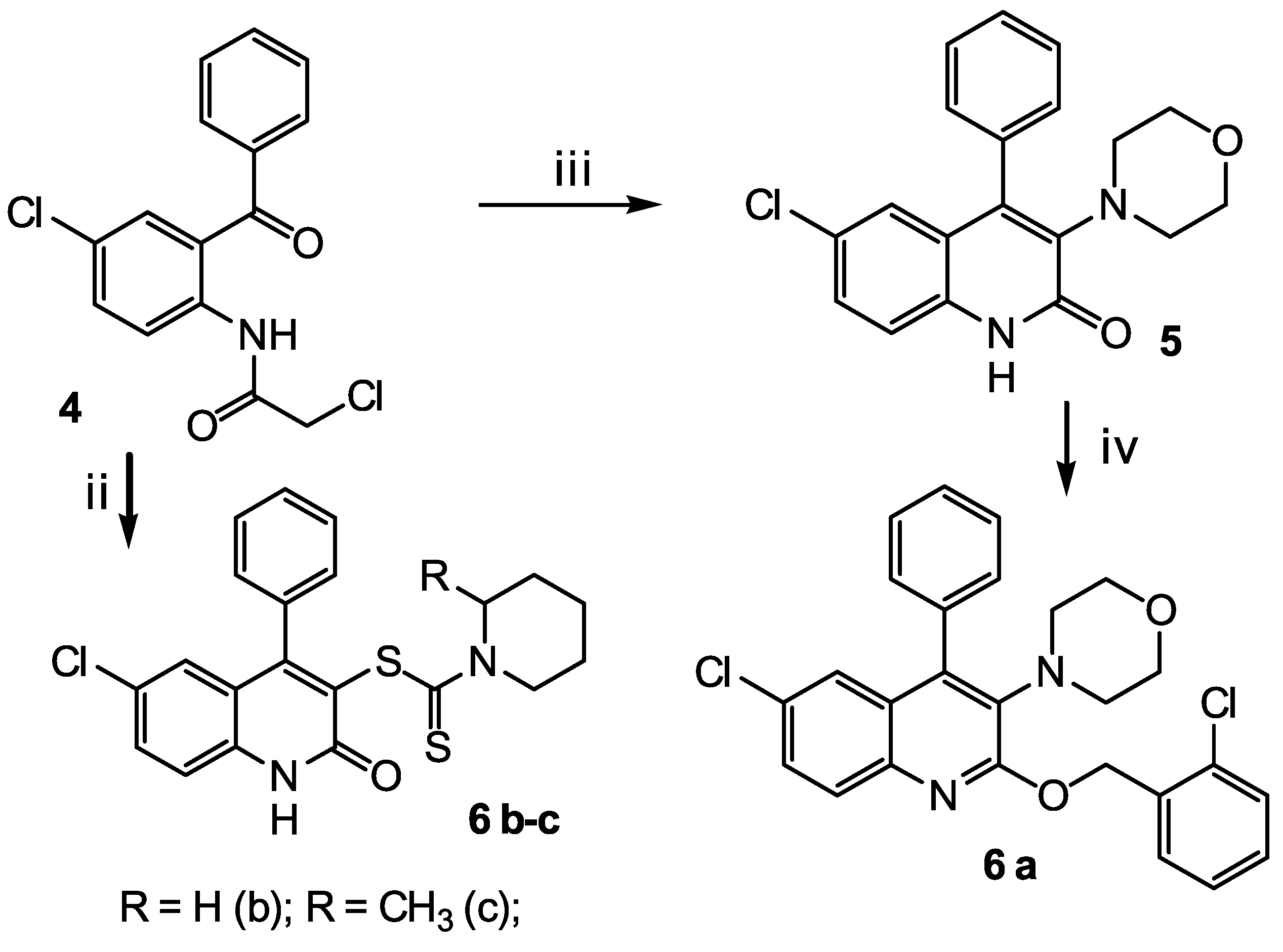
2.2. Synthesis
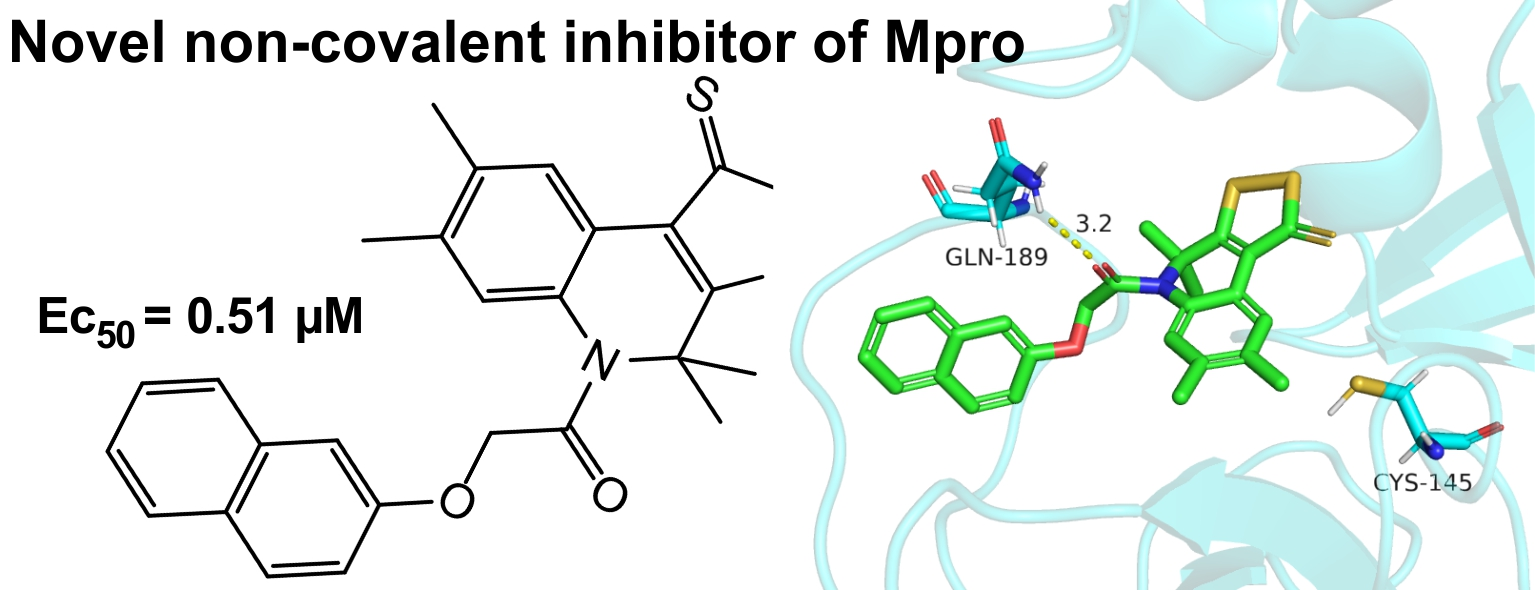
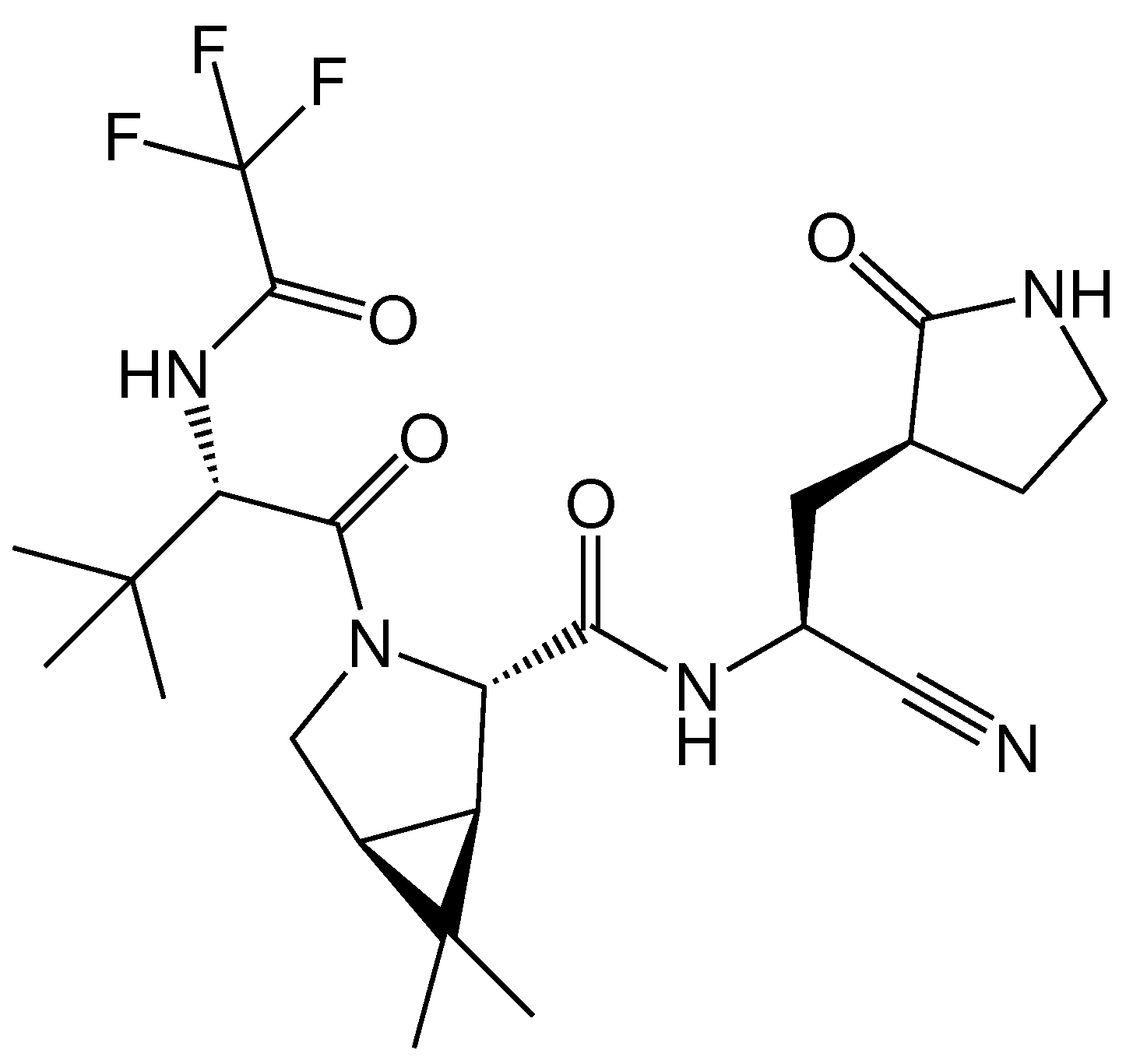
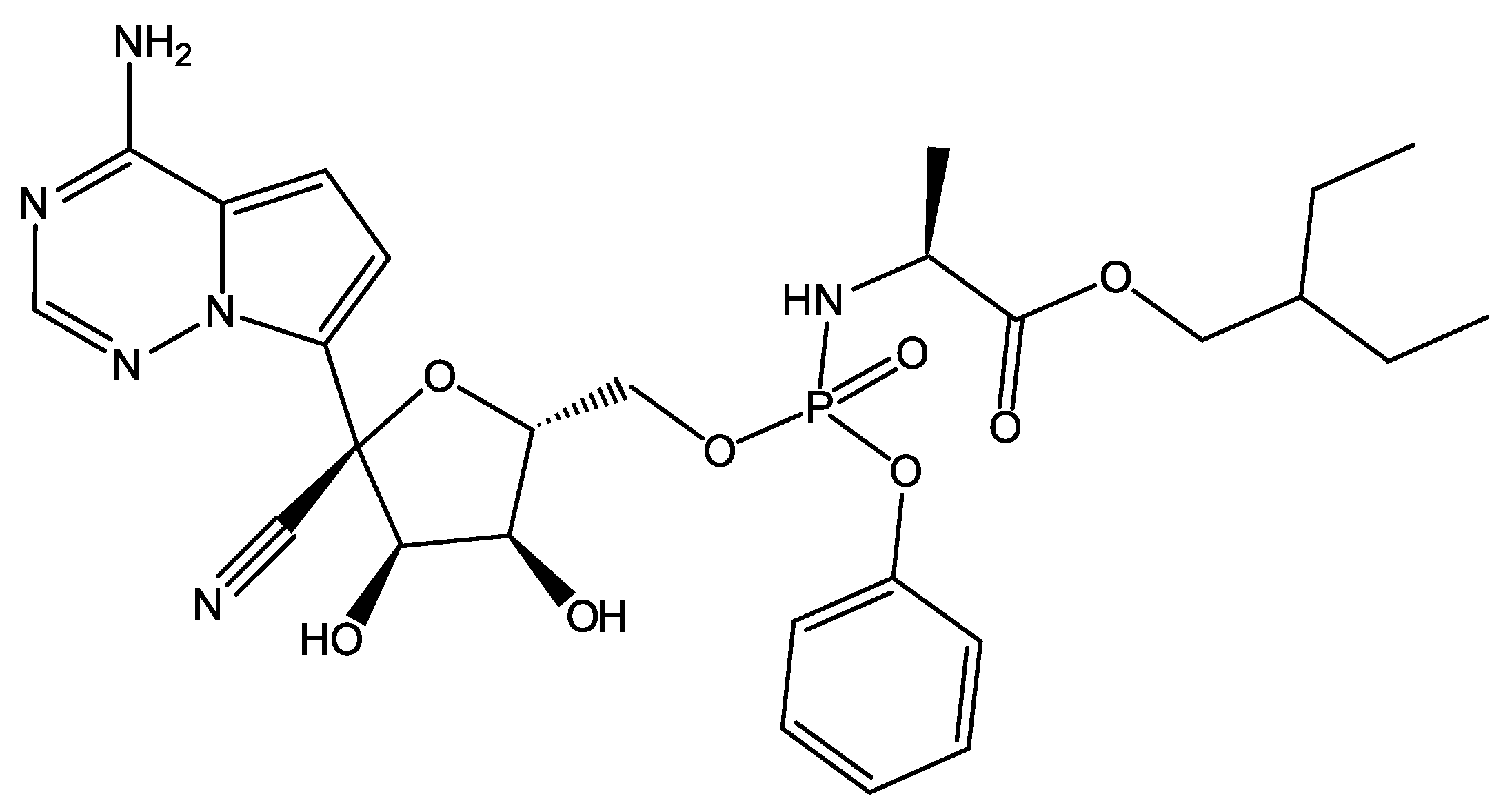
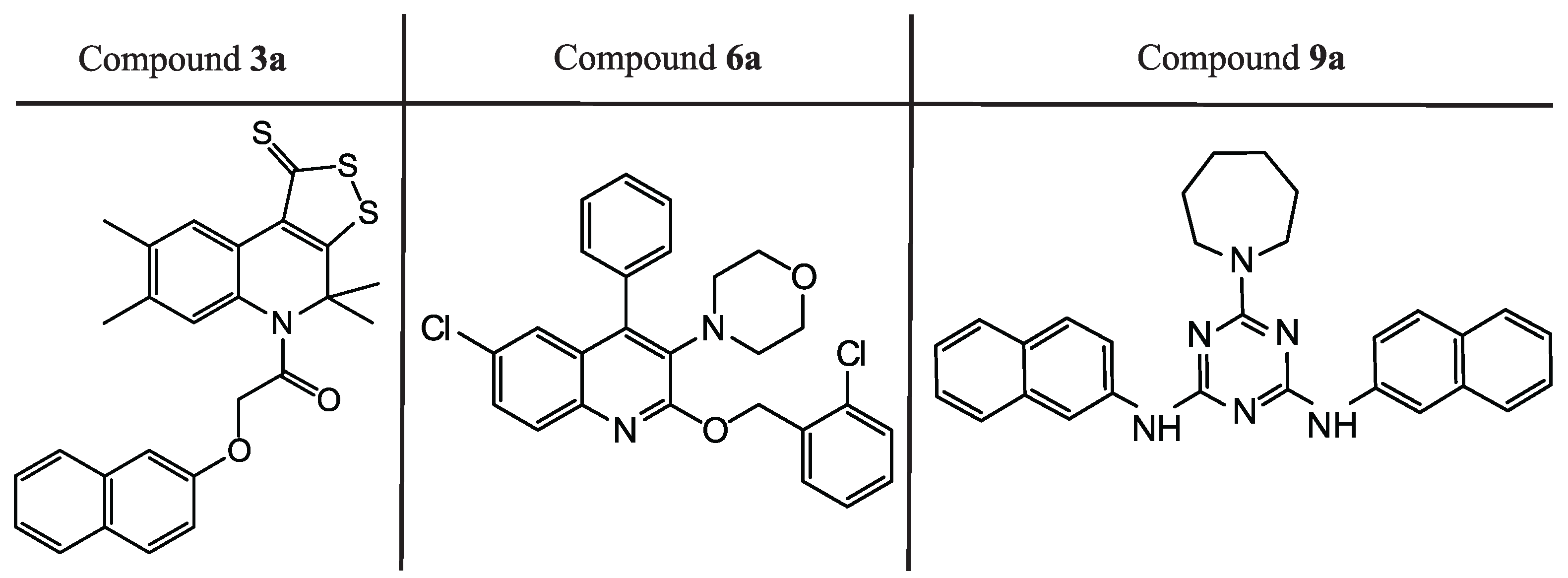
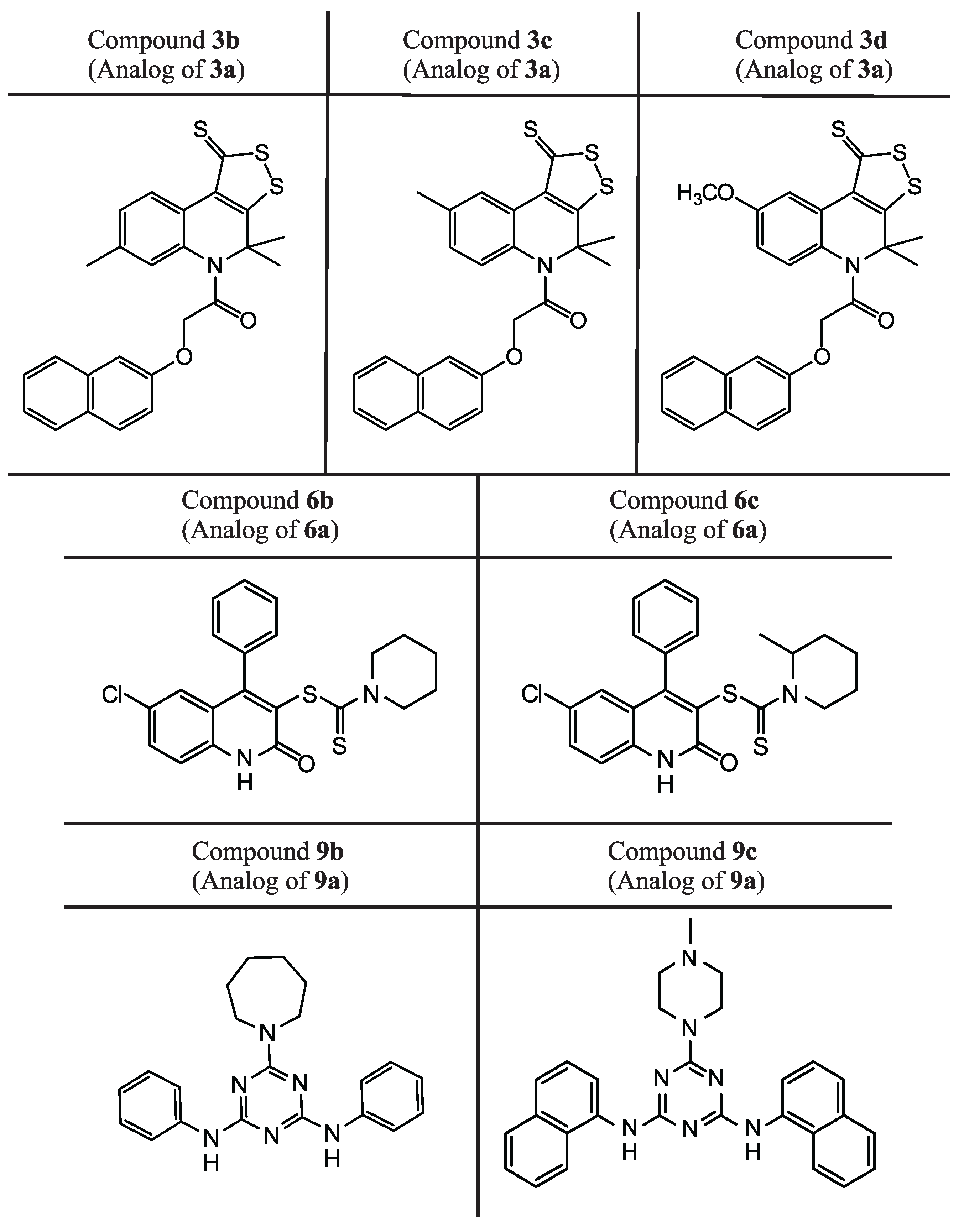
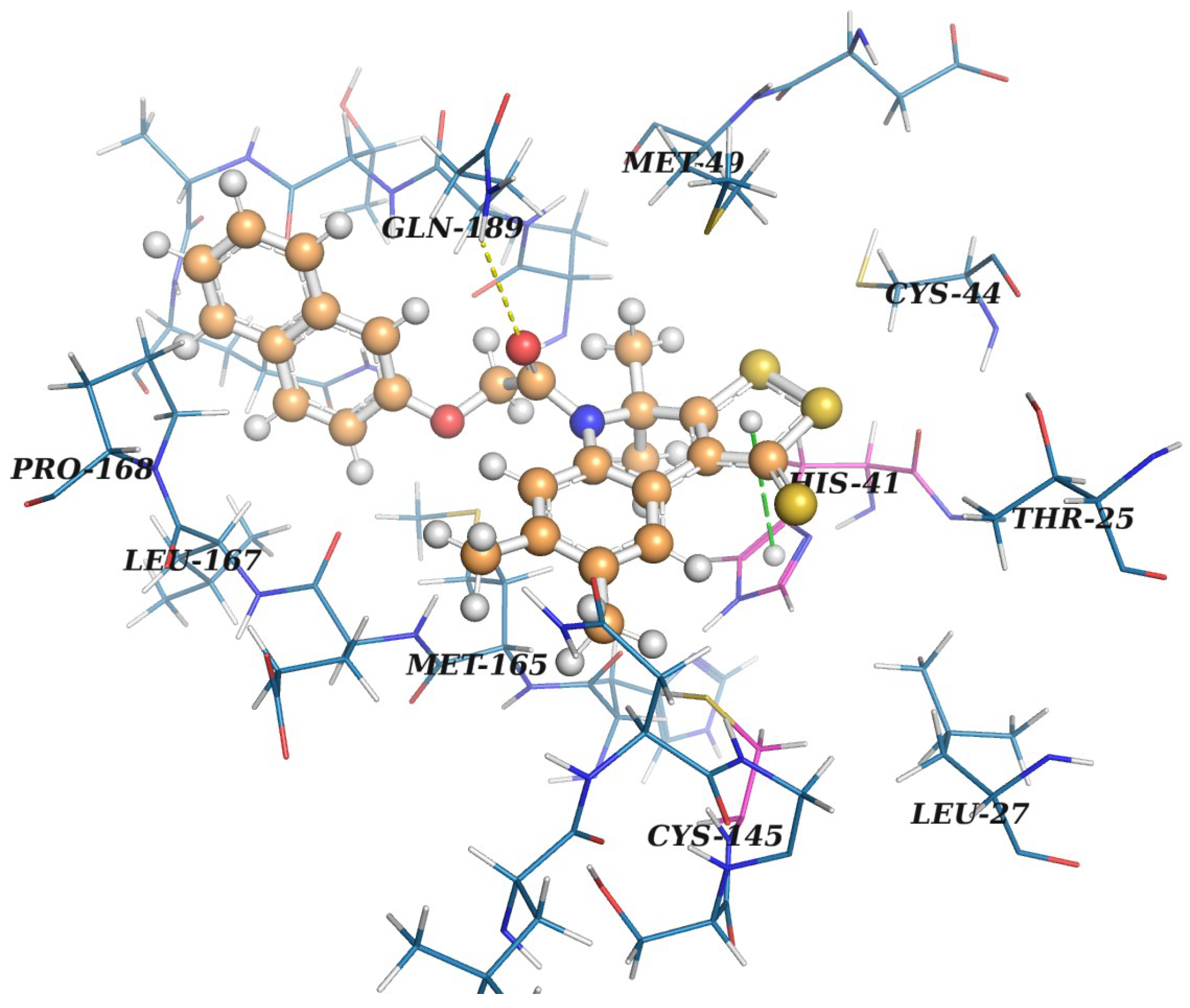
2.3. Protein–Ligand Binding: Modeling and In Vitro Testing
3. Conclusions
4. Materials and Methods
4.1. The Target Protein Model
4.2. The Database of Organic Compounds
4.3. Docking and Postprocessing
4.4. Antiviral Activity (Wild-Type SARS-CoV-2)
4.5. Chemistry
4.5.1. General
4.5.2. General Procedure for Synthesis of Substituted
4,4-dimethyl-5-[(naphthalen-2-yloxy)acetyl]-7-R2-8-R1-4,5-dihydro-1H-[1,2]dithiolo[3,4-c]quinoline-1-thiones 3 a-d
4,4,7,8-tetramethyl-5-[(naphthalen-2-yloxy)acetyl-4,5-dihydro-1H-[1,2]dithiolo[3,4-c]quinoline-1-thione 3 a
4,4,7-trimethyl-5-[(naphthalen-2-yloxy)acetyl-4,5-dihydro-1H-[1,2]dithiolo[3,4-c]quinoline-1-thione 3 b
4,4,8-trimethyl-5-[(naphthalen-2-yloxy)acetyl-4,5-dihydro-1H-[1,2]dithiolo[3,4-c]quinoline-1-thione 3 c
8-methoxy-4,4-dimethyl-5-[(naphthalen-2-yloxy)acetyl-4,5-dihydro-1H-[1,2]dithiolo[3,4-c]quinoline-1-thione 3 d
4.5.3. Synthesis of Substituted 6-Chloro-4-phenylquinolines 6 a-c
6-chloro-2-[(2-chlorobenzyl)oxy]-3-(morpholin-4-yl)-4-phenylquinoline 6 a
6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl piperidine-1-carboditioate 6 b
6-chloro-2-oxo-4-phenyl-1,2-dihydroquinolin-3-yl 2-methylpiperidine-1-carboditioate 6 c
4.5.4. General Procedure for Synthesis of Substituted
6-R-N,N-diaryl-1,3,5-triazine-2,4-diamine 9 a-c
6-(azepan-1-yl)-N,N-di(naphthalen-2-yl)-1,3,5-triazine-2,4-diamine 9 a
6-(azepan-1-yl)-N,N’-diphenyl-1,3,5-triazine-2,4-diamine 9 b
6-(4-methylpiperazin-1-yl)-N,N-di(naphthalen-1-yl)-1,3,5-triazine-2,4-amine 9 c
