
Sulodexide Increases Glutathione Synthesis and Causes Pro-Reducing Shift in Glutathione-Redox State in HUVECs Exposed to Oxygen–Glucose Deprivation: Implication for Protection of Endothelium against Ischemic Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
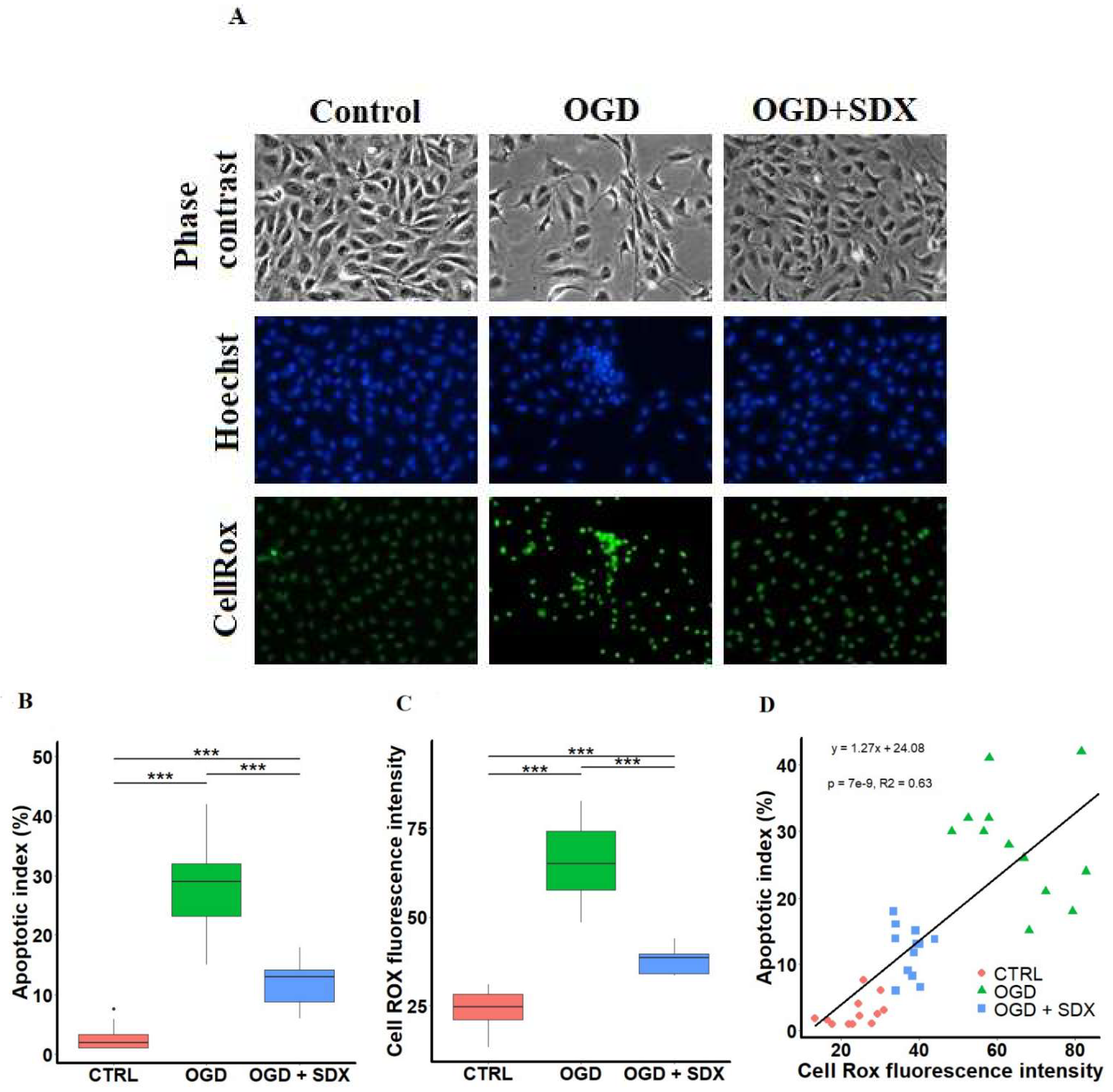
2.1. Effect of SDX on Apoptosis
2.2. Effect of SDX on Intracellular ROS Accumulation
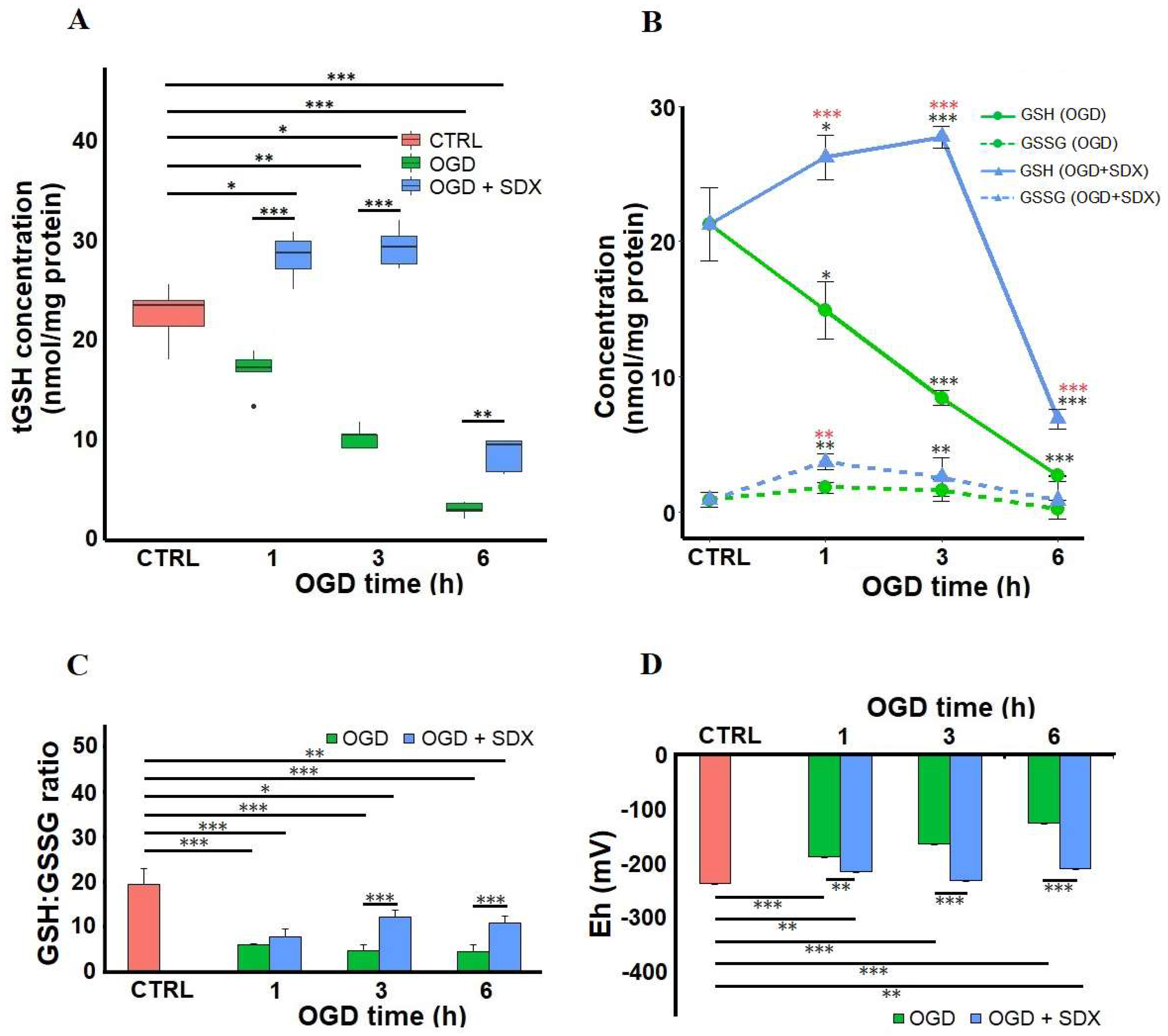
2.3. Effect of SDX on Intracellular GSH Content, GSH:GSSG Ratio and Redox Potential
2.4. Effect of SDX on GCLc and GSS Protein Levels
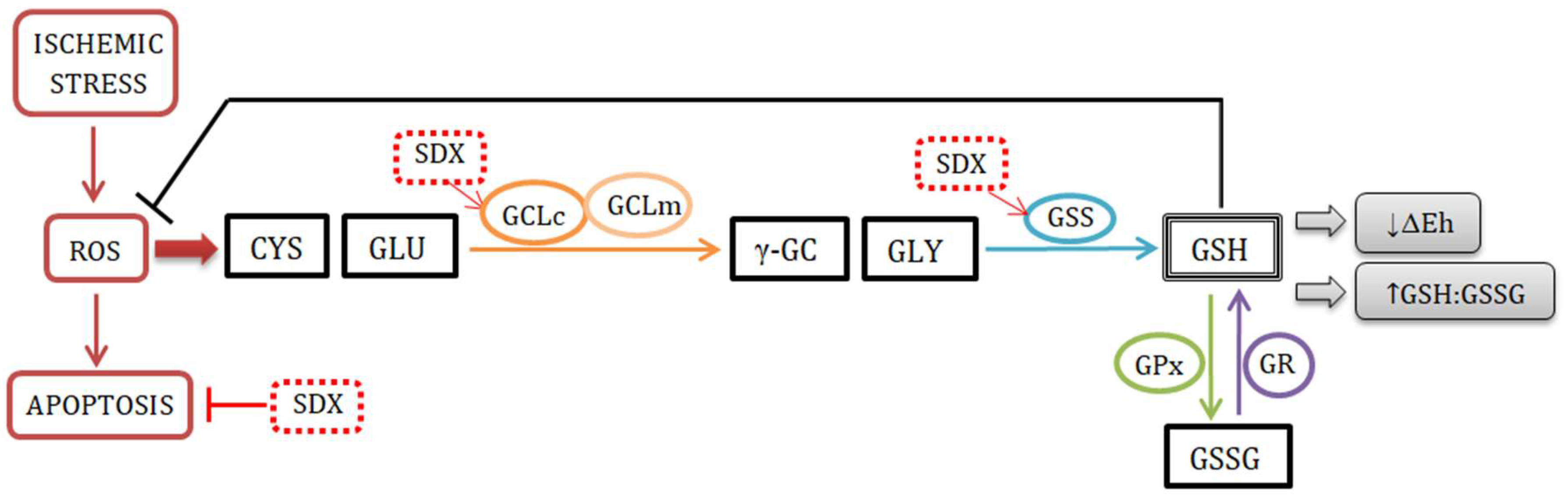
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. In Vitro Model of Simulated Ischemia
4.3. Drug Administration and Experimental Groups
4.4. Apoptosis Assay
4.5. Detection of Intracellular ROS
4.6. Measurement of GSH
4.7. Redox Potential Calculations
4.8. Determination of Protein Content
4.9. Enzyme Linked Immunosorbent Assay (ELISA)
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hirase, T.; Node, K. Endothelial dysfunction as a cellular mechanism for vascular failure. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Vemulapalli, S.; Patel, M.R.; Jones, W.S. Limb Ischemia: Cardiovascular Diagnosis and Management from Head to Toe. Curr. Cardiol. Rep. 2015, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; He, G.-W.; Underwood, M.J.; Yu, C.-M. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: Perspectives and implications for postischemic myocardial protection. Am. J. Transl. Res. 2016, 8, 765–777. [Google Scholar] [PubMed]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; El Yazbi, A.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci. 2022, 27, 105. [Google Scholar] [CrossRef]
- Higashi, Y.; Maruhashi, T.; Noma, K.; Kihara, Y. Oxidative stress and endothelial dysfunction: Clinical evidence and therapeutic implications. Trends Cardiovasc. Med. 2014, 24, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Matuz-Mares, D.; Riveros-Rosas, H.; Vilchis-Landeros, M.; Vázquez-Meza, H. Glutathione Participation in the Prevention of Cardiovascular Diseases. Antioxidants 2021, 10, 1220. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Glutathione and modulation of cell apoptosis. Biochim. Biophys. Acta 2012, 1823, 1767–1777. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia–reperfusion injury. Cardiovasc. Res. 2000, 47, 446–456. