
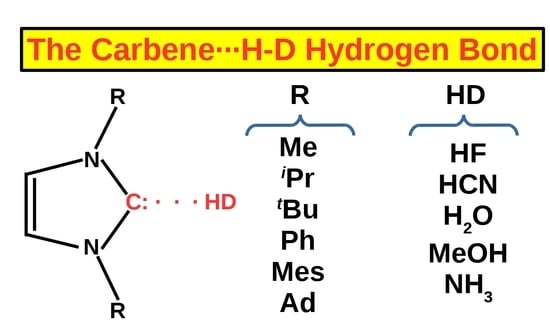
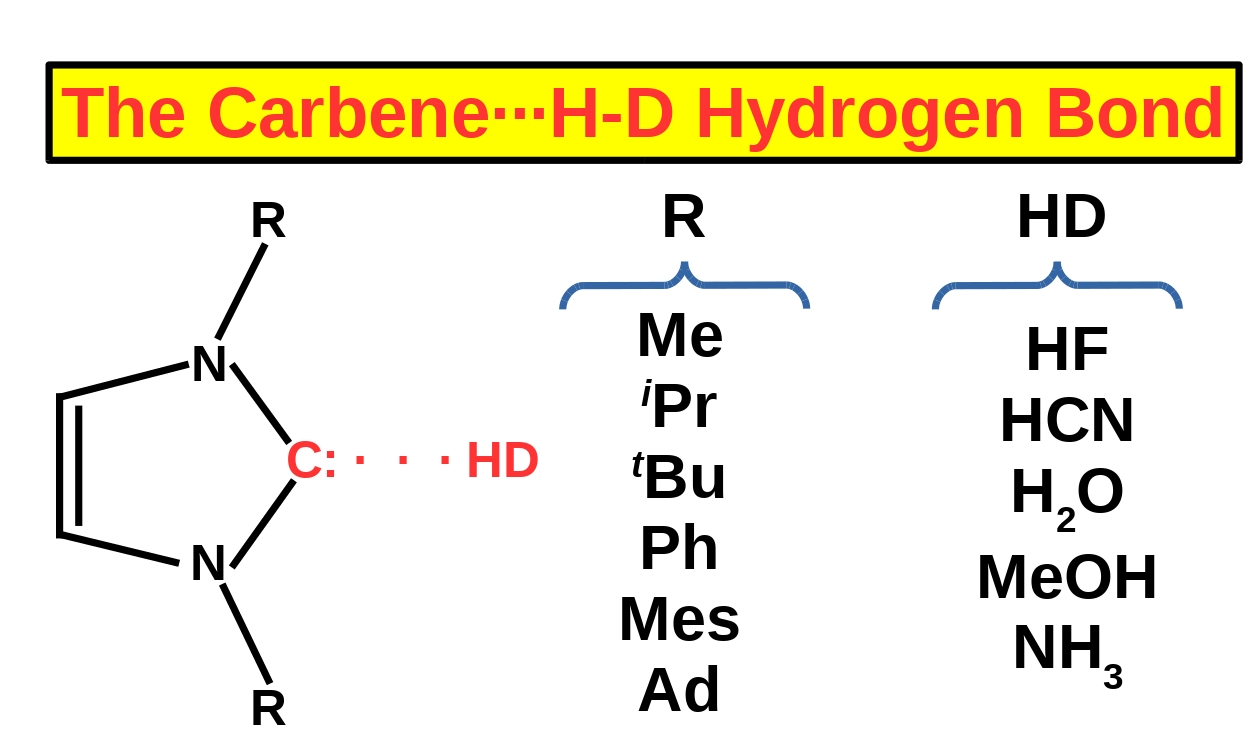
On the Coexistence of the Carbene⋯H-D Hydrogen Bond and Other Accompanying Interactions in Forty Dimers of N-Heterocyclic-Carbenes (I, IMe2, IiPr2, ItBu2, IMes2, IDipp2, IAd2; I = imidazol-2-ylidene) and Some Fundamental Proton Donors (HF, HCN, H2O, MeOH, NH3)


Abstract
:
1. Introduction
2. Theoretical Methods