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gurung, A.; Ajmal Ali, M.; Lee, J.; Farah, M.; Alanazi, K. An Updated Review of Computer-Aided Drug Design and Its Application to COVID-19. Biomed Res. Int. 2021, 2021, 8853056. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.J.; Chen, T.H. NSP16 2’-O-MTase in Coronavirus Pathogenesis: Possible Prevention and Treatments Strategies. Viruses 2021, 13, 538. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.A.; Urquiza, J.; Ramirez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.R.; da Silva Santos-Júnior, P.F.; de Andrade Brandão, J.; Anderson, L.; Bassi, Ê.J.; Xavier de Araújo-Júnior, J.; Cardoso, S.H.; da Silva-Júnior, E.F. Druggable targets from coronaviruses for designing new antiviral drugs. Bioorganic Med. Chem. 2020, 28, 115745. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Sulimov, V.B.; Ilin, I.S.; Kutov, D.C.; Sulimov, A.V. Development of docking programs for Lomonosov supercomputer. J. Turk. Chem. Soc. Sect. Chem. 2019, 7, 259–276. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A.V. Advances in Docking. Curr. Med. Chem. 2019, 26, 7555–7580. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Kutov, D.C.; Taschilova, A.S.; Ilin, I.S.; Tyrtyshnikov, E.E.; Sulimov, A.V. Docking paradigm in Drug Design. Curr. Top. Med. Chem. 2021, 21, 507–546. [Google Scholar] [CrossRef]
- Sulimov, A.V.; Kutov, D.C.; Katkova, E.V.; Kondakova, O.A.; Sulimov, V.B. Search for approaches to improving the calculation accuracy of the protein-ligand binding energy by docking. Russ. Chem. Bull. 2017, 66, 1913–1924. [Google Scholar] [CrossRef]
- Yuriev, E.; Holien, J.K.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 28, 581–604. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15 – Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- ZINC Catalog DrugBank-Approved. Available online: https://zinc.docking.org/catalogs/dbap/ (accessed on 16 August 2022).
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2013, 42, D1091–D1097. [Google Scholar] [CrossRef]