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Akaboshi, T.; Yamanishi, R. Certain carotenoids enhance the intracellular glutathione level in a murine cultured macrophage cell line by inducing glutamate-cysteine-ligase. Mol. Nutr. Food Res. 2014, 58, 1291–1300. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Mannello, F.; Ligi, D.; Canale, M.; Raffetto, J.D. Sulodexide Down-Regulates the Release of Cytokines, Chemokines, and Leukocyte Colony Stimulating Factors from Human Macrophages: Role of Glycosaminoglycans in Inflammatory Pathways of Chronic Venous Disease. Curr. Vasc. Pharmacol. 2014, 12, 173–185. [Google Scholar] [CrossRef]
- Coccheri, S.; Mannello, F. Development and use of sulodexide in vascular diseases: Implications for treatment. Drug Des. Dev. Ther. 2013, 8, 49–65. [Google Scholar] [CrossRef]
- Coccheri, S. Biological and clinical effects of sulodexide in arterial disorders and diseases. Int. Angiol. 2014, 33, 263–274. [Google Scholar]
- Carroll, B.J.; Piazza, G.; Goldhaber, S.Z. Sulodexide in venous disease. J. Thromb. Haemost. 2019, 17, 31–38. [Google Scholar] [CrossRef]
- Chaitidis, N.; Kokkinidis, D.G.; Papadopoulou, Z.; Hasemaki, N.; Attaran, R.; Bakoyiannis, C. Management of Post-Thrombotic Syndrome: A Comprehensive Review. Curr. Pharm. Des. 2022, 28, 550–559. [Google Scholar] [CrossRef]
- Yongwatana, K.; Supasyndh, O.; Satirapoj, B. Renal Effects of Sulodexide in Type 2 Diabetic Patients without Nephrotic Range Proteinuria. J. Diabetes Res. 2020, 2020, 2984680. [Google Scholar] [CrossRef]
- Sosińska, P.; Baum, E.; Maćkowiak, B.; Maj, M.; Sumińska-Jasińska, K.; Staniszewski, R.; Bręborowicz, A. Sulodexide Reduces the Proinflammatory Effect of Serum from Patients with Peripheral Artery Disease in Human Arterial Endothelial Cells. Cell. Physiol. Biochem. 2016, 40, 1005–1012. [Google Scholar] [CrossRef]
- Liu, Y.N.; Zhou, J.; Li, T.; Wu, J.; Xie, S.H.; Liu, H.-F.; Liu, Z.; Park, T.S.; Wang, Y.; Liu, W.J. Sulodexide Protects Renal Tubular Epithelial Cells from Oxidative Stress-Induced Injury via Upregulating Klotho Expression at an Early Stage of Diabetic Kidney Disease. J. Diabetes Res. 2017, 2017, 4989847. [Google Scholar] [CrossRef] [Green Version]
