
Indomethacin: The Interplay between Structural Relaxation, Viscous Flow and Crystal Growth

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Procedure
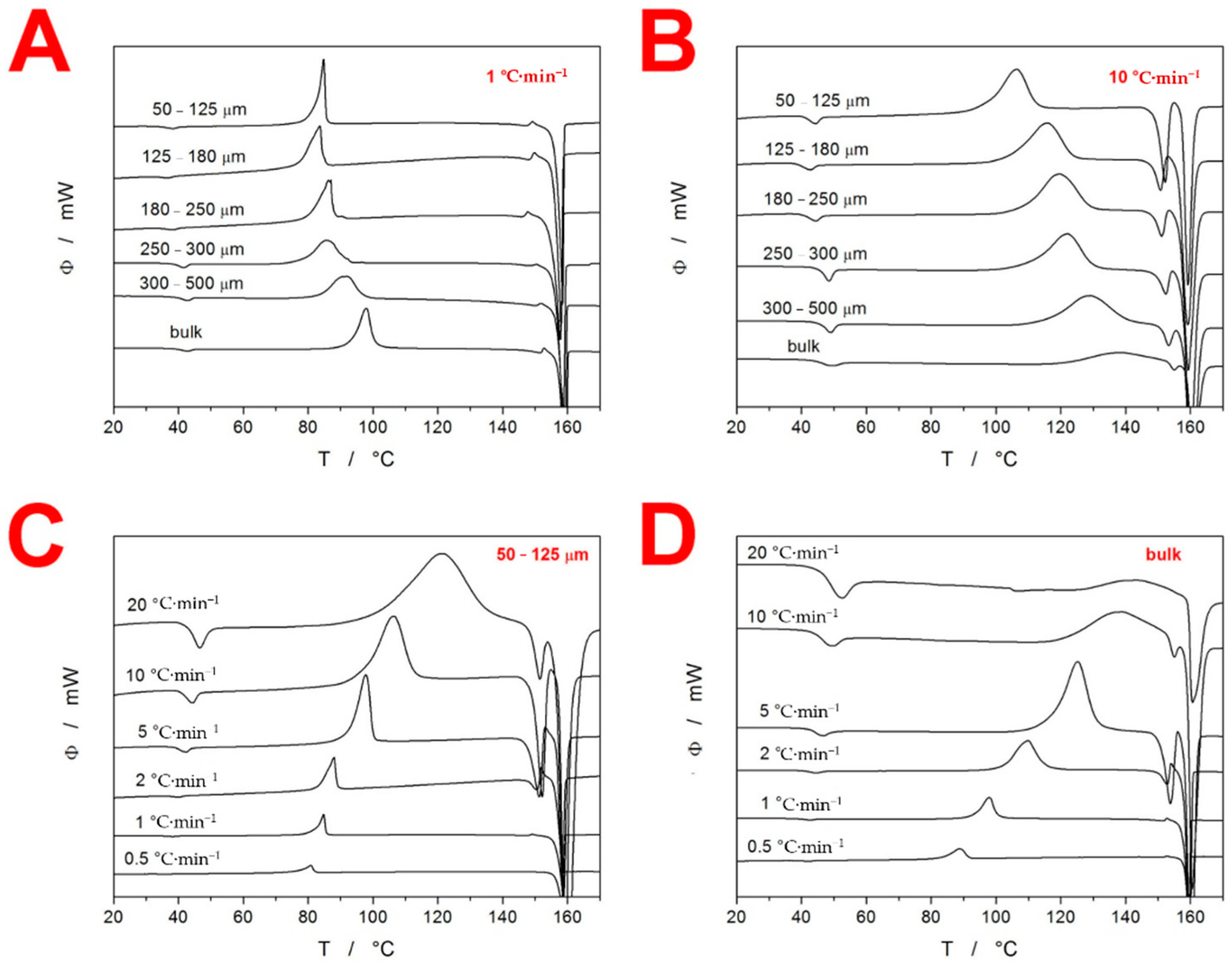
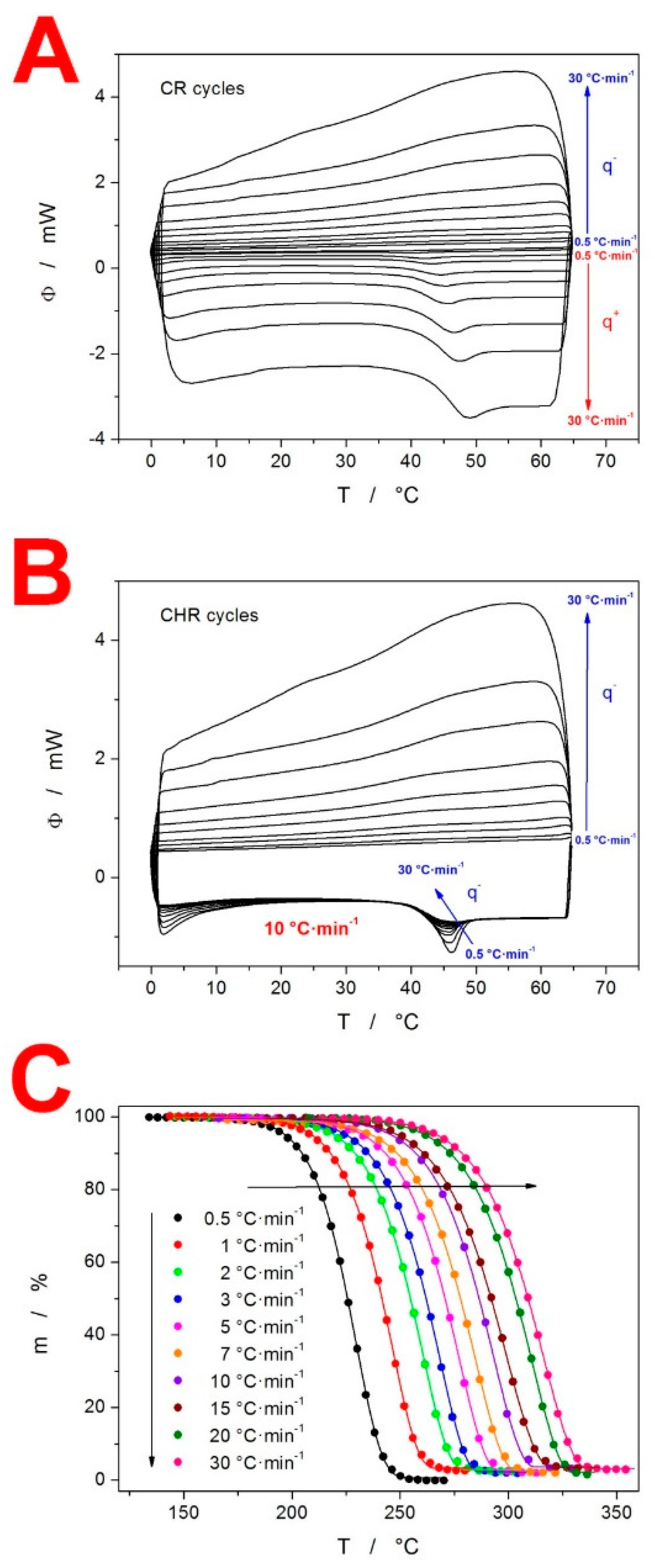
3. Results
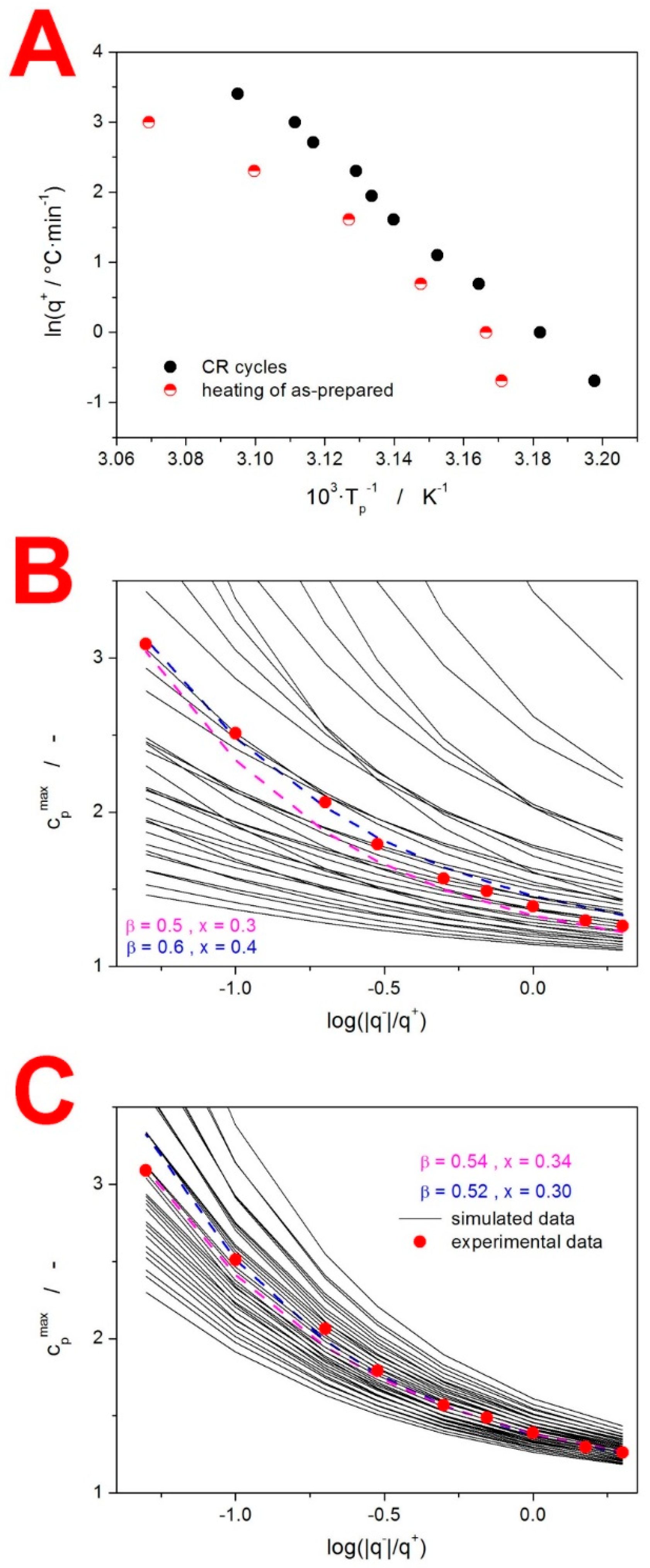
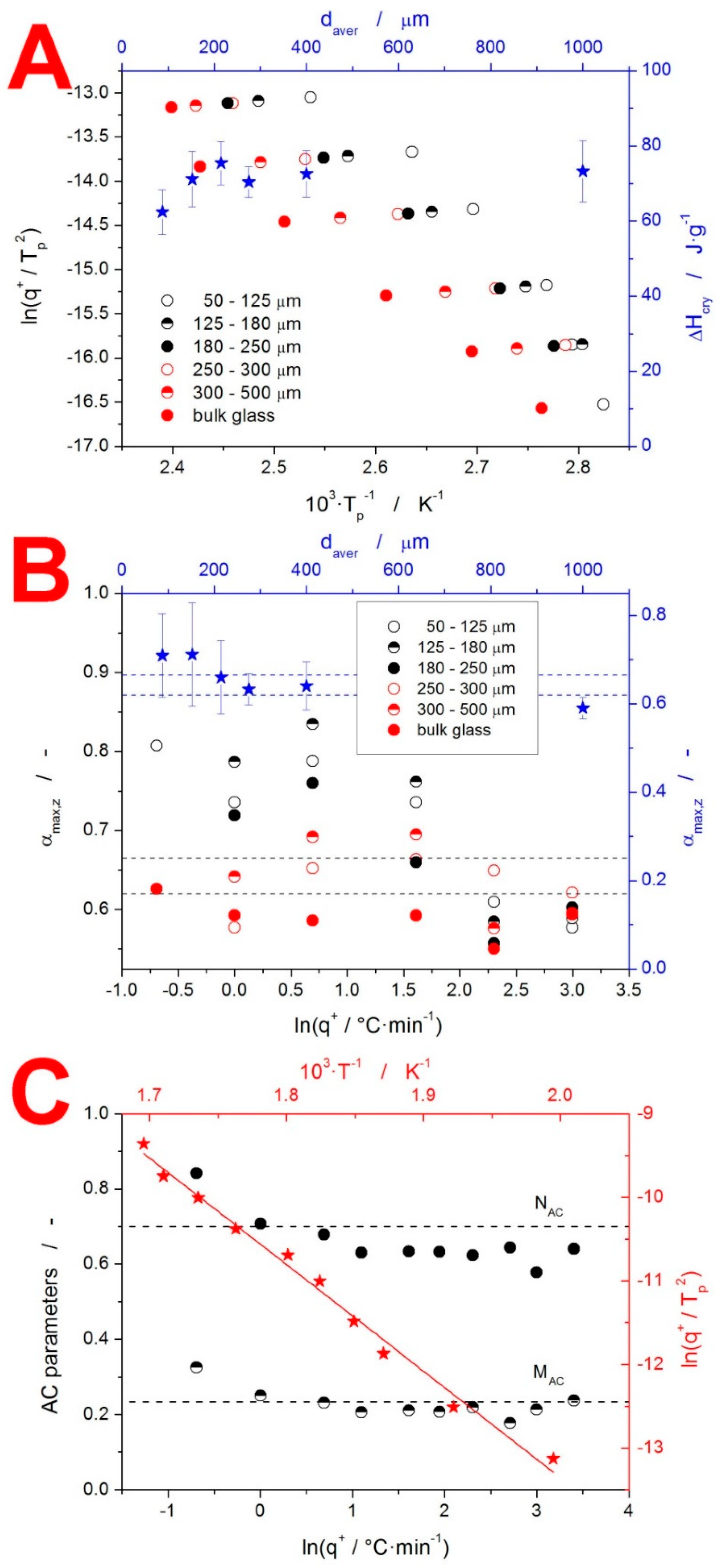
4. Discussion
4.1. Kinetics of the Thermo-Analytically Observed Phenomena
4.1.1. Glass Transition Kinetics
4.1.2. Crystallization Kinetics
4.1.3. Decomposition Kinetics
4.2. Mutual Relationships between the Structural Relaxation, Crystal Growth and Viscous Flow