- Ghahremanpour, M.M.; Tirado-Rives, J.; Deshmukh, M.; Ippolito, J.A.; Zhang, C.H.; Cabeza de Vaca, I.; Liosi, M.E.; Anderson, K.S.; Jorgensen, W.L. Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020, 11, 2526–2533. [Google Scholar] [CrossRef]
- Zhang, C.H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Song, L.F.; Merz, K.M. Evolution of Alchemical Free Energy Methods in Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5308–5318. [Google Scholar] [CrossRef]
- Shcherbakov, D.; Baev, D.; Kalinin, M.; Dalinger, A.; Chirkova, V.; Belenkaya, S.; Khvostov, A.; Krut’ko, D.; Medved’ko, A.; Volosnikova, E.; et al. Design and Evaluation of Bispidine-Based SARS-CoV-2 Main Protease Inhibitors. ACS Med. Chem. Lett. 2022, 13, 140–147. [Google Scholar] [CrossRef]
- Zhu, W.; Xu, M.; Chen, C.Z.; Guo, H.; Shen, M.; Hu, X.; Shinn, P.; Klumpp-Thomas, C.; Michael, S.G.; Zheng, W. Identification of SARS-CoV-2 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacol. Transl. Sci. 2020, 3, 1008–1016. [Google Scholar] [CrossRef]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M<sup>pro</sup> inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Foo, C.S.; Jochmans, D.; Vangeel, L.; De Jonghe, S.; Augustijns, P.; Mols, R.; Weynand, B.; Wattanakul, T.; Hoglund, R.M.; et al. The oral protease inhibitor (PF-07321332) protects Syrian hamsters against infection with SARS-CoV-2 variants of concern. Nat. Commun. 2022, 13, 719. [Google Scholar] [CrossRef]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021, 12, 6055. [Google Scholar] [CrossRef] [PubMed]
- Ali Khan, S.; Zia, K.; Ashraf, S.; Uddin, R.; Ul-Haq, Z. Identification of Chymotrypsin-like Protease Inhibitors of SARS-CoV-2 Via Integrated Computational Approach. J. Biomol. Struct. Dyn. 2020, 39, 1–13. [Google Scholar] [CrossRef]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, M. Potential anti-SARS-CoV-2 drug candidates identified through virtual screening of the ChEMBL database for compounds that target the main coronavirus protease. FEBS Open Bio 2020, 10, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I. The race to treat COVID-19: Potential therapeutic agents for the prevention and treatment of SARS-CoV-2. Eur. J. Med. Chem. 2021, 213, 113157. [Google Scholar] [CrossRef]