3. Results and Discussion
3.1. Hydrogen Bonds to Carbenes Observed in the Solid State
3.2. Investigated Systems
3.3. Imidazol-2-ylidene Complexes
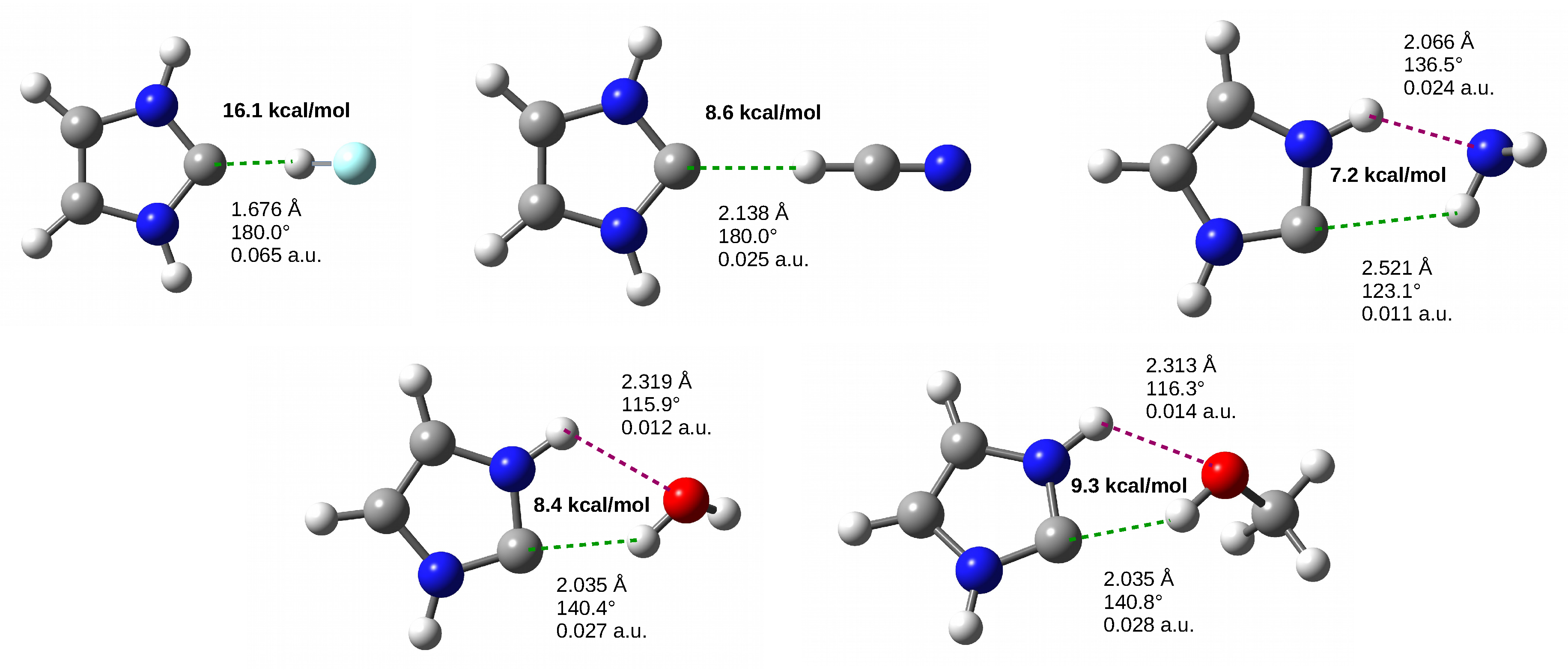
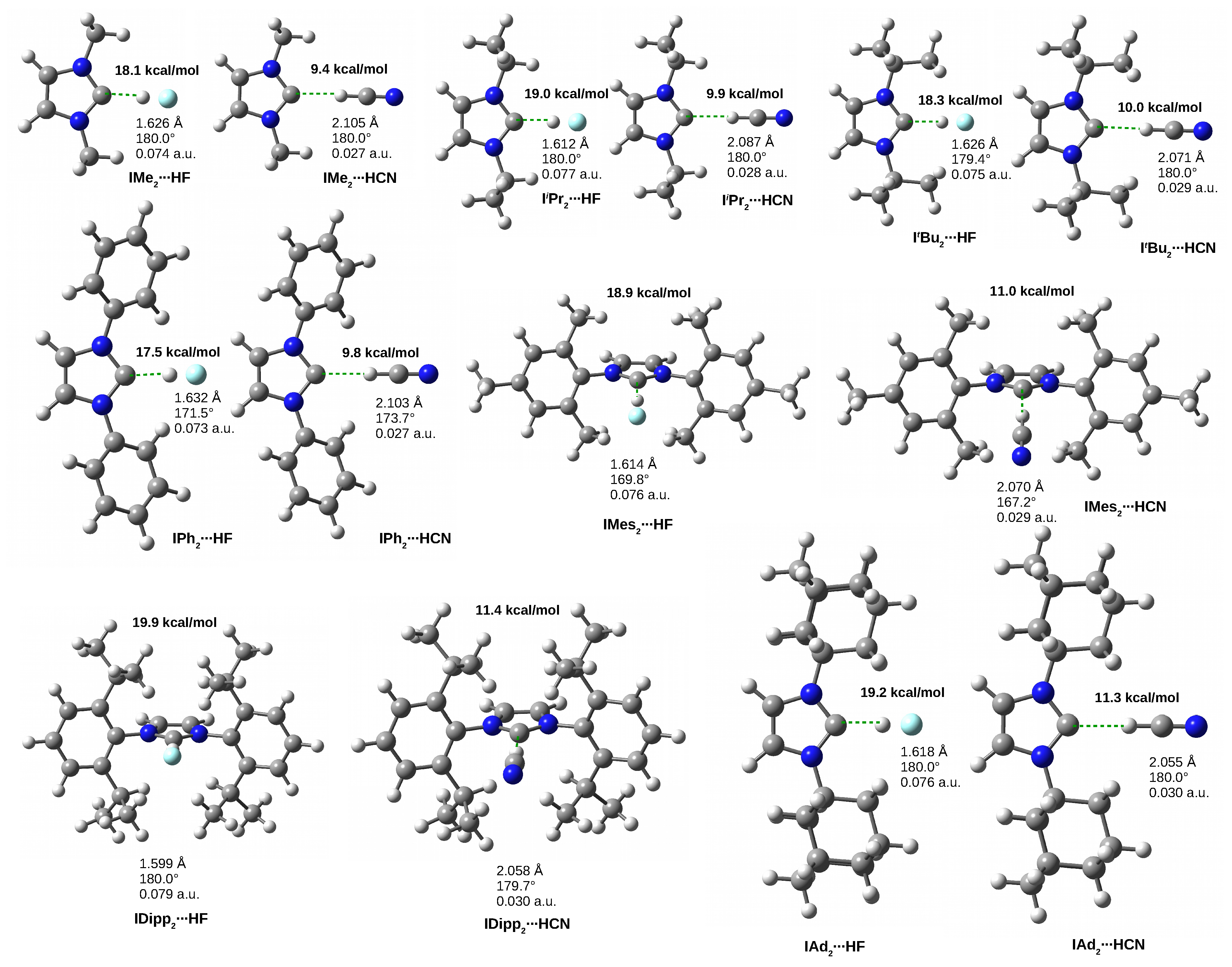
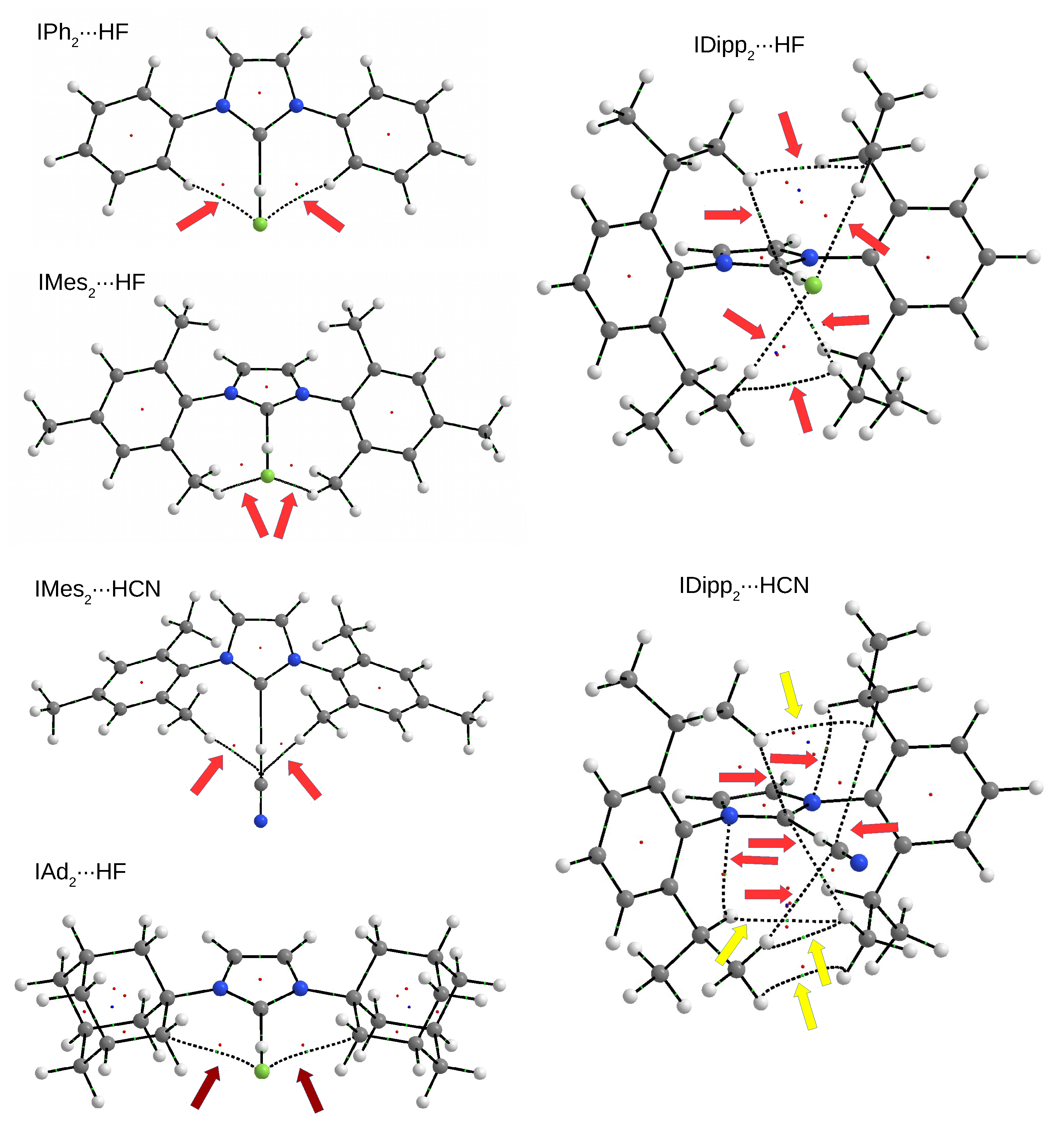
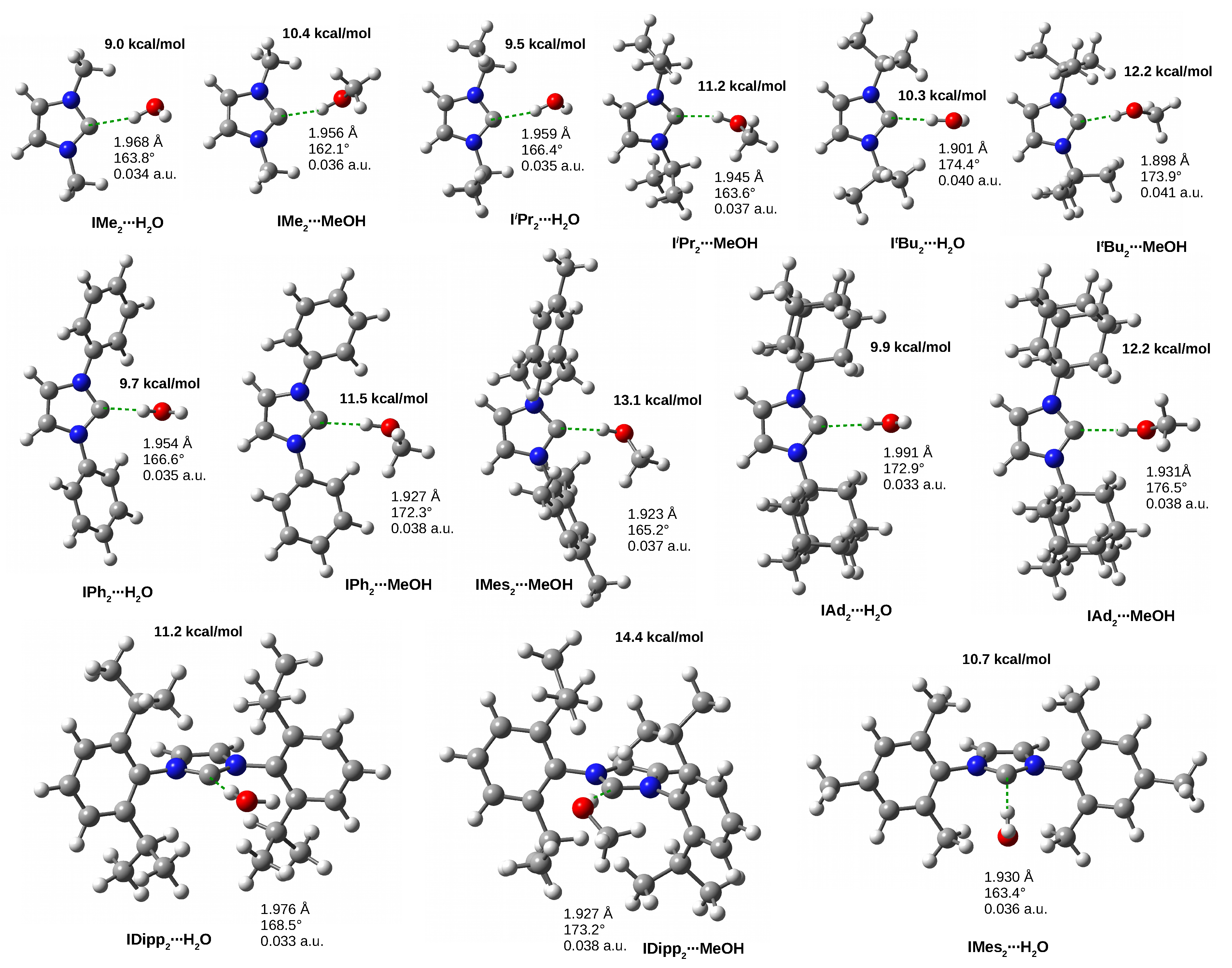
3.4. The IRHD Dimers
3.4.1. The IRHF and IRHCN Dimers
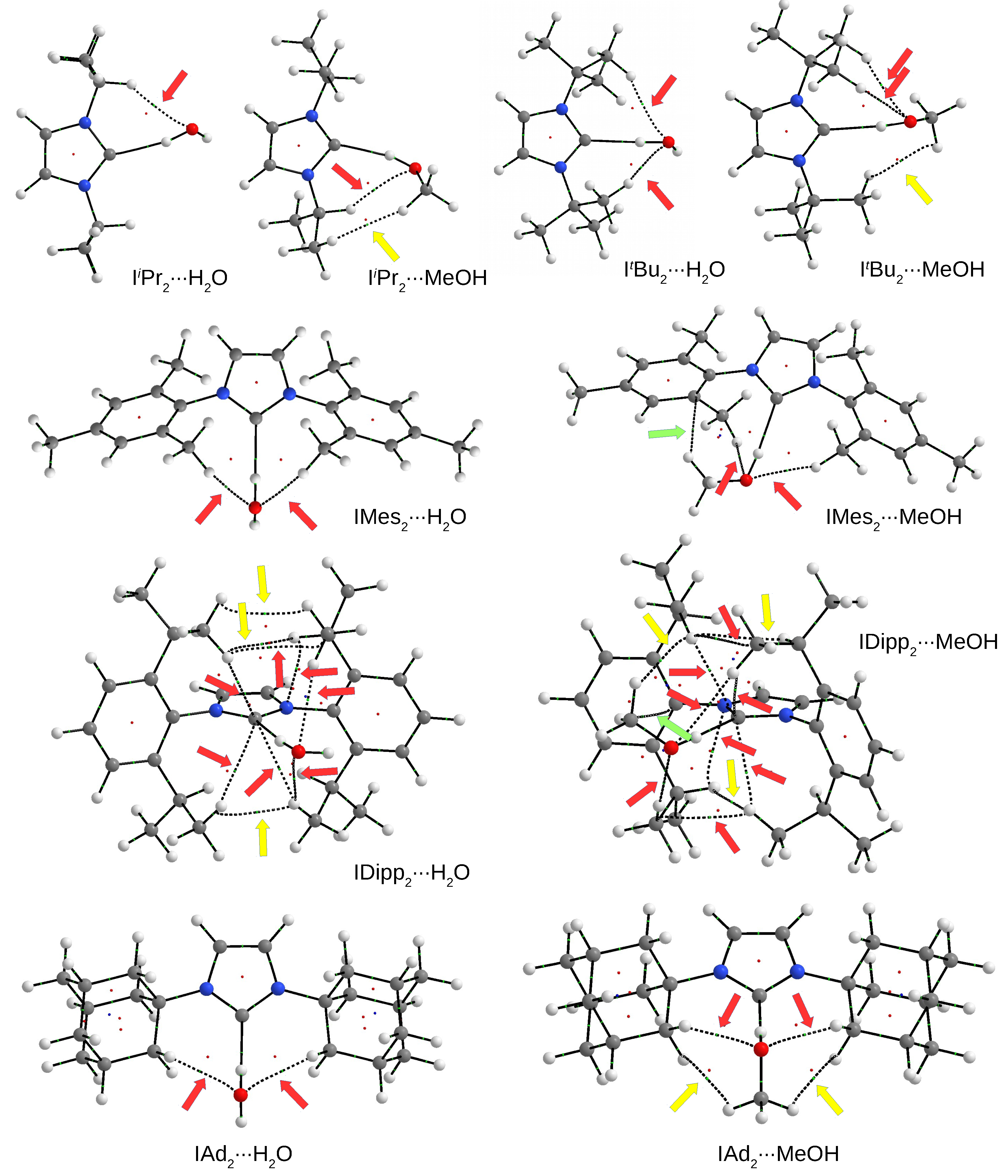