- Pletinck, A.; Van Landschoot, M.; Steppan, S.; Laukens, D.; Passlick-Deetjen, J.; Vanholder, R.; Van Biesen, W. Oral supplementation with sulodexide inhibits neo-angiogenesis in a rat model of peritoneal perfusion. Nephrol. Dial. Transplant. 2012, 27, 548–556. [Google Scholar] [CrossRef]
- Park, H.Y.; Kang, S.; Kim, G.Y.; Jang, Y.; Kwon, H.M.; Shim, W.H.; Cho, S.Y.; Cho, S.H. Inhibition of neointimal proliferation of rat carotid artery by sulodexide. J. Korean Med. Sci. 1997, 12, 210–214. [Google Scholar] [CrossRef]
- Mannello, F.; Medda, V.; Ligi, D.; Raffetto, J.D. Glycosaminoglycan sulodexide inhibition of MMP-9 gelatinase secretion and activity: Possible pharmacological role against collagen degradation in vascular chronic diseases. Curr. Vasc. Pharmacol. 2013, 11, 354–365. [Google Scholar] [CrossRef]
- Dobiaš, L.; Petrová, M.; Vojtko, R.; Uličná, O.; Vančová, O.; Kristová, V. Effect of Sulodexide on Vascular Responses and Liver Mitochondrial Function in Diabetic Rats. Physiol. Res. 2015, 64, S497–S505. [Google Scholar] [CrossRef]
- Li, T.; Liu, X.; Zhao, Z.; Ni, L.; Liu, C. Sulodexide recovers endothelial function through reconstructing glycocalyx in the balloon-injury rat carotid artery model. Oncotarget 2017, 8, 91350–91361. [Google Scholar] [CrossRef]
- Kristová, V.; Liskova, S.; Sotníková, R.; Vojtko, R.; Kurtanský, A. Sulodexide improves endothelial dysfunction in streptozotocin-induced diabetes in rats. Physiol. Res. 2008, 57, 491–494. [Google Scholar] [CrossRef]
- Urbanek, T.; Krasinski, Z.; Suminska-Jasinska, K.; Baum, E.; Borej-Nowicka, G.; Begier-Krasinska, B.; Brȩborowicz, A. Sulo-dexide Reduces the Inflammatory Reaction and Senescence of Endothelial Cells in Conditions Involving Chronic Venous Disease. Int. Angiol. 2016, 35, 140–147. [Google Scholar] [PubMed]
- Sosińska-Zawierucha, P.; Mackowiak, B.; Staniszewski, R.; Sumińska-Jasińska, K.; Maj, M.; Krasiński, Z.; Bręborowicz, A. Sulodexide Slows Down the Senescence of Aortic Endothelial Cells Exposed to Serum from Patients with Peripheral Artery Diseases. Cell. Physiol. Biochem. 2018, 45, 2225–2232. [Google Scholar] [CrossRef] [PubMed]
- Skrha, J.; Perusicová, J.; Kvasnička, J.; Hilgertová, J. The effect of glycosaminoglycan sulodexide on oxidative stress and fibrinolysis in diabetes mellitus. Sb. Lek. 1998, 99, 103–109. [Google Scholar] [PubMed]