- Cui, J.; Jia, J. Discovery of juglone and its derivatives as potent SARS-CoV-2 main proteinase inhibitors. Eur. J. Med. Chem. 2021, 225, 113789. [Google Scholar] [CrossRef]
- Di Sarno, V.; Lauro, G.; Musella, S.; Ciaglia, T.; Vestuto, V.; Sala, M.; Scala, M.C.; Smaldone, G.; Di Matteo, F.; Novi, S.; et al. Identification of a dual acting SARS-CoV-2 proteases inhibitor through in silico design and step-by-step biological characterization. Eur. J. Med. Chem. 2021, 226, 113863. [Google Scholar] [CrossRef]
- Vuong, W.; Fischer, C.; Khan, M.B.; van Belkum, M.J.; Lamer, T.; Willoughby, K.D.; Lu, J.; Arutyunova, E.; Joyce, M.A.; Saffran, H.A.; et al. Improved SARS-CoV-2 Mpro inhibitors based on feline antiviral drug GC376: Structural enhancements, increased solubility, and micellar studies. Eur. J. Med. Chem. 2021, 222, 113584. [Google Scholar] [CrossRef] [PubMed]
- Gupta, Y.; Maciorowski, D.; Zak, S.E.; Jones, K.A.; Kathayat, R.S.; Azizi, S.A.; Mathur, R.; Pearce, C.M.; Ilc, D.J.; Husein, H.; et al. Bisindolylmaleimide IX: A novel anti-SARS-CoV2 agent targeting viral main protease 3CLpro demonstrated by virtual screening pipeline and in vitro validation assays. Methods 2021, 195, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Yañez, O.; Osorio, M.I.; Uriarte, E.; Areche, C.; Tiznado, W.; Pérez-Donoso, J.M.; García-Beltrán, O.; González-Nilo, F. In Silico Study of Coumarins and Quinolines Derivatives as Potent Inhibitors of SARS-CoV-2 Main Protease. Front. Chem. 2021, 8, 595097. [Google Scholar] [CrossRef] [PubMed]
- Alexpandi, R.; De Mesquita, J.F.; Pandian, S.K.; Ravi, A.V. Quinolines-Based SARS-CoV-2 3CLpro and RdRp Inhibitors and Spike-RBD-ACE2 Inhibitor for Drug-Repurposing Against COVID-19: An in silico Analysis. Front. Microbiol. 2020, 11, 1796. [Google Scholar] [CrossRef]
- Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19. J. Med. Chem. 2022, 65, 6499–6512. [Google Scholar] [CrossRef]
- Voevodin, V.V.; Antonov, A.S.; Nikitenko, D.A.; Shvets, P.A.; Sobolev, S.I.; Sidorov, I.Y.; Stefanov, K.S.; Voevodin, V.V.; Zhumatiy, S.A. Supercomputer Lomonosov-2: Large Scale, Deep Monitoring and Fine Analytics for the User Community. Supercomput. Front. Innov. 2019, 6, 4–11. [Google Scholar] [CrossRef]
- Sulimov, A.V.; Kutov, D.C.; Taschilova, A.S.; Ilin, I.S.; Stolpovskaya, N.V.; Shikhaliev, K.S.; Sulimov, V.B. In Search of Non-covalent Inhibitors of SARS-CoV-2 Main Protease: Computer Aided Drug Design Using Docking and Quantum Chemistry. Supercomput. Front. Innov. 2020, 7, 41–56. [Google Scholar] [CrossRef]
- Brown, J.P. Reactions of 2,2-dialkyl-1,2-dihydroquinolines. Part IV. 4,5-Dihydro-4,4-dimethyl-1H-1,2-dithiolo[3,4-c]quinoline-1-thiones. J. Chem. Soc. C 1968, 1074–1075. [Google Scholar] [CrossRef]
- Kartsev, V.; Shikhaliev, K.S.; Geronikaki, A.; Medvedeva, S.M.; Ledenyova, I.V.; Krysin, M.Y.; Petrou, A.; Ciric, A.; Glamoclija, J.; Sokovic, M. Appendix A. dithioloquinolinethiones as new potential multitargeted antibacterial and antifungal agents: Synthesis, biological evaluation and molecular docking studies. Eur. J. Med. Chem. 2019, 175, 201–214. [Google Scholar] [CrossRef]
- Craig, D.; Gregg, E.C. 2,2,4-Trimethyl-1,2-dihydroquinoline. J. Am. Chem. Soc. 1953, 75, 2252. [Google Scholar] [CrossRef]
- Singh, R.K.; Prasad, D.; Bhardwaj, T.R. Design, synthesis and evaluation of aminobenzophenone derivatives containing nitrogen mustard moiety as potential central nervous system antitumor agent. Med. Chem. Res. 2013, 22, 5901–5911. [Google Scholar] [CrossRef]
- Kubota, H.; Suzuki, T.; Miura, M.; Nakai, E.; Yahiro, K.; Miyake, A.; Mochizuki, S.; Nakato, K. 2,4,6-Triamino-1,3,5-triazine Derivative. No. EP1479397A4, 7 January 2009. [Google Scholar]