3.4.2. The IRHO and IRMeOH Dimers
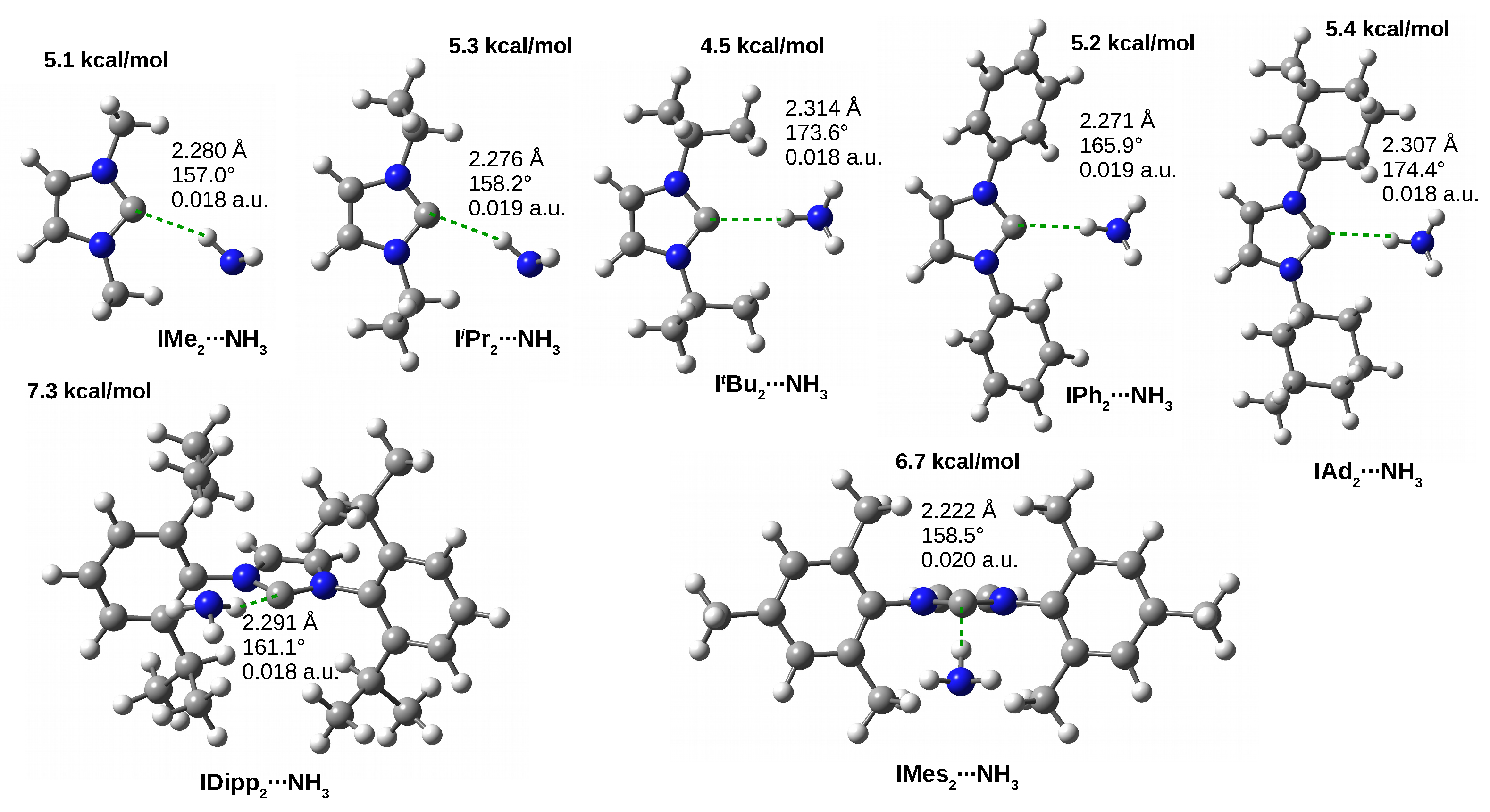
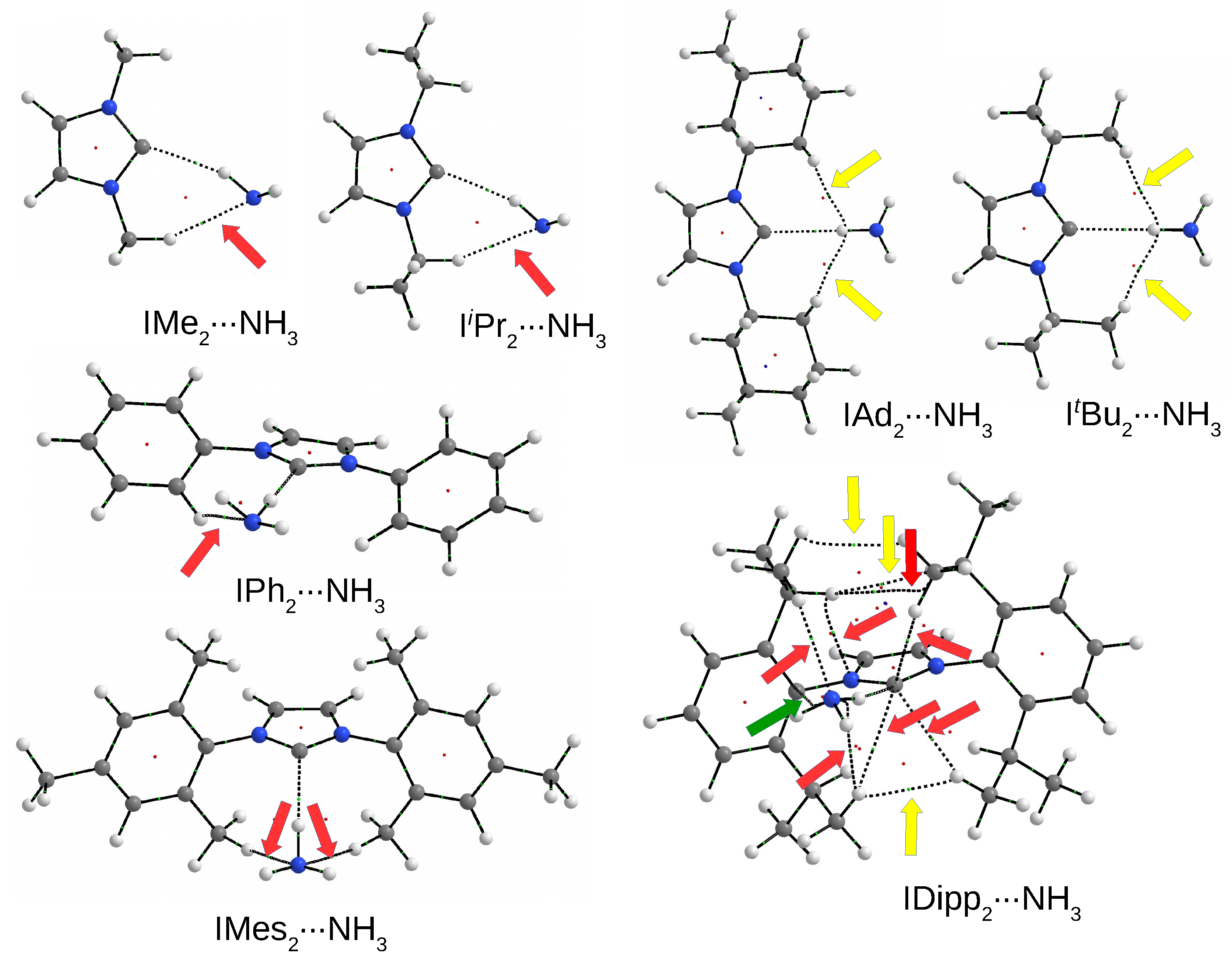
3.4.3. The IRNH Dimers
3.5. Relationship between Dimer Strength and the C⋯H Distance
4. Conclusions
- For a given carbene, dissociation energies of the IRHD dimers increase in the following order: NH< HO < HCN ≤ MeOH ≪ HF.
- For a given HD molecule (HF, HCN, HO, MeOH, or NH), IDipp, i.e., 1,3-bis[2,6-diisopropylphenyl]imidazol-2-ylidene, has been found to form the strongest dimers. This has been attributed to the multiplicity of various interactions accompanying the dominant C⋯H-D hydrogen bond.
- The substitution of hydrogen atoms in both N-H bonds of the imidazol-2-ylidene molecule by Me, Pr, Bu, Ph, Mes, Dipp or Ad groups leads to stronger dimers with HF, HCN, HO or MeOH.