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Harrigan, M.R.; Tuteja, S.; Neudeck, B.L. Indomethacin in the Management of Elevated Intracranial Pressure: A Review. J. Neurotrauma 1997, 14, 637–650. [Google Scholar] [CrossRef]
- Macones, G.A.; Marder, S.J.; Clothier, B.; Stamilio, D.M. The controversy surrounding indomethacin for tocolysis. Am. J. Obstet. Gynecol. 2001, 184, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S. The Pharmacology of Indomethacin. Headache J. Head Face Pain 2016, 56, 436–446. [Google Scholar] [CrossRef]
- Draper, M.P.; Martell, R.L.; Levy, S.B. Indomethacin-mediated reversal of multidrug resistance and drug efflux in human and murine cell lines overexpressing MRP, but not P-glycoprotein. Br. J. Cancer 1997, 75, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Amici, C.; La Frazia, S.; Brunelli, C.; Balsamo, M.; Angelini, M.; Santoro, M.G. Inhibition of viral protein translation by indomethacin in vesicular stomatitis virus infection: Role of eIF2α kinase PKR. Cell. Microbiol. 2015, 17, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Yalkowsky, S.H.; Dannenfelser, R.M. Aquasol Database of Aqueous Solubility; Version 5; College of Pharmacy, University of Arizona-Tucson: Tucson, AZ, USA, 1992. [Google Scholar]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef]
- Kawakami, K. Crystallization Tendency of Pharmaceutical Glasses: Relevance to Compound Properties, Impact of Formulation Process, and Implications for Design of Amorphous Solid Dispersions. Pharmaceutics 2019, 11, 202. [Google Scholar] [CrossRef]
- Kawakami, K.; Harada, T.; Yoshihashi, Y.; Yonemochi, E.; Terada, K.; Moriyama, H. Correlation between Glass-Forming Ability and Fragility of Pharmaceutical Compounds. J. Phys. Chem. B 2015, 119, 4873–4880. [Google Scholar] [CrossRef]
- A. Einstein. Ann. der Physik 17 (1905) 549.