- Fujii, S.; Kobayashi, T.; Nakatsu, A.; Miyazawa, H.; Kagechika, H. Synthesis and Structure–Activity Relationship Study of Triazine-Based Inhibitors of the DNA Binding of NF-kB. Chem. Pharm. Bull. 2014, 62, 700–708. [Google Scholar] [CrossRef] [PubMed]
- León, F.; Elizalde, P.; Prieto, P.; Sánchez-Migallón, A.; Rodriguez Garcia, A.; Hoz, A. Bistriazine-based streptocyanines. Preparation, structural determination and optoelectronic properties. Dye. Pigment. 2016, 131, 307–319. [Google Scholar] [CrossRef]
- Solankee, A.; Kapadia, K.; Ćirić, A.; Soković, M.; Doytchinova, I.; Geronikaki, A. Synthesis of some new S-triazine based chalcones and their derivatives as potent antimicrobial agents. Eur. J. Med. Chem. 2010, 45, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- Delano, W.L. The PyMOL Molecular Graphics System. 2002. Available online: http://www.pymol.org (accessed on 14 January 2021).
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Guo, S.; Xie, H.; Lei, Y.; Liu, B.; Zhang, L.; Xu, Y.; Zuo, Z. Discovery of novel inhibitors against main protease (Mpro) of SARS-CoV-2 via virtual screening and biochemical evaluation. Bioorganic Chem. 2021, 110, 104767. [Google Scholar] [CrossRef] [PubMed]
- Vuong, W.; Khan, M.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.; Mckay, R.; Belkum, M.; Joyce, M.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef]
- Kitamura, N.; Sacco, M.D.; Ma, C.; Hu, Y.; Townsend, J.A.; Meng, X.; Zhang, F.; Zhang, X.; Ba, M.; Szeto, T.; et al. Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2848–2865. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sulimov, A.V.; Kutov, D.C.; Oferkin, I.V.; Katkova, E.V.; Sulimov, V.B. Application of the Docking Program SOL for CSAR Benchmark. J. Chem. Inf. Model. 2013, 53, 1946–1956. [Google Scholar] [CrossRef]
- The Department of Organic Chemistry of Voronezh State University. Available online: http://www.vsu.ru/english/depts/chem.html (accessed on 16 August 2022).