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| NHC | N-heterocyclic carbene |
| I | imidazol-2-ylidene |
| IR | R-substituted imidazol-2-ylidene |
| Me | methyl group |
| Pr | isopropyl group |
| Bu | tert-butyl group |
| Ph | phenyl group |
| Mes | mesityl group |
| Dipp | 2,6-diisopropylphenyl group |
| Ad | adamantyl group |
| QTAIM | quantum theory of atoms in molecules |
References
- Wanzlick, H.W. Aspects of Nucleophilic Carbene Chemistry. Angew. Chem. Int. Ed. Engl. 1962, 1, 75–80. [Google Scholar] [CrossRef]
- Kirmse, W. Carbene Chemistry; Academic Press: Cambridge, MA, USA, 1964. [Google Scholar]
- Hoffmann, R.; Zeiss, G.D.; Van Dine, G.W. The Electronic Structure of Methylenes. J. Am. Chem. Soc. 1968, 90, 1485–1499. [Google Scholar] [CrossRef]
- Gleiter, R.; Hoffmann, R. On Stabilizing a Singlet Methylene. J. Am. Chem. Soc. 1968, 90, 5457–5460. [Google Scholar] [CrossRef]
- Baird, N.C.; Taylor, K.F. Multiplicity of the Ground State and Magnitude of the T1–S0 gap in Substituted Carbenes. J. Am. Chem. Soc. 1978, 100, 1333–1338. [Google Scholar] [CrossRef]
- Harrison, J.F.; Liedtke, R.C.; Liebman, J.F. The Multiplicity of Substituted Acyclic Carbenes and Related Molecules. J. Am. Chem. Soc. 1979, 101, 7162–7168. [Google Scholar] [CrossRef]
- Schoeller, W.W. Electrophilicity and nucleophilicity in singlet carbenes. II. Electrophilic selectivity. Tetrahedron Lett. 1980, 21, 1509–1510. [Google Scholar] [CrossRef]
- Mueller, P.H.; Rondan, N.G.; Houk, K.N.; Harrison, J.F.; Hooper, D.; Willen, B.H.; Liebman, J.F. Carbene Singlet–Triplet Gaps. Linear Correlations with Substituent π Donation. J. Am. Chem. Soc. 1981, 103, 5049–5052. [Google Scholar] [CrossRef]
- Pople, J.A.; Raghavachari, K.; Frisch, M.J.; Binkley, J.S.; Schleyer, P.v.R. Comprehensive Theoretical Study of Isomers and Rearrangement Barriers of Even-Electron Polyatomic Molecules HmABHn (A, B = C, N, O, and F). J. Am. Chem. Soc. 1983, 105, 6389–6398. [Google Scholar] [CrossRef]
- Pople, J.A. A theoretical search for the methylenefluoronium ylide. Chem. Phys. Lett. 1986, 132, 144–146. [Google Scholar] [CrossRef]
- Schubert, U. Advances in Metal Carbene Chemistry; Springer: Dordrecht, The Netherlands, 1989. [Google Scholar]
- Arduengo, A.J., III; Kline, M.; Calabrese, J.C.; Davidson, F. Synthesis of a Reverse Ylide from a Nucleophilic Carbene. J. Am. Chem. Soc. 1991, 113, 9704–9705. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Dias, H.V.R.; Calabrese, J.C.; Davidson, F. A Stable Carbene-Alane Adduct. J. Am. Chem. Soc. 1992, 114, 9724–9725. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Rasika Dias, H.V.; Davidson, F.; Harlow, R.L. Carbene adducts of magnesium and zinc. J. Organomet. Chem. 1993, 462, 13–18. [Google Scholar] [CrossRef]
- Kuhn, N.; Kratz, T.; Henkel, G. A Stable Carbene Iodine Adduct: Secondary Bonding in 1,3-Diethyl-2-iodo-4,5-dimethylimidazolium Iodide. J. Chem. Soc. Chem. Commun. 1993, 1778–1779. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Gamper, S.F.; Tamm, M.; Calabrese, J.C.; Davidson, F.; Craig, H.A. A Bis(carbene)–Proton Complex: Structure of a C–H–C Hydrogen Bond. J. Am. Chem. Soc. 1995, 117, 572–573. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Runte, O.; Artus, G. Synthesis and structure of an ionic beryllium–“carbene” complex. J. Organomet. Chem. 1995, 501, Cl–C4. [Google Scholar] [CrossRef]
- Boehme, C.; Frenking, G. Electronic Structure of Stable Carbenes, Silylenes, and Germylenes. J. Am. Chem. Soc. 1996, 118, 2039–2046. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Carbenes and Silylenes as Hydrogen Bond Acceptors. J. Phys. Chem. 1996, 100, 19367–19370. [Google Scholar] [CrossRef]
- Li, X.-W.; Su, J.; Robinson, G.H. Syntheses and molecular structure of organo-group 13 metal carbene complexes. Chem. Commun. 1996, 2683–2684. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Köcher, C. N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. Engl. 1997, 36, 2162–2187. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Davidson, F.; Krafczyk, R.; Marshall, W.J.; Tamm, M. Adducts of Carbenes with Group II and XII Metallocenes. Organometallics 1998, 17, 3375–3382. [Google Scholar] [CrossRef]
- Hibbs, D.E.; Hursthouse, M.B.; Jones, C.; Smithies, N.A. Synthesis, crystal and molecular structure of the first indium trihydride complex, [InH3CN(Pri)C2Me2N(Pri)]. Chem. Commun. 1998, 869–870. [Google Scholar] [CrossRef]
- Goumri-Magnet, S.; Polishchuck, O.; Gornitzka, H.; Marsden, C.J.; Baceiredo, A.; Bertrand, G. The Electrophilic Behavior of Stable Phosphanylcarbenes Towards Phosphorus Lone Pairs. Angew. Chem. Int. Ed. 1999, 38, 3727–3729. [Google Scholar] [CrossRef]
- Bourissou, D.; Guerret, O.; Gabbaï, F.P.; Bertrand, G. Stable Carbenes. Chem. Rev. 2000, 100, 39–91. [Google Scholar] [CrossRef] [PubMed]
- Bertrande, G. Carbene Chemistry: From Fleeting Intermediates to Powerful Reagents; FontisMedia S.A.: Lausanne, Switzerland; Marcel Dekker, Inc.: New York, NY, USA, 2002. [Google Scholar]
- Merceron, N.; Miqueu, K.; Baceiredo, A.; Bertrand, G. Stable (Amino)(phosphino)carbenes: Difunctional Molecules. J. Am. Chem. Soc. 2002, 124, 6806–6807. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wurst, K.; Buchmeiser, M.R. N-heterocyclic carbene complexes of Zn(II): Synthesis, X-ray structures and reactivity. J. Organomet. Chem. 2004, 689, 2123–2130. [Google Scholar] [CrossRef]
- Moss, R.A.; Platz, M.S.; Jones, M., Jr. (Eds.) Reactive Intermediate Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Scott, N.M.; Nolan, S.P. Stabilization of Organometallic Species Achieved by the Use of N-Heterocyclic Carbene (NHC) Ligands. Eur. J. Inorg. Chem. 2005, 1815–1828. [Google Scholar] [CrossRef]
- Frenking, G.; Solà, M.; Vyboishchikov, S.F. Chemical bonding in transition metal carbene complexes. J. Organomet. Chem. 2005, 690, 6178–6204. [Google Scholar] [CrossRef]
- Garrison, J.C.; Youngs, W.J. Ag(I) N-Heterocyclic Carbene Complexes: Synthesis, Structure, and Application. Chem. Rev. 2005, 105, 3978–4008. [Google Scholar] [CrossRef]
- Nolan, S.P. N-Heterocyclic Carbenes in Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Carey, F.A.; Sundberg, R.J. Carbenes, Part B: Reactions and Synthesis. Advanced Organic Chemistry; Springer: New York, NY, USA, 2007. [Google Scholar]