- Swallen, S.F.; Ediger, M.D. Self-diffusion of the amorphous pharmaceutical indomethacin near Tg. Soft Matter 2011, 7, 10339–10344. [Google Scholar] [CrossRef]
- Ediger, M.D.; Harrowell, P.; Yu, L. Crystal growth kinetics exhibit a fragility-dependent decoupling from viscosity. J. Chem. Phys. 2008, 128, 034709. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, D.; Hasebe, M.; Yu, L. Crystallization of Organic Glasses: How Does Liquid Flow Damage Surface Crystal Growth? Cryst. Growth Des. 2016, 16, 2931–2936. [Google Scholar] [CrossRef]
- Yoshioka, M.; Hancock, B.C.; Zografi, G. Crystallization of Indomethacin from the Amorphous State below and above Its Glass Transition Temperature. J. Pharm. Sci. 1994, 83, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Komaki, H.; Hirai, A.; Goto, S.; Hashimoto, Y.; Uchiro, H.; Terada, H. Decarboxylation of indomethacin induced by heat treatment. Int. J. Pharm. 2018, 545, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, R. Utilization of “q+/q−=const.” DSC cycles for physical aging studies. Eur. Polym. J. 2014, 59, 180–188. [Google Scholar] [CrossRef]
- Svoboda, R. Utilization of constant heating rate DSC cycles for enthalpy relaxation studies and their influenceability by erroneous experimental operations. J. Non-Cryst. Solids 2015, 408, 115–122. [Google Scholar] [CrossRef]
- Angell, C.A. Formation of Glasses from Liquids and Biopolymers. Science 1995, 267, 1924–1935. [Google Scholar] [CrossRef]
- Planinsek, O.; Zadnik, J.; Kunaver, M.; Srcic, S.; Godec, A. Structural evolution of indomethacin particles upon milling: Time-resolved quantification and localization of disordered structure studied by IGC and DSC. J. Pharm. Sci. 2010, 99, 1968–1981. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Favergeon, L.; Koga, N.; Moukhina, E.; Pérez-Maqueda, L.A.; Sbirrazzuoli, N. ICTAC Kinetics Committee recommendations for analysis of multi-step kinetics. Thermochim. Acta 2020, 689, 178597. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of Phase Change. I General Theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of Phase Change. II Transformation-Time Relations for Random Distribution of Nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Granulation, Phase Change, and Microstructure Kinetics of Phase Change. III. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Šesták, J. Science of Heat and Thermophysical Studies: A Generalized Approach to Thermal Analysis; Elsevier: Amsterdam, the Netherlands, 2005. [Google Scholar]
- Tarasov, A.V.; Tikhonov, N.A.; Makarenko, I.V.; Dunaev, A.V.; Arhangelskii, I.V. Non-Isothermal Kinetic Methods; Open Access Edition—Max Planck Research Library for the History and Development and Knowledge: Berlin, Germany, 2013. [Google Scholar]
- Van Duong, T.; Lüdeker, D.; Van Bockstal, P.-J.; De Beer, T.; Van Humbeeck, J.; Van den Mooter, G. Polymorphism of Indomethacin in Semicrystalline Dispersions: Formation, Transformation, and Segregation. Mol. Pharm. 2018, 15, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Surwase, S.A.; Boetker, J.; Saville, D.; Boyd, B.; Gordon, K.; Peltonen, L.; Strachan, C.J. Indomethacin: New Polymorphs of an Old Drug. Mol. Pharm. 2013, 10, 4472–4480. [Google Scholar] [CrossRef] [PubMed]
- Rusu, M.; Olea, M.; Rusu, D. Kinetic study of the indomethacin synthesis and thermal decomposition reactions. J. Pharm. Biomed. Anal. 2000, 24, 19–24. [Google Scholar] [CrossRef]
- Tita, B.; Fulias, A.; Rusu, G.; Tita, D. Thermal behaviour of indomethacin—active substance and tablets. Rev. Chim. 2009, 60, 1210–1215. [Google Scholar]
- Tita, B.; Fulias, A.; Tita, D. Kinetic study of indomethacin under isothermal conditions. Rev. Chim-Buchar. 2010, 61, 1037–1041. [Google Scholar]