- Ilin, I.; Lipets, E.; Sulimov, A.; Kutov, D.; Shikhaliev, K.; Potapov, A.; Krysin, M.; Zubkov, F.; Sapronova, L.; Ataullakhanov, F.; et al. New factor Xa inhibitors based on 1,2,3,4-tetrahydroquinoline developed by molecular modelling. J. Mol. Graph. Model. 2019, 89, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, S.; Potapov, A.; Gribkova, I.; Katkova, E.; Sulimov, V.; Shikhaliev, K. Synthesis, Docking, and Anticoagulant Activity of New Factor-Xa Inhibitors in a Series of Pyrrolo[3,2,1-ij]Quinoline-1,2-Diones. Pharm. Chem. J. 2018, 51, 975–979. [Google Scholar] [CrossRef]
- Novichikhina, N.; Ilin, I.; Tashchilova, A.; Sulimov, A.; Kutov, D.; Ledenyova, I.; Krysin, M.; Shikhaliev, K.; Gantseva, A.; Gantseva, E.; et al. Synthesis, Docking, and In Vitro Anticoagulant Activity Assay of Hybrid Derivatives of Pyrrolo[3,2,1-ij]Quinolin-2(1H)-one as New Inhibitors of Factor Xa and Factor XIa. Molecules 2020, 25, 1889. [Google Scholar] [CrossRef]
- Novichikhina, N.; Shestakov, A.; Potapov, A.; Kosheleva, E.; Shatalov, G.; Verezhnikov, V.; Vandyshev, D.; Ledeneva, I.; Shikhaliev, K. Synthesis of 4H-pyrrolo[3,2,1-ij]quinoline-1,2-diones containing a piperazine fragment and study of their inhibitory properties against protein kinases. Russ. Chem. Bull. 2020, 69, 787–792. [Google Scholar] [CrossRef]
- Stolpovskaya, N.; Kruzhilin, A.; Zorina, V.Z.; Shikhaliev, K.; Ledeneva, I.; Kosheleva, E.; Vandyshev, D. Synthesis of Substituted Aminopyrimidines as Novel Promising Tyrosine Kinase Inhibitors. Russ. J. Org. Chem. 2019, 55, 1322–1328. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Gribkova, I.V.; Kochugaeva, M.P.; Katkova, E.V.; Sulimov, A.V.; Kutov, D.C.; Shikhaliev, K.S.; Medvedeva, S.M.; Krysin, M.; Sinauridze, E.I.; et al. Application of Molecular Modeling to Development of New Factor Xa Inhibitors. Biomed Res. Int. 2015, 2015, 120802. [Google Scholar] [CrossRef]
- Vostrikova, T.; Kalaev, V.; Medvedeva, S.; Novichkina, N.; Shikhaliev, K. Synthesized organic compounds as growth stimulators for woody plants. Period. Tche Quim. 2020, 17, 327–337. [Google Scholar] [CrossRef]
- Kalaev, V.N.; Potapov, A.Y.; Potapov, M.A.; Shikhaliev, K.S. Use of new compounds of the quinoline series as effective stimulants of growth processes. Period. Tche Quim. 2020, 17, 781–790. [Google Scholar] [CrossRef]
- O’Boyle, N.; Banck, M.; James, C.; Morley, C.; Vandermeersch, T.; Hutchison, G. Open Babel: An Open Chemical Toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Marvin Was Used for Drawing, Displaying and Characterizing Chemical Structures, Substructures and Reactions, Marvin 21.3.0, 2021, ChemAxon. Available online: https://chemaxon.com/products/marvin (accessed on 16 August 2022).
- Romanov, A.N.; Kondakova, O.A.; Grigoriev, F.V.; Sulimov, A.V.; Luschekina, S.V.; Martynov, Y.B.; Sulimov, V.B. The SOL docking package for computer-aided drug design. Numer. Methods Program. (Vychislitel’Nye Metod. Program.) 2008, 9, 213–233. (In Russian) [Google Scholar]
- Sulimov, A.V.; Sulimov, V.B.; Romanov, A.N.; Grigoriev, F.V.; Kondakova, O.A.; Bryzgalov, P.A.; Ostapenko, D.A. Web-oriented system Keenbase for new drugs design. In Proceedings of the 4th International Symposium on Computational Methods in Toxicology and Pharmacology Integrating Internet Resourses, Moscow, Russia, 1–5 September 2007; p. 158. [Google Scholar]
- Sinauridze, E.I.; Romanov, A.N.; Gribkova, I.V.; Kondakova, O.A.; Surov, S.S.; Gorbatenko, A.S.; Butylin, A.A.; Monakov, M.Y.; Bogolyubov, A.A.; Kuznetsov, Y.V.; et al. New Synthetic Thrombin Inhibitors: Molecular Design and Experimental Verification. PLoS ONE 2011, 6, e19969. [Google Scholar] [CrossRef] [PubMed]
- Beloglazova, I.; Plekhanova, O.; Katkova, E.; Rysenkova, K.; Stambol’skii, D.; Sulimov, V.; Tkachuk, V. Molecular Modeling as a New Approach to the Development of Urokinase Inhibitors. Bull. Exp. Biol. Med. 2015, 158, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Sulimov, V.; Katkova, E.; Oferkin, I.; Sulimov, A.; Romanov, A.; Roschin, A.; Beloglazova, I.; Plekhanova, O.; Tkachuk, V.; Sadovnichiy, V. Application of Molecular Modeling to Urokinase Inhibitors Development. Biomed Res. Int. 2014, 2014, 625176. [Google Scholar] [CrossRef] [PubMed]
- Romanov, A.N.; Jabin, S.N.; Martynov, Y.B.; Sulimov, A.V.; Grigoriev, F.V.; Sulimov, V.B. Surface Generalized Born Method: A Simple, Fast, and Precise Implicit Solvent Model beyond the Coulomb Approximation. J. Phys. Chem. 2004, 108, 9323–9327. [Google Scholar] [CrossRef]
- Stewart, J. MOPAC2016. Available online: http://openmopac.net/MOPAC2016.html (accessed on 16 August 2022).
