
New Insights on Glutathione’s Supramolecular Arrangement and Its In Silico Analysis as an Angiotensin-Converting Enzyme Inhibitor

 , , ,
, , ,  and
and
Abstract
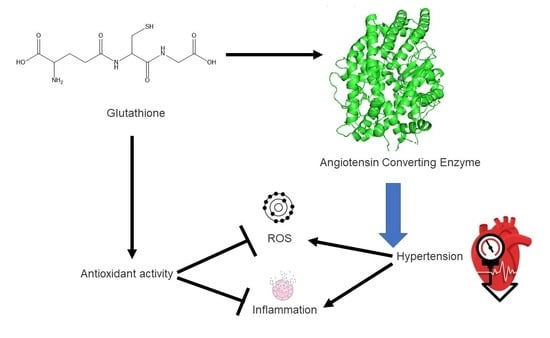
1. Introduction
2. Results and Discussion
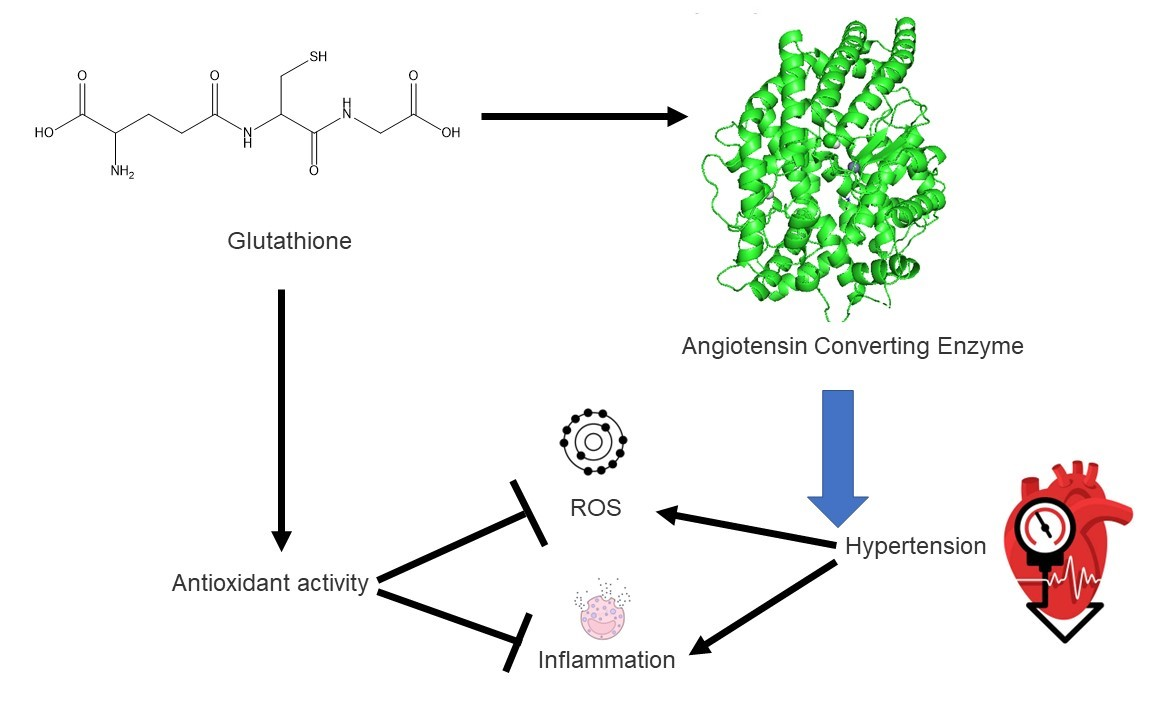