- Ueda, H.; Ida, Y.; Kadota, K.; Tozuka, Y. Raman mapping for kinetic analysis of crystallization of amorphous drug based on distributional images. Int. J. Pharm. 2013, 462, 115–122. [Google Scholar] [CrossRef]
- Otsuka, M.; Kato, F.; Matsuda, Y.; Ozaki, Y. Comparative determination of polymorphs of indomethacin in powders and tablets by chemometrical near-infrared spectroscopy and X-ray powder diffractometry. AAPS PharmSciTech 2003, 4, 58–69. [Google Scholar] [CrossRef]
- Tool, A.Q. Relation between inelastic deformability and thermal expansion of glass in its annealing range. J. Am. Ceram. Soc. 1946, 29, 240–253. [Google Scholar] [CrossRef]
- Narayanaswamy, O.S. A Model of Structural Relaxation in Glass. J. Am. Ceram. Soc. 1971, 54, 491–498. [Google Scholar] [CrossRef]
- Moynihan, C.T.; Easteal, A.J.; De Bolt, M.A.; Tucker, J. Dependence of the Fictive Temperature of Glass on Cooling Rate. J. Am. Ceram. Soc. 1976, 59, 12–16. [Google Scholar] [CrossRef]
- Svoboda, R.; Málek, J. Description of enthalpy relaxation dynamics in terms of TNM model. J. Non-Cryst. Solids 2013, 378, 186–195. [Google Scholar] [CrossRef]
- Svoboda, R. Novel equation to determine activation energy of enthalpy relaxation. J. Therm. Anal. 2015, 121, 895–899. [Google Scholar] [CrossRef]
- Svoboda, R.; Čičmanec, P.; Málek, J. Kissinger equation versus glass transition phenomenology. J. Therm. Anal. 2013, 114, 285–293. [Google Scholar] [CrossRef]
- Svoboda, R.; Málek, J. Glass transition in polymers: (In)correct determination of activation energy. Polymer 2013, 54, 1504–1511. [Google Scholar] [CrossRef]
- Hodge, I.M.; Berens, A.R. Effects of annealing and prior history on enthalpy relaxation in glassy polymers. 2. Mathematical modeling. Macromolecules 1982, 15, 762–770. [Google Scholar] [CrossRef]
- Svoboda, R.; Málek, J. Enthalpy relaxation in Ge–Se glassy system. J. Therm. Anal. 2012, 113, 831–842. [Google Scholar] [CrossRef]
- Šesták, J. Thermophysical Properties of Solids, Their Measurements and Theoretical Analysis; Elsevier: Amsterdam, the Netherlands, 1984. [Google Scholar]
- Kissinger, H.E. Reaction Kinetics in Differential Thermal Analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Svoboda, R.; Málek, J. Is the original Kissinger equation obsolete today? J. Therm. Anal. 2013, 115, 1961–1967. [Google Scholar] [CrossRef]
- Málek, J.; Svoboda, R. Kinetic Processes in Amorphous Materials Revealed by Thermal Analysis: Application to Glassy Selenium. Molecules 2019, 24, 2725. [Google Scholar] [CrossRef]
- Rautaniemi, K.; Vuorimaa-Laukkanen, E.; Strachan, C.J.; Laaksonen, T. Crystallization Kinetics of an Amorphous Pharmaceutical Compound Using Fluorescence-Lifetime-Imaging Microscopy. Mol. Pharm. 2018, 15, 1964–1971. [Google Scholar] [CrossRef]
- Díaz-Díaz, A.M.; López-Beceiro, J.; Li, Y.; Cheng, Y.; Artiaga, R. Crystallization kinetics of a commercial poly(lactic acid) based on characteristic crystallization time and optimal crystallization temperature. J. Therm. Anal. 2020, 145, 3125–3132. [Google Scholar] [CrossRef]
- Svoboda, R. Usage of masterplots in kinetic analysis of complex surface/volume crystallization processes in Se-Te glasses. J. Non-Cryst. Solids 2020, 541, 120068. [Google Scholar] [CrossRef]
- Svoboda, R. Crystallization of glasses—When to use the Johnson-Mehl-Avrami kinetics? J. Eur. Ceram. Soc. 2021, 41, 7862–7867. [Google Scholar] [CrossRef]
- Svoboda, R.; Chovanec, J.; Slang, S.; Beneš, L.; Konrád, P. Single-curve multivariate kinetic analysis: Application to the crystallization of commercial Fe-Si-Cr-B amorphous alloys. J. Alloys Compd. 2021, 889, 161672. [Google Scholar] [CrossRef]
- Andronis, V.; Zografi, G. Crystal nucleation and growth of indomethacin polymorphs from the amorphous state. J. Non-Cryst. Solids 2000, 271, 236–248. [Google Scholar] [CrossRef]
- Cohen, M.H.; Turnbull, D. Molecular Transport in Liquids and Glasses. J. Chem. Phys. 1959, 31, 1164–1169. [Google Scholar] [CrossRef]