- Díez-González, S.; Nolan, S.P. Stereoelectronic parameters associated with N-heterocyclic carbene (NHC) ligands: A quest for understanding. Coord. Chem. Rev. 2007, 251, 874–883. [Google Scholar] [CrossRef]
- Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Understanding the M–(NHC) (NHC = N-heterocyclic carbene) bond. Coord. Chem. Rev. 2009, 253, 687–703. [Google Scholar] [CrossRef]
- de Frémont, P.; Marion, N.; Nolan, S.P. Carbenes: Synthesis, properties, and organometallic chemistry. Coord. Chem. Rev. 2009, 253, 862–892. [Google Scholar] [CrossRef]
- Jabłoński, M.; Palusiak, M. Divalent carbon atom as the proton acceptor in hydrogen bonding. Phys. Chem. Chem. Phys. 2009, 11, 5711–5719. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, H.; Liu, Z.; Li, W.; Cheng, J.; Gong, B.; Sun, J. Ab Initio Study of Lithium-Bonded Complexes with Carbene as an Electron Donor. J. Phys. Chem. A 2009, 113, 14156–14160. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, M.; Hill, M.S.; MacDougall, D.J.; Mahon, M.F. A Hydride-Rich Magnesium Cluster. Angew. Chem. Int. Ed. 2009, 48, 4013–4016. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Casely, I.J.; Turner, Z.R.; Bellabarba, R.; Tooze, R.B. Magnesium and zinc complexes of functionalised, saturated N-heterocyclic carbene ligands: Carbene lability and functionalisation, and lactide polymerisation catalysis. Dalton Trans. 2009, 35, 7236–7247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Robinson, G.H. Unique homonuclear multiple bonding in main group compounds. Chem. Commun. 2009, 5201–5213. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Y.; Liu, Z.; Li, W.; Cheng, J.; Gong, B.; Sun, J. An unconventional halogen bond with carbene as an electron donor: An ab initio study. Chem. Phys. Lett. 2009, 469, 48–51. [Google Scholar] [CrossRef]
- Hindi, K.M.; Panzner, M.J.; Tessier, C.A.; Cannon, C.L.; Youngs, W.J. The Medicinal Applications of Imidazolium Carbene–Metal Complexes. Chem. Rev. 2009, 109, 3859–3884. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, Y.; Abraham, M.Y.; Wei, P.; Schaefer, H.F., III; Schleyer, P.v.R.; Robinson, G.H. A Viable Anionic N-Heterocyclic Dicarbene. J. Am. Chem. Soc. 2010, 132, 14370–14372. [Google Scholar] [CrossRef]
- Li, Z.-F.; Yang, S.; Li, H.-X. Theoretical prediction characters of unconventional weak bond with carbene as electron donors and Li-Y (Y = OH, H, F, NC and CN) as electron acceptors. J. Mol. Struct. THEOCHEM 2010, 952, 56–60. [Google Scholar]
- Wang, Y.; Xie, Y.; Abraham, M.Y.; Gilliard, R.J., Jr.; Wei, P.; Schaefer, H.F., III; Schleyer, P.v.R.; Robinson, G.H. Carbene-Stabilized Parent Phosphinidene. Organometallics 2010, 29, 4778–4780. [Google Scholar] [CrossRef]
- Abraham, M.Y.; Wang, Y.; Xie, Y.; Wei, P.; Schaefer, H.F., III; Schleyer, P.v.R.; Robinson, G.H. Carbene Stabilization of Diarsenic: From Hypervalency to Allotropy. Chem. Eur. J. 2010, 16, 432–435. [Google Scholar] [CrossRef]
- Mercs, L.; Albrecht, M. Beyond catalysis: N-Heterocyclic Carbene Complexes as Components for Medicinal, Luminescent, and Functional Materials Applications. Chem. Soc. Rev. 2010, 39, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.S.; Bharatam, P.V. Divalent N(I) Compounds with Two Lone Pairs on Nitrogen. J. Phys. Chem. A 2011, 115, 7645–7655. [Google Scholar] [CrossRef] [PubMed]
- Giffin, N.A.; Makramalla, M.; Hendsbee, A.D.; Robertson, K.N.; Sherren, C.; Pye, C.C.; Masuda, J.D.; Clyburne, J.A.C. Anhydrous TEMPO-H: Reactions of a good hydrogen atom donor with low-valent carbon centres. Org. Biomol. Chem. 2011, 9, 3672–3680. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Feng, D.; Sun, Y.; Hao, J.; Cai, Z. Theoretical Investigations on the Weak Nonbonded C=S⋯CH2 Interactions: Chalcogen-Bonded Complexes With Singlet Carbene as an Electron Donor. Int. J. Quant. Chem. 2011, 111, 3881–3887. [Google Scholar] [CrossRef]
- Gilliard, R.J., Jr.; Abraham, M.Y.; Wang, Y.; Wei, P.; Xie, Y.; Quillian, B.; Schaefer, H.F., III; Schleyer, P.v.R.; Robinson, G.H. Carbene-Stabilized Beryllium Borohydride. J. Am. Chem. Soc. 2012, 134, 9953–9955. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Hill, M.S.; Kociok-Köhn, G.; MacDougall, D.J.; Mahon, M.F. Beryllium-Induced C–N Bond Activation and Ring Opening of an N-Heterocyclic Carbene. Angew. Chem. Int. Ed. 2012, 51, 2098–2100. [Google Scholar] [CrossRef]
- Moss, R.A.; Doyle, M.P. Contemporary Carbene Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Gerbig, D.; Ley, D. Computational methods for contemporary carbene chemistry. WIREs Comput. Mol. Sci. 2013, 3, 242–272. [Google Scholar] [CrossRef]
- Nelson, D.J.; Nolan, S.P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 6723–6753. [Google Scholar] [CrossRef]
- Samanta, R.C.; De Sarkar, S.; Fröhlich, R.; Grimme, S.; Studer, A. N-Heterocyclic carbene (NHC) catalyzed chemoselective acylation of alcohols in the presence of amines with various acylating reagents. Chem. Sci. 2013, 4, 2177–2184. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadirad, N. Insights into the strength and nature of carbene⋯halogen bond interactions: A theoretical perspective. J. Mol. Model. 2013, 19, 2559–2566. [Google Scholar] [CrossRef] [PubMed]
- Budagumpi, S.; Endud, S. Group XII Metal–N-Heterocyclic Carbene Complexes: Synthesis, Structural Diversity, Intramolecular Interactions, and Applications. Organometallics 2013, 32, 1537–1562. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Bellemin-Laponnaz, S.; Dagorne, S. Group 1 and 2 and Early Transition Metal Complexes Bearing N-Heterocyclic Carbene Ligands: Coordination Chemistry, Reactivity, and Applications. Chem. Rev. 2014, 114, 8747–8774. [Google Scholar] [CrossRef]
- Visbal, R.; Concepción Gimeno, M. N-Heterocyclic Carbene Metal Complexes: Photoluminescence and Applications. Chem. Soc. Rev. 2014, 43, 3551–3574. [Google Scholar] [CrossRef]
- Santoro, O.; Nahra, F.; Cordes, D.B.; Slawin, A.M.Z.; Nolan, S.P.; Cazin, C.S.J. Synthesis, characterization and catalytic activity of stable [(NHC)H][ZnXY2] (NHC = N-Heterocyclic carbene, X, Y = Cl, Br) species. J. Mol. Catal. 2016, 423, 85–91. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Carbon–Carbon Bonding between Nitrogen Heterocyclic Carbenes and CO2. J. Phys. Chem. A 2017, 121, 8136–8146. [Google Scholar] [CrossRef]