2.1. Solid State Analysis
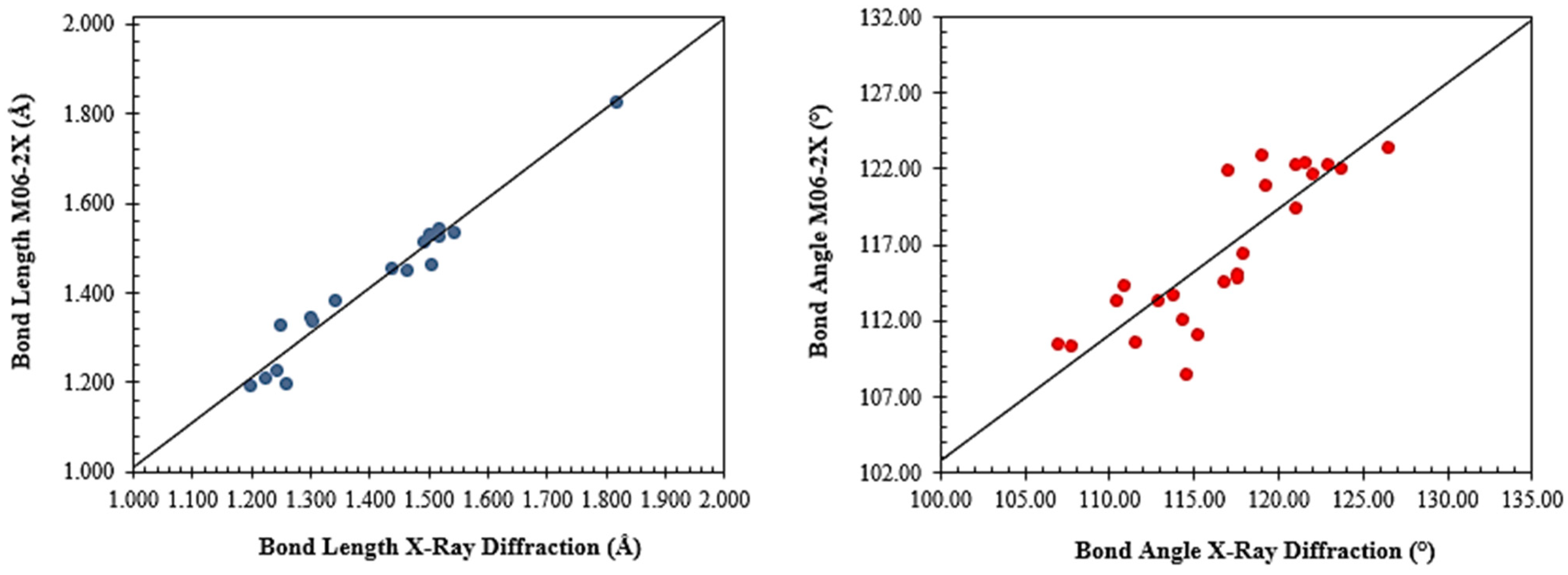
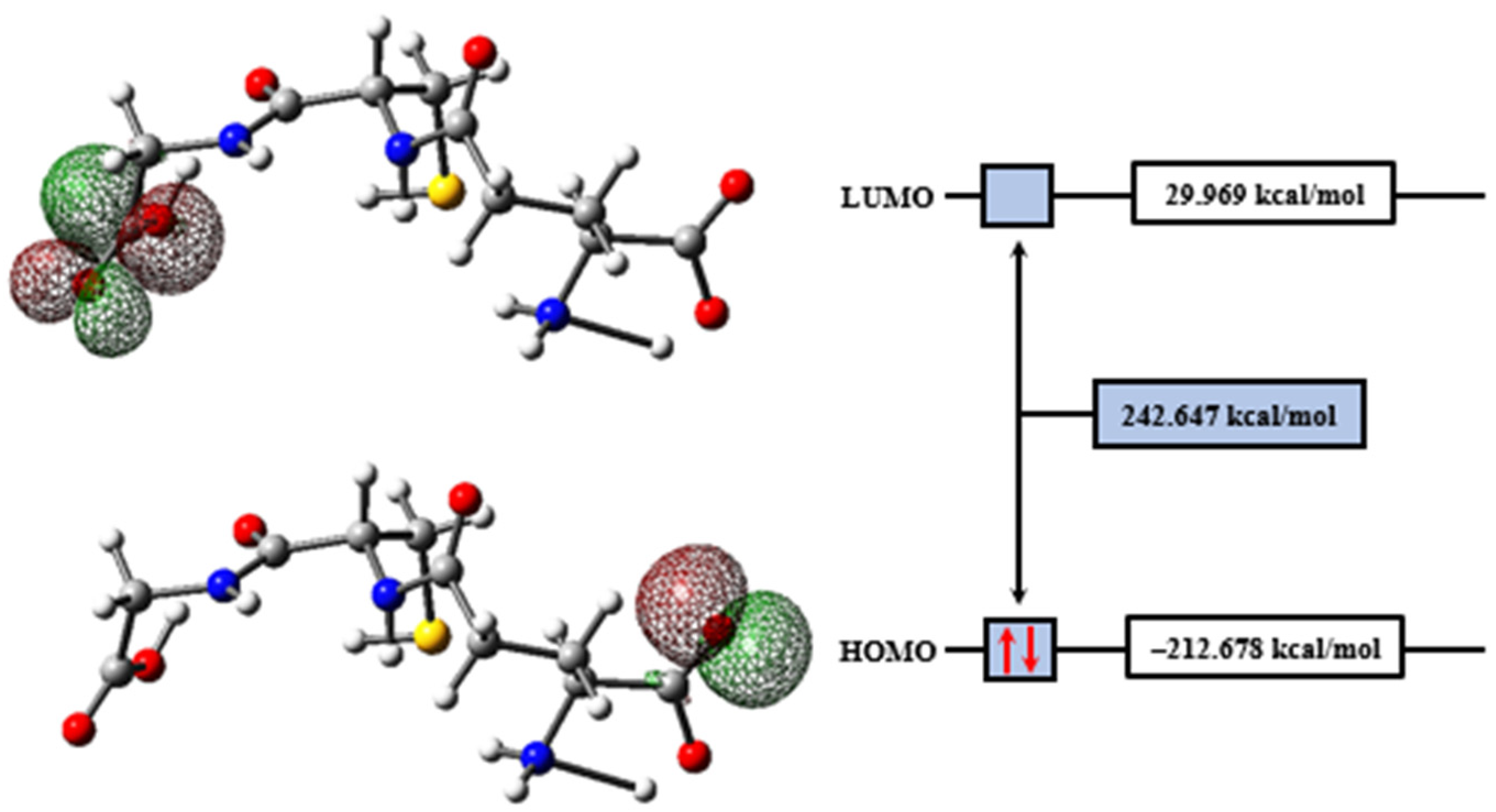