- Angell, C. Relaxation in liquids, polymers and plastic crystals—strong/fragile patterns and problems. J. Non-Cryst. Solids 1991, 131–133, 13–31. [Google Scholar] [CrossRef]
- Wu, T.; Yu, L. Origin of Enhanced Crystal Growth Kinetics near Tg Probed with Indomethacin Polymorphs. J. Phys. Chem. B 2006, 110, 15694–15699. [Google Scholar] [CrossRef]
- Hancock, B.C.; Dupuis, Y.; Thibert, R. Determination of the viscosity of an amorphous drug using thermomechanical analysis (TMA). Pharm. Res. 1999, 16, 672–675. [Google Scholar] [CrossRef]
- Málek, J.; Svoboda, R.; Pustková, P.; Čičmanec, P. Volume and enthalpy relaxation of a-Se in the glass transition region. J. Non-Cryst. Solids 2009, 355, 264–272. [Google Scholar] [CrossRef]
- Hodge, I. Adam-Gibbs formulation of non-linear enthalpy relaxation. J. Non-Cryst. Solids 1991, 131-133, 435–441. [Google Scholar] [CrossRef]
- Chromčiková, M.; Liška, M. Simple relaxation model of the reversible part of the StepScan® DSC record of glass transition. J. Therm. Anal. 2006, 84, 703–708. [Google Scholar] [CrossRef]
- Mullin, J.W. Crystallization, 4th ed.; Elsevier: Boston, MA, USA, 2001. [Google Scholar]
- Alonzo, D.E.; Zhang, G.G.Z.; Zhou, D.; Gao, Y.; Taylor, L.S. Understanding the Behavior of Amorphous Pharmaceutical Systems during Dissolution. Pharm. Res. 2010, 27, 608–618. [Google Scholar] [CrossRef]
- Lee, A.Y.; Erdemir, D.; Myerson, A.S. Crystal Polymorphism in Chemical Process Development. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, R.; Brandová, D. Crystal growth from mechanically induced defects. J. Therm. Anal. 2016, 127, 799–808. [Google Scholar] [CrossRef]
- Gunn, E.M.; Guzei, I.A.; Yu, L. Does Crystal Density Control Fast Surface Crystal Growth in Glasses? A Study with Polymorphs. Cryst. Growth Des. 2011, 11, 3979–3984. [Google Scholar] [CrossRef]
- Hasebe, M.; Musumeci, D.; Powell, C.T.; Cai, T.; Gunn, E.; Zhu, L.; Yu, L. Fast Surface Crystal Growth on Molecular Glasses and Its Termination by the Onset of Fluidity. J. Phys. Chem. B 2014, 118, 7638–7646. [Google Scholar] [CrossRef]
- Wu, T.; Sun, Y.; Li, N.; de Villiers, M.M.; Yu, L. Inhibiting Surface Crystallization of Amorphous Indomethacin by Nanocoating. Langmuir 2007, 23, 5148–5153. [Google Scholar] [CrossRef]
- Newman, A.; Zografi, G. What We Need to Know about Solid-State Isothermal Crystallization of Organic Molecules from the Amorphous State below the Glass Transition Temperature. Mol. Pharm. 2020, 17, 1761–1777. [Google Scholar] [CrossRef]
- Zhang, W.; Brian, C.W.; Yu, L. Fast Surface Diffusion of Amorphous o-Terphenyl and Its Competition with Viscous Flow in Surface Evolution. J. Phys. Chem. B 2015, 119, 5071–5078. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhu, L.; Kearns, K.L.; Ediger, M.D.; Yu, L. Glasses crystallize rapidly at free surfaces by growing crystals upward. Proc. Natl. Acad. Sci. USA 2011, 108, 5990–5995. [Google Scholar] [CrossRef] [Green Version]
- Romanová, J.; Svoboda, R.; Obadalová, I.; Beneš, L.; Pekárek, T.; Krejčík, L.; Komersová, A. Amorphous Enzalutamide—Non-isothermal recrystallization kinetics and thermal stability. Thermochim. Acta 2018, 665, 134–141. [Google Scholar] [CrossRef]
- Svoboda, R.; Romanová, J.; Šlang, S.; Obadalová, I.; Komersová, A. Influence of particle size and manufacturing conditions on the recrystallization of amorphous Enzalutamide. Eur. J. Pharm. Sci. 2020, 153, 105468. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, R.; Prikryl, J.; Provotorov, P.; Kolobov, A.V.; Krbal, M. Next-gen approach to the combined micro/macro-scopic measurements of crystal growth in chalcogenide thin films: The case of Se90Te10. J. Alloys Compd. 2022, 923, 166389. [Google Scholar] [CrossRef]