- Ciszewicz, M.; PoLubinska, A.; Antoniewicz, A.; Suminska-Jasinska, K.; BrĘborowicz, A. Sulodexide suppresses inflammation in human endothelial cells and prevents glucose cytotoxicity. Transl. Res. 2009, 153, 118–123. [Google Scholar] [CrossRef]
- Bilinska, M.; Wolszakiewicz, J.; Duda, M.; Janas, J.; Beręsewicz, A.; Piotrowicz, R. Antioxidative activity of sulodexide, a glycosaminoglycan, in patients with stable coronary artery disease: A pilot study. Med. Sci. Monit. 2009, 15, 618–623. [Google Scholar]
- Jin, H.Y.; Lee, K.A.; Song, S.K.; Liu, W.J.; Choi, J.H.; Song, C.H.; Baek, H.S.; Park, T.S. Sulodexide prevents peripheral nerve damage in streptozotocin induced diabetic rats. Eur. J. Pharmacol. 2012, 674, 217–226. [Google Scholar] [CrossRef]
- Gabryel, B.; Jarząbek, K.; Machnik, G.; Adamczyk, J.; Belowski, D.; Obuchowicz, E.; Urbanek, T. Superoxide dismutase 1 and glutathione peroxidase 1 are involved in the protective effect of sulodexide on vascular endothelial cells exposed to oxygen–glucose deprivation. Microvasc. Res. 2016, 103, 26–35. [Google Scholar] [CrossRef]
- Yin, J.; Chen, W.; Ma, F.; Lu, Z.; Wu, R.; Zhang, G.; Wang, N.; Wang, F. Sulodexide pretreatment attenuates renal ischemia-reperfusion injury in rats. Oncotarget 2017, 8, 9986–9995. [Google Scholar] [CrossRef]
- Gabryel, B.; Bontor, K.; Jarząbek, K.; Plato, M.; Pudełko, A.; Machnik, G.; Urbanek, T. Sulodexide up-regulates glutathione S-transferase P1 by enhancing Nrf2 expression and translocation in human umbilical vein endothelial cells injured by oxygen glucose deprivation. Arch. Med. Sci. 2019, 16, 957–963. [Google Scholar] [CrossRef]
- Gabryel, B.; Bontor, K.; Urbanek, T. Sulodexide increases mRNA expression of glutathione-related genes in human umbilical endothelial cells exposed to oxygen-glucose deprivation. Arch. Med. Sci. 2019, 16, 1444–1447. [Google Scholar] [CrossRef]
- Shen, D.; Chen, R.; Zhang, L.; Rao, Z.; Ruan, Y.; Li, L.; Chu, M.; Zhang, Y. Sulodexide attenuates endoplasmic reticulum stress induced by myocardial ischaemia/reperfusion by activating the PI3K/Akt pathway. J. Cell. Mol. Med. 2019, 23, 5063–5075. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, N.; Bi, W.; Zhang, J.; Li, X.; Shi, L.; Liu, Y.; Gao, X. Protective Role of Sulodexide on Renal Injury Induced by Limb Ischemia-Reperfusion. Evid. Based Complement. Altern. Med. 2021, 2021, 6629718. [Google Scholar] [CrossRef]