- Liu, M.; Li, Q.; Li, W.; Cheng, J. Carbene tetrel-bonded complexes. Struct. Chem. 2017, 28, 823–831. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Sabouri, A. Carbene–aerogen bonds: An ab initio study. Mol. Phys. 2017, 115, 971–980. [Google Scholar] [CrossRef]
- Moss, R.A.; Wang, L.; Cang, H.; Krogh-Jespersen, K. Extremely reactive carbenes: Electrophiles and nucleophiles. J. Phys. Org. Chem. 2017, 30, e3555. [Google Scholar] [CrossRef]
- Nesterov, V.; Reiter, D.; Bag, P.; Frisch, P.; Holzner, R.; Porzelt, A.; Inoue, S. NHCs in Main Group Chemistry. Chem. Rev. 2018, 118, 9678–9842. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J. A LFER analysis of the singlet-triplet gap in a series of sixty-six carbenes. Chem. Phys. Lett. 2018, 691, 33–36. [Google Scholar] [CrossRef]
- Jabłoński, M. The first theoretical proof of the existence of a hydride-carbene bond. Chem. Phys. Lett. 2018, 710, 78–83. [Google Scholar] [CrossRef]
- Dagorne, S. Recent Developments on N-Heterocyclic Carbene Supported Zinc Complexes: Synthesis and Use in Catalysis. Synthesis 2018, 50, 3662–3670. [Google Scholar] [CrossRef]
- Walley, J.E.; Wong, Y.-O.; Freeman, L.A.; Dickie, D.A.; Gilliard, R.J., Jr. N-Heterocyclic Carbene-Supported Aryl- and Alk- oxides of Beryllium and Magnesium. Catalysts 2019, 9, 934. [Google Scholar] [CrossRef]
- Jabłoński, M. In search for a hydride-carbene bond. J. Phys. Org. Chem. 2019, 32, e3949. [Google Scholar] [CrossRef]
- Yourdkhani, S.; Jabłoński, M. Physical nature of silane⋯carbene dimers revealed by state-of-the-art ab initio calculations. J. Comput. Chem. 2019, 40, 2643–2652. [Google Scholar] [CrossRef]
- Procter, R.J.; Uzelac, M.; Cid, J.; Rushworth, P.J.; Ingleson, M.J. Low-Coordinate NHC–Zinc Hydride Complexes Catalyze Alkyne C–H Borylation and Hydroboration Using Pinacolborane. ACS Catal. 2019, 9, 5760–5771. [Google Scholar] [CrossRef]
- Dzieszkowski, K.; Barańska, I.; Mroczyńska, K.; Słotwiński, M.; Rafiński, Z. Organocatalytic Name Reactions Enabled by NHCs. Materials 2020, 13, 3574. [Google Scholar] [CrossRef]
- Specklin, D.; Fliedel, C.; Dagorne, S. Recent Representative Advances on the Synthesis and Reactivity of N-Heterocyclic-Carbene-Supported Zinc Complexes. Chem. Rec. 2021, 21, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. Study of Beryllium, Magnesium, and Spodium Bonds to Carbenes and Carbodiphosphoranes. Molecules 2021, 26, 2275. [Google Scholar] [CrossRef]
- Jabłoński, M. Theoretical Study of N-Heterocyclic-Carbene–ZnX2 (X = H, Me, Et) Complexes. Materials 2021, 14, 6147. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: New York, NY, USA, 1960. [Google Scholar]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; W.H. Freeman & Co.: San Francisco, CA, USA, 1960. [Google Scholar]
- Hamilton, W.C.; Ibers, J.A. Hydrogen Bonding in Solids; W. A. Benjamin: New York, NY, USA, 1968. [Google Scholar]
- Vinogradov, S.N.; Linnell, R.H. Hydrogen Bonding; Van Nostrand-Reinhold: Princeton, NJ, USA, 1971. [Google Scholar]
- Schuster, P.; Zundel, G.; Sandorfy, C. (Eds.) The Hydrogen Bond. Recent Developments in Theory and Experiments; North Holland: Amsterdam, The Netherlands, 1976; Volumes I–III. [Google Scholar]
- Schuster, P. Intermolecular Interactions: From Diatomics to Biopolymers; Pullman, B., Ed.; John Wiley: New York, NY, USA, 1978. [Google Scholar]
- Hobza, P.; Zahradník, R. Weak Intermolecular Interactions in Chemistry and Biology; Academia: Prague, Czech Republic, 1980. [Google Scholar]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Hadži, D. (Ed.) Theoretical Treatments of Hydrogen Bonding; John Wiley: Chichester, UK, 1997. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Scheiner, S. (Ed.) Molecular Interactions. From van der Waals to Strongly Bound Complexes; Wiley: Chichester, UK, 1997. [Google Scholar]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Grabowski, S.J. (Ed.) Hydrogen Bonding—New Insights; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Maréchal, Y. The Hydrogen Bond and the Water Molecule; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond. Outline of a Comprehensive Hydrogen Bond Theory; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Scheiner, S. Evaluation of DFT Methods to Study Reactions of Benzene With OH Radical. Int. J. Quant. Chem. 2012, 112, 1879–1886. [Google Scholar] [CrossRef]
- Li, Q.-Z.; Li, H.-B. Hydrogen Bonds Involving Radical Species. In Noncovalent Forces; Scheiner, S., Ed.; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion correction. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Curtiss, L.A.; McGrath, M.P.; Blandeau, J.-P.; Davis, N.E.; Binning, R.C., Jr.; Radom, L. Extension of Gaussian-2 theory to molecules containing third-row atoms Ga–Kr. J. Chem. Phys. 1995, 103, 6104–6113. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.v.R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 19, 2315–2372. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules. An Introduction; Longman: Singapore, 2000. [Google Scholar]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Keith, T.A. AIMAll (Version 15.05.18); TK Gristmill Software: Overland Park, KS, USA, 2015; Available online: aim.tkgristmill.com (accessed on 26 July 2022).
- Gatti, C. Chemical bonding in crystals: New directions. Z. Kristallogr. 2005, 220, 399–457. [Google Scholar] [CrossRef]
- Gatti, C.; Macchi, P. (Eds.) A Guided Tour Through Modern Charge-Density Analysis; Springer: Dordrecht, The Netherlands, 2011. [Google Scholar]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Bläser, D.; Boese, R.; Göhner, M.; Herrmann, F.; Kuhn, N.; Ströbele, M. 2,3-Dihydro-1,3,4,5-tetraisopropylimidazol-2-yliden / 2,3-Dihydro-1,3,4,5-tetraisopropylimidazol-2-ylidene. Z. Naturforsch. B 2014, 69, 71–76. [Google Scholar] [CrossRef]
- Cowan, J.A.; Clyburne, J.A.C.; Davidson, M.G.; Harris, R.L.W.; Howard, J.A.K.; Küpper, P.; Leech, M.A.; Richards, S.P. On the Interaction between N-Heterocyclic Carbenes and Organic Acids: Structural Authentication of the First N–H⋯C Hydrogen Bond and Remarkably Short C–H⋯O Interactions. Angew. Chem., Int. Ed. 2002, 41, 1432–1434. [Google Scholar] [CrossRef]