- Descamps, M.; Dudognon, E. Crystallization from the Amorphous State: Nucleation–Growth Decoupling, Polymorphism Interplay, and the Role of Interfaces. J. Pharm. Sci. 2014, 103, 2615–2628. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Cheng, J.; Chen, H.; Xu, J.; Liu, Z.; Shi, Q.; Zhang, C. Recent Advances in the Application of Characterization Techniques for Studying Physical Stability of Amorphous Pharmaceutical Solids. Crystals 2021, 11, 1440. [Google Scholar] [CrossRef]
- Andronis, V.; Zografi, G. The molecular mobility of supercooled amorphous indomethacin as a function of temperature and relative humidity. Pharm. Res. 1998, 15, 835–842. [Google Scholar] [CrossRef]
- Nascimento, M.L.F.; Zanotto, E.D. Does viscosity describe the kinetic barrier for crystal growth from the liquidus to the glass transition? J. Chem. Phys. 2010, 133, 174701. [Google Scholar] [CrossRef]
- Schmelzer, J.W.; Abyzov, A.S.; Fokin, V.M.; Schick, C.; Zanotto, E.D. Crystallization in glass-forming liquids: Effects of decoupling of diffusion and viscosity on crystal growth. J. Non-Cryst. Solids 2015, 429, 45–53. [Google Scholar] [CrossRef]
- Svoboda, R.; Prikryl, J.; Cicmancova, V.; Prokop, V.; Kolobov, A.V.; Krbal, M. Crystal Growth in Amorphous Selenium Thin Films─Reviewed and Revisited: Direct Comparison of Microscopic and Calorimetric Measurements. Cryst. Growth Des. 2021, 21, 7087–7097. [Google Scholar] [CrossRef]
- Martin, J.D.; Hillis, B.G.; Hou, F. Transition Zone Theory Compared to Standard Models: Reexamining the Theory of Crystal Growth from Melts. J. Phys. Chem. C 2020, 124, 18724–18740. [Google Scholar] [CrossRef]
- Adam, G.; Gibbs, J.H. On the Temperature Dependence of Cooperative Relaxation Properties in Glass-Forming Liquids. J. Chem. Phys. 1965, 43, 139–146. [Google Scholar] [CrossRef]
- Mallamace, F.; Branca, C.; Corsaro, C.; Leone, N.; Spooren, J.; Chen, S.-H.; Stanley, H.E. Transport properties of glass-forming liquids suggest that dynamic crossover temperature is as important as the glass transition temperature. Proc. Natl. Acad. Sci. USA 2010, 107, 22457–22462. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Ragoonanan, V.; McKenna, G.B.; Suryanarayanan, R. Correlation between Molecular Mobility and Physical Stability in Pharmaceutical Glasses. Mol. Pharm. 2016, 13, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Szamel, G.; Douglas, J.F. Nature of the breakdown in the Stokes-Einstein relationship in a hard sphere fluid. J. Chem. Phys. 2006, 124, 214501. [Google Scholar] [CrossRef] [PubMed]
- Berthier, L.; Chandler, D.; Garrahan, J.P. Length scale for the onset of Fickian diffusion in supercooled liquids. Eur. Lett. 2005, 69, 320–326. [Google Scholar] [CrossRef]
- Zou, Q.-Z.; Li, Z.-W.; Zhu, Y.-L.; Sun, Z.-Y. Coupling and decoupling between translational and rotational dynamics in supercooled monodisperse soft Janus particles. Soft Matter 2019, 15, 3343–3352. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svoboda, R.; Košťálová, D.; Krbal, M.; Komersová, A. Indomethacin: The Interplay between Structural Relaxation, Viscous Flow and Crystal Growth. Molecules 2022, 27, 5668. https://doi.org/10.3390/molecules27175668
Svoboda R, Košťálová D, Krbal M, Komersová A. Indomethacin: The Interplay between Structural Relaxation, Viscous Flow and Crystal Growth. Molecules. 2022; 27(17):5668. https://doi.org/10.3390/molecules27175668
Chicago/Turabian StyleSvoboda, Roman, Daniela Košťálová, Miloš Krbal, and Alena Komersová. 2022. "Indomethacin: The Interplay between Structural Relaxation, Viscous Flow and Crystal Growth" Molecules 27, no. 17: 5668. https://doi.org/10.3390/molecules27175668
APA StyleSvoboda, R., Košťálová, D., Krbal, M., & Komersová, A. (2022). Indomethacin: The Interplay between Structural Relaxation, Viscous Flow and Crystal Growth. Molecules, 27(17), 5668. https://doi.org/10.3390/molecules27175668