- Li, C.; Zhang, W.-J.; Choi, J.; Frei, B. Quercetin affects glutathione levels and redox ratio in human aortic endothelial cells not through oxidation but formation and cellular export of quercetin-glutathione conjugates and upregulation of glutamate-cysteine ligase. Redox Biol. 2016, 9, 220–228. [Google Scholar] [CrossRef]
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar] [CrossRef]
- Ha, K.-N.; Chen, Y.; Cai, J.; Sternberg, P. Increased Glutathione Synthesis through an ARE-Nrf2–Dependent Pathway by Zinc in the RPE: Implication for Protection against Oxidative Stress. Investig. Opthalmol. Vis. Sci. 2006, 47, 2709–2715. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Zhang, Z.; Li, L.; Song, W. Resveratrol affects the expression of glutamate cysteine ligase in the kidneys of aged rats. Exp. Ther. Med. 2014, 7, 1762–1766. [Google Scholar] [CrossRef]
- Yang, Y.; Li, L.; Hang, Q.; Fang, Y.; Dong, X.; Cao, P.; Yin, Z.; Luo, L. γ-glutamylcysteine exhibits anti-inflammatory effects by increasing cellular glutathione level. Redox Biol. 2019, 20, 157–166. [Google Scholar] [CrossRef]
- Yang, H.; Zeng, Y.; Lee, T.D.; Yang, Y.; Ou, X.; Chen, L.; Haque, M.; Rippe, R.; Lu, S.C. Role of AP-1 in the Coordinate Induction of Rat Glutamate-cysteine Ligase and Glutathione Synthetase bytert-Butylhydroquinone. J. Biol. Chem. 2002, 277, 35232–35239. [Google Scholar] [CrossRef]
- Lee, T.D.; Yang, H.; Whang, J.; Lu, S.C. Cloning and characterization of the human glutathione synthetase 5′-flanking region. Biochem. J. 2005, 390, 521–528. [Google Scholar] [CrossRef]
- Song, P.; Zou, M.-H. Redox regulation of endothelial cell fate. Cell. Mol. Life Sci. 2014, 71, 3219–3239. [Google Scholar] [CrossRef]
- Daiber, A.; Chlopicki, S. Revisiting pharmacology of oxidative stress and endothelial dysfunction in cardiovascular disease: Evidence for redox-based therapies. Free Radic. Biol. Med. 2020, 157, 15–37. [Google Scholar] [CrossRef]
- Masola, V.; Zaza, G.; Onisto, M.; Lupo, A.; Gambaro, G. Glycosaminoglycans, proteoglycans and sulodexide and the endothelium: Biological roles and pharmacological effects. Int. Angiol. 2014, 33, 243–254. [Google Scholar]
- Połubińska, A.; Staniszewski, R.; Baum, E.; Sumińska-Jasińska, K.; Bręborowicz, A. Sulodexide modifies intravascular homeostasis what affects function of the endothelium. Adv. Med. Sci. 2013, 58, 304–310. [Google Scholar] [CrossRef]