- Kinney, Z.J.; Rheingold, A.L.; Protasiewicz, J.D. Preferential N–H⋯:C< hydrogen bonding involving ditopic NH-containing systems and N-heterocyclic carbenes. RSC Adv. 2020, 10, 42164–42171. [Google Scholar]
- Li, C.-Y.; Kuo, Y.-Y.; Tsai, J.-H.; Yap, G.P.A.; Ong, T.-G. Amine-Linked N-Heterocyclic Carbenes: The Importance of an Pendant Free-Amine Auxiliary in Assisting the Catalytic Reaction. Chem. Asian J. 2011, 6, 1520–1524. [Google Scholar] [CrossRef]
- Movassaghi, M.; Schmidt, M.A. N-Heterocyclic Carbene-Catalyzed Amidation of Unactivated Esters with Amino Alcohols. Org. Lett. 2005, 7, 2453–2456. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Moneuse, R.; Petit, J.; Pavard, P.-A.; Dardun, V.; Rivat, M.; Schiltz, P.; Solari, M.; Jeanneau, E.; Veyre, L.; et al. Early/Late Heterobimetallic Tantalum/Rhodium Species Assembled Through a Novel Bifunctional NHC-OH Ligand. Chem. Eur. J. 2018, 24, 4361–4370. [Google Scholar] [CrossRef]
- Guo, R.; Huang, X.; Zhao, M.; Lei, Y.; Ke, Z.; Kong, L. Bifurcated Hydrogen-Bond-Stabilized Boron Analogues of Carboxylic Acids. Inorg. Chem. 2019, 58, 13370–13375. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. Does the Presence of a Bond Path Really Mean Interatomic Stabilization? The Case of the Ng@Superphane (Ng = He, Ne, Ar, and Kr) Endohedral Complexes. Symmetry 2021, 13, 2241. [Google Scholar] [CrossRef]
- Jabłoński, M. Endo- and exohedral complexes of superphane with cations. J. Comput. Chem. 2022, 43, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. The physical nature of the ultrashort spike–ring interaction in iron maiden molecules. J. Comput. Chem. 2022, 43, 1206–1220. [Google Scholar] [CrossRef] [PubMed]
- Mallinson, P.R.; Smith, G.T.; Wilson, C.C.; Grech, E.; Wozniak, K. From Weak Interactions to Covalent Bonds: A Continuum in the Complexes of 1,8-Bis(dimethylamino)naphthalene. J. Am. Chem. Soc. 2003, 125, 4259–4270. [Google Scholar] [CrossRef]
- Dominiak, P.M.; Makal, A.; Mallinson, P.R.; Trzcinska, K.; Eilmes, J.; Grech, E.; Chruszcz, M.; Minor, W.; Woźniak, K. Continua of Interactions between Pairs of Atoms in Molecular Crystals. Chem. Eur. J. 2006, 12, 1941–1949. [Google Scholar] [CrossRef]
- Jabłoński, M.; Palusiak, M. Basis Set and Method Dependence in Atoms in Molecules Calculations. J. Phys. Chem. A 2010, 114, 2240–2244. [Google Scholar] [CrossRef]
- Jabłoński, M.; Solà, M. Influence of Confinement on Hydrogen Bond Energy. The Case of the FH⋯NCH Dimer. J. Phys. Chem. A 2010, 114, 10253–10260. [Google Scholar] [CrossRef]












{kind=link}
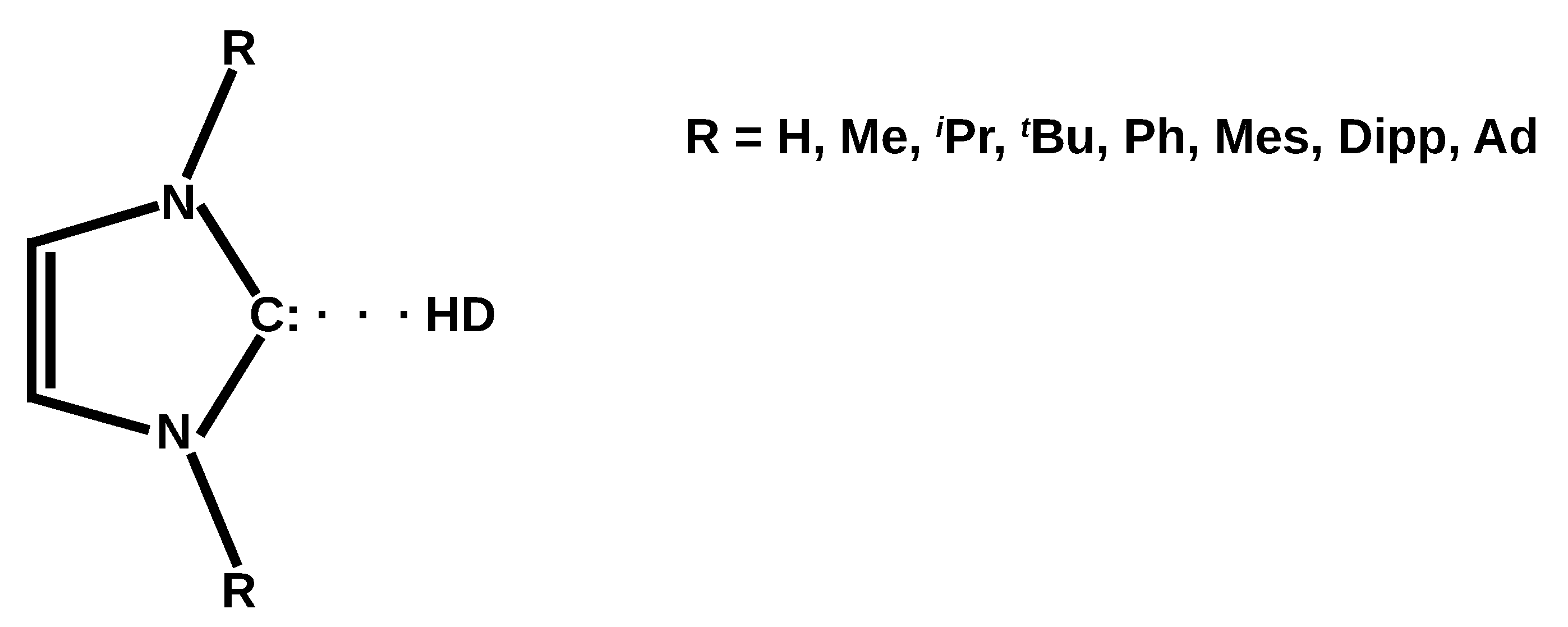{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HD | Property | Carbene | |||||||
|---|---|---|---|---|---|---|---|---|---|
| I | IMe | IPr | IBu | IPh | IMes | IDipp | IAd | ||
| HF | 16.1 | 18.1 | 19.0 | 18.3 | 17.5 | 18.9 | 19.9 | 19.2 | |
| 0.065 | 0.074 | 0.077 | 0.075 | 0.073 | 0.076 | 0.079 | 0.076 | ||
| −13.7 | −15.7 | −16.3 | −15.9 | −15.5 | −16.1 | −16.8 | −16.3 | ||
| 1.676 | 1.626 | 1.612 | 1.626 | 1.632 | 1.614 | 1.599 | 1.618 | ||
| 180.0 | 180.0 | 180.0 | 179.4 | 171.5 | 169.8 | 180.0 | 180.0 | ||
| 0.061 | 0.075 | 0.080 | 0.084 | 0.075 | 0.080 | 0.083 | 0.088 | ||
| HCN | 8.6 | 9.4 | 9.9 | 10.0 | 9.8 | 11.0 | 11.4 | 11.3 | |
| 0.025 | 0.027 | 0.028 | 0.029 | 0.027 | 0.029 | 0.030 | 0.030 | ||
| −4.8 | −5.3 | −5.5 | −5.8 | −5.2 | −5.6 | −5.9 | −6.0 | ||
| 2.138 | 2.105 | 2.087 | 2.071 | 2.103 | 2.070 | 2.058 | 2.055 | ||
| 180.0 | 180.0 | 180.0 | 180.0 | 173.7 | 167.2 | 179.7 | 180.0 | ||
| 0.029 | 0.033 | 0.035 | 0.038 | 0.031 | 0.034 | 0.035 | 0.040 | ||
| HO | 8.4 | 9.0 | 9.5 | 10.3 | 9.7 | 10.7 | 11.2 | 9.9 | |
| 0.027 | 0.034 | 0.035 | 0.040 | 0.035 | 0.036 | 0.033 | 0.033 | ||
| −5.3 | −6.8 | −7.0 | −8.3 | −7.1 | −7.3 | −6.7 | −6.6 | ||
| 2.035 | 1.968 | 1.959 | 1.901 | 1.954 | 1.930 | 1.976 | 1.991 | ||
| 140.4 | 163.8 | 166.4 | 174.4 | 166.6 | 163.4 | 168.5 | 172.9 | ||
| 0.019 | 0.025 | 0.026 | 0.033 | 0.026 | 0.028 | 0.023 | 0.027 | ||
| MeOH | 9.3 | 10.4 | 11.2 | 12.2 | 11.5 | 13.1 | 14.4 | 12.2 | |
| 0.028 | 0.036 | 0.037 | 0.041 | 0.038 | 0.037 | 0.038 | 0.038 | ||
| −5.5 | −7.2 | −7.4 | −8.4 | −7.6 | −7.6 | −7.7 | −7.6 | ||
| 2.035 | 1.956 | 1.945 | 1.898 | 1.927 | 1.923 | 1.927 | 1.931 | ||
| 140.8 | 162.1 | 163.6 | 173.9 | 172.3 | 165.2 | 173.2 | 176.5 | ||
| 0.018 | 0.025 | 0.026 | 0.032 | 0.026 | 0.026 | 0.026 | 0.030 | ||
| NH | 7.2 | 5.1 | 5.3 | 4.5 | 5.2 | 6.7 | 7.3 | 5.4 | |
| 0.011 | 0.018 | 0.019 | 0.018 | 0.019 | 0.020 | 0.018 | 0.018 | ||
| −1.6 | −3.3 | −3.4 | −3.2 | −3.5 | −3.7 | −3.2 | −3.2 | ||
| 2.521 | 2.280 | 2.276 | 2.314 | 2.271 | 2.222 | 2.291 | 2.307 | ||
| 123.1 | 157.0 | 158.2 | 173.6 | 165.9 | 158.5 | 161.1 | 174.4 | ||
| 0.007 | 0.011 | 0.011 | 0.011 | 0.010 | 0.012 | 0.009 | 0.011 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabłoński, M. On the Coexistence of the Carbene⋯H-D Hydrogen Bond and Other Accompanying Interactions in Forty Dimers of N-Heterocyclic-Carbenes (I, IMe2, IiPr2, ItBu2, IMes2, IDipp2, IAd2; I = imidazol-2-ylidene) and Some Fundamental Proton Donors (HF, HCN, H2O, MeOH, NH3). Molecules 2022, 27, 5712. https://doi.org/10.3390/molecules27175712
Jabłoński M. On the Coexistence of the Carbene⋯H-D Hydrogen Bond and Other Accompanying Interactions in Forty Dimers of N-Heterocyclic-Carbenes (I, IMe2, IiPr2, ItBu2, IMes2, IDipp2, IAd2; I = imidazol-2-ylidene) and Some Fundamental Proton Donors (HF, HCN, H2O, MeOH, NH3). Molecules. 2022; 27(17):5712. https://doi.org/10.3390/molecules27175712
Chicago/Turabian StyleJabłoński, Mirosław. 2022. "On the Coexistence of the Carbene⋯H-D Hydrogen Bond and Other Accompanying Interactions in Forty Dimers of N-Heterocyclic-Carbenes (I, IMe2, IiPr2, ItBu2, IMes2, IDipp2, IAd2; I = imidazol-2-ylidene) and Some Fundamental Proton Donors (HF, HCN, H2O, MeOH, NH3)" Molecules 27, no. 17: 5712. https://doi.org/10.3390/molecules27175712
APA StyleJabłoński, M. (2022). On the Coexistence of the Carbene⋯H-D Hydrogen Bond and Other Accompanying Interactions in Forty Dimers of N-Heterocyclic-Carbenes (I, IMe2, IiPr2, ItBu2, IMes2, IDipp2, IAd2; I = imidazol-2-ylidene) and Some Fundamental Proton Donors (HF, HCN, H2O, MeOH, NH3). Molecules, 27(17), 5712. https://doi.org/10.3390/molecules27175712