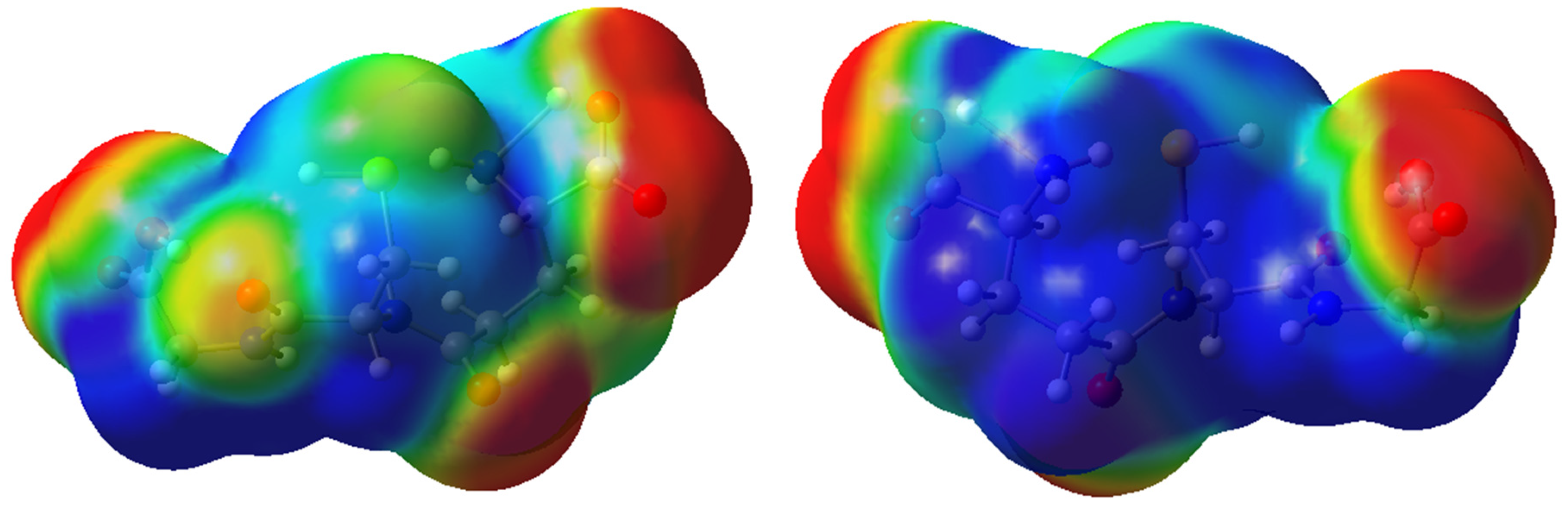
2.2. Molecular Modeling Analysis
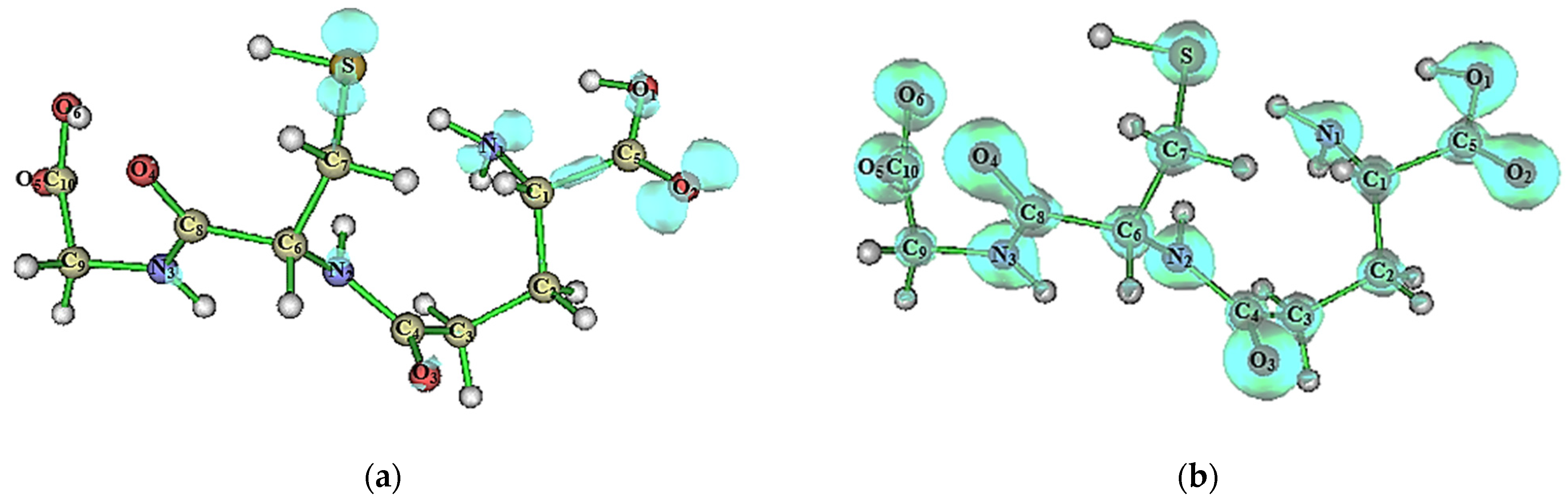
2.3. Supramolecular Arrangement