- Gericke, A.; Suminska-Jasińska, K.; Bręborowicz, A. Sulodexide reduces glucose induced senescence in human retinal endothelial cells. Sci. Rep. 2021, 11, 11532. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.; Prasai, P.K.; Kaskas, A.M.; Khanna, A.; Letchuman, V.; Letchuman, S.; Alexander, J.S.; Orr, A.W.; Woolard, M.D.; Pattillo, C.B. Differential arterial and venous endothelial redox responses to oxidative stress. Microcirculation 2018, 25, e12486. [Google Scholar] [CrossRef] [PubMed]
- Szasz, T.; Thakali, K.; Fink, G.D.; Watts, S.W. A comparison of arteries and veins in oxidative stress: Producers, destroyers, function, and disease. Exp. Biol. Med. 2007, 232, 27–37. [Google Scholar]
- Wang, T.; Gotoh, Y.; Jennings, M.H.; Rhoads, C.A.; Aw, T.Y. Lipid hydroperoxide-induced apoptosis in human colonic CaCo-2 cells is associated with an early loss of cellular redox balance. FASEB J. 2000, 14, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Anathy, V.; Roberson, E.C.; Guala, A.S.; Godburn, K.E.; Budd, R.C.; Janssen-Heininger, Y.M. Redox-Based Regulation of Apoptosis: S-Glutathionylation As a Regulatory Mechanism to Control Cell Death. Antioxid. Redox Signal. 2012, 16, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Rashdan, N.A.; Shrestha, B.; Pattillo, C.B. S-glutathionylation, friend or foe in cardiovascular health and disease. Redox Biol. 2020, 37, 101693. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.K.; Dubick, M.A.; Omaye, S.T. γ-Glutamylcysteine inhibits oxidative stress in human endothelial cells. Life Sci. 2012, 90, 116–121. [Google Scholar] [CrossRef]
- Tian, L.; Shi, M.M.; Forman, H.J. Increased Transcription of the Regulatory Subunit of γ-Glutamylcysteine Synthetase in Rat Lung Epithelial L2 Cells Exposed to Oxidative Stress or Glutathione Depletion. Arch. Biochem. Biophys. 1997, 342, 126–133. [Google Scholar] [CrossRef]
- Krejsa, C.M.; Franklin, C.C.; White, C.C.; Ledbetter, J.A.; Schieven, G.L.; Kavanagh, T.J. Rapid Activation of Glutamate Cysteine Ligase following Oxidative Stress. J. Biol. Chem. 2010, 285, 16116–16124. [Google Scholar] [CrossRef]
- Ibbotson, K.; Yell, J.; Ronaldson, P.T. Nrf2 signaling increases expression of ATP-binding cassette subfamily C mRNA transcripts at the blood-brain barrier following hypoxia-reoxygenation stress. Fluids Barriers CNS 2017, 14, 51. [Google Scholar] [CrossRef]