- Stewart, J. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar] [CrossRef]
- Sulimov, A.; Kutov, D.; Gribkova, A.; Ilin, I.; Tashchilova, A.; Sulimov, V. Search for Approaches to Supercomputer Quantum-Chemical Docking. In Supercomputing. RuSCDays 2019; Communications in Computer and Information Science; Voevodin, V., Sobolev, S., Eds.; Springer: Cham, Switzerland, 2019; Volume 1129, pp. 363–378. [Google Scholar] [CrossRef]
- Sulimov, A.; Kutov, D.; Ilin, I.; Sulimov, V. Quantum-Chemical Quasi-Docking for Molecular Dynamics Calculations. Nanomaterials 2022, 12, 274. [Google Scholar] [CrossRef]



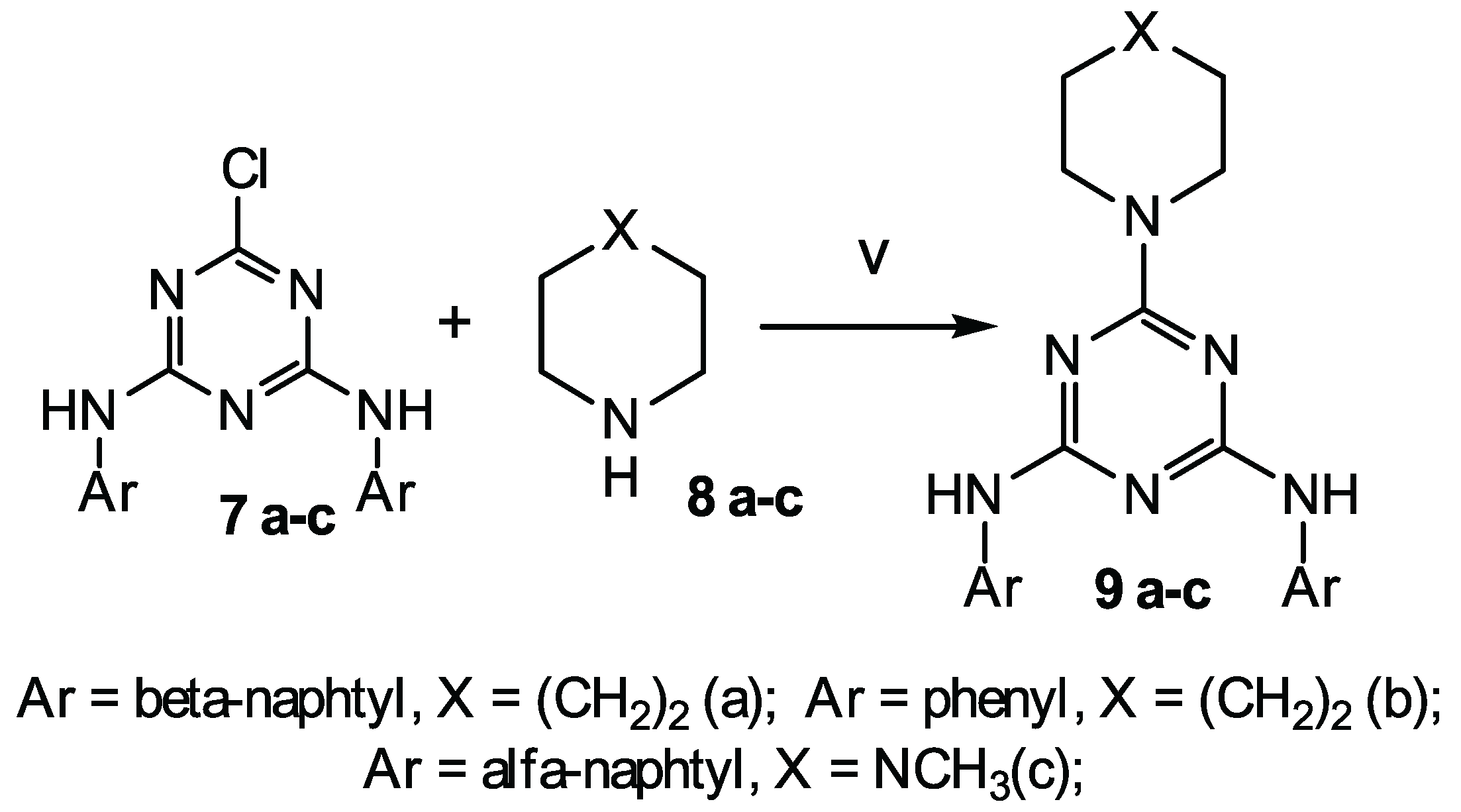



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | SOL Score, kcal/mol | , kcal/mol | EC, µM | SI |
|---|---|---|---|---|
| 3a | −7.54 | −33.1 | 0.51 ± 0.41 | >411.5 |
| 6a | −6.91 | −50.8 | 4.77 ± 1.87 | >45.1 |
| 9a | −6.94 | −55.5 | 18.19 ± 4.20 | >11.9 |
| remdesivir | - | - | 2.94 ± 0.67 | 56.5 |
| Compound | SOL Score, kcal/mol | , kcal/mol | EC, µM | SI |
|---|---|---|---|---|
| 9b | −5.76 | −53.4 | 1.04 ± 0.26 | >7.57 |
| 3b | −7.27 | −34.5 | 1.16 ± 0.23 | >86.21 |
| 3c | −7.36 | −36.1 | 2.71 ± 0.91 | >36.86 |
| 6b | −5.57 | −28.6 | 7.62 ± 1.84 | >4.75 |
| 3d | −7.25 | −35.0 | 9.40 ± 1.67 | >10.64 |
| 9c | −6.66 | −47.9 | 19.62 ± 2.94 | >1.62 |
| 6c | −6.03 | −45.1 | 22.40 ± 2.58 | >1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulimov, A.; Ilin, I.; Kutov, D.; Shikhaliev, K.; Shcherbakov, D.; Pyankov, O.; Stolpovskaya, N.; Medvedeva, S.; Sulimov, V. New Chemicals Suppressing SARS-CoV-2 Replication in Cell Culture. Molecules 2022, 27, 5732. https://doi.org/10.3390/molecules27175732
Sulimov A, Ilin I, Kutov D, Shikhaliev K, Shcherbakov D, Pyankov O, Stolpovskaya N, Medvedeva S, Sulimov V. New Chemicals Suppressing SARS-CoV-2 Replication in Cell Culture. Molecules. 2022; 27(17):5732. https://doi.org/10.3390/molecules27175732
Chicago/Turabian StyleSulimov, Alexey, Ivan Ilin, Danil Kutov, Khidmet Shikhaliev, Dmitriy Shcherbakov, Oleg Pyankov, Nadezhda Stolpovskaya, Svetlana Medvedeva, and Vladimir Sulimov. 2022. "New Chemicals Suppressing SARS-CoV-2 Replication in Cell Culture" Molecules 27, no. 17: 5732. https://doi.org/10.3390/molecules27175732
APA StyleSulimov, A., Ilin, I., Kutov, D., Shikhaliev, K., Shcherbakov, D., Pyankov, O., Stolpovskaya, N., Medvedeva, S., & Sulimov, V. (2022). New Chemicals Suppressing SARS-CoV-2 Replication in Cell Culture. Molecules, 27(17), 5732. https://doi.org/10.3390/molecules27175732