- Brzica, H.; Abdullahi, W.; Ibbotson, K.; Ronaldson, P.T. Role of Transporters in Central Nervous System Drug Delivery and Blood-Brain Barrier Protection: Relevance to Treatment of Stroke. J. Cent. Nerv. Syst. Dis. 2017, 9, 9380. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.F.H.; Widder, J.D.; McNally, J.S.; McCann, L.; Jones, D.P.; Harrison, D.G. The Role of the Multidrug Resistance Protein-1 in Modulation of Endothelial Cell Oxidative Stress. Circ. Res. 2005, 97, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Glutathione Efflux and Cell Death. Antioxid. Redox Signal. 2012, 17, 1694–1713. [Google Scholar] [CrossRef] [Green Version]
- Krawczenko, A.; Bielawska-Pohl, A.; Wojtowicz, K.; Jura, R.; Paprocka, M.; Wojdat, E.; Kozlowska, U.; Klimczak, A.; Grillon, C.; Kieda, C.; et al. Expression and activity of multidrug resistance proteins in mature endothelial cells and their precursors: A challenging correlation. PLoS ONE 2017, 12, e0172371. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bontor, K.; Gabryel, B. Sulodexide Increases Glutathione Synthesis and Causes Pro-Reducing Shift in Glutathione-Redox State in HUVECs Exposed to Oxygen–Glucose Deprivation: Implication for Protection of Endothelium against Ischemic Injury. Molecules 2022, 27, 5465. https://doi.org/10.3390/molecules27175465
Bontor K, Gabryel B. Sulodexide Increases Glutathione Synthesis and Causes Pro-Reducing Shift in Glutathione-Redox State in HUVECs Exposed to Oxygen–Glucose Deprivation: Implication for Protection of Endothelium against Ischemic Injury. Molecules. 2022; 27(17):5465. https://doi.org/10.3390/molecules27175465
Chicago/Turabian StyleBontor, Klaudia, and Bożena Gabryel. 2022. "Sulodexide Increases Glutathione Synthesis and Causes Pro-Reducing Shift in Glutathione-Redox State in HUVECs Exposed to Oxygen–Glucose Deprivation: Implication for Protection of Endothelium against Ischemic Injury" Molecules 27, no. 17: 5465. https://doi.org/10.3390/molecules27175465
APA StyleBontor, K., & Gabryel, B. (2022). Sulodexide Increases Glutathione Synthesis and Causes Pro-Reducing Shift in Glutathione-Redox State in HUVECs Exposed to Oxygen–Glucose Deprivation: Implication for Protection of Endothelium against Ischemic Injury. Molecules, 27(17), 5465. https://doi.org/10.3390/molecules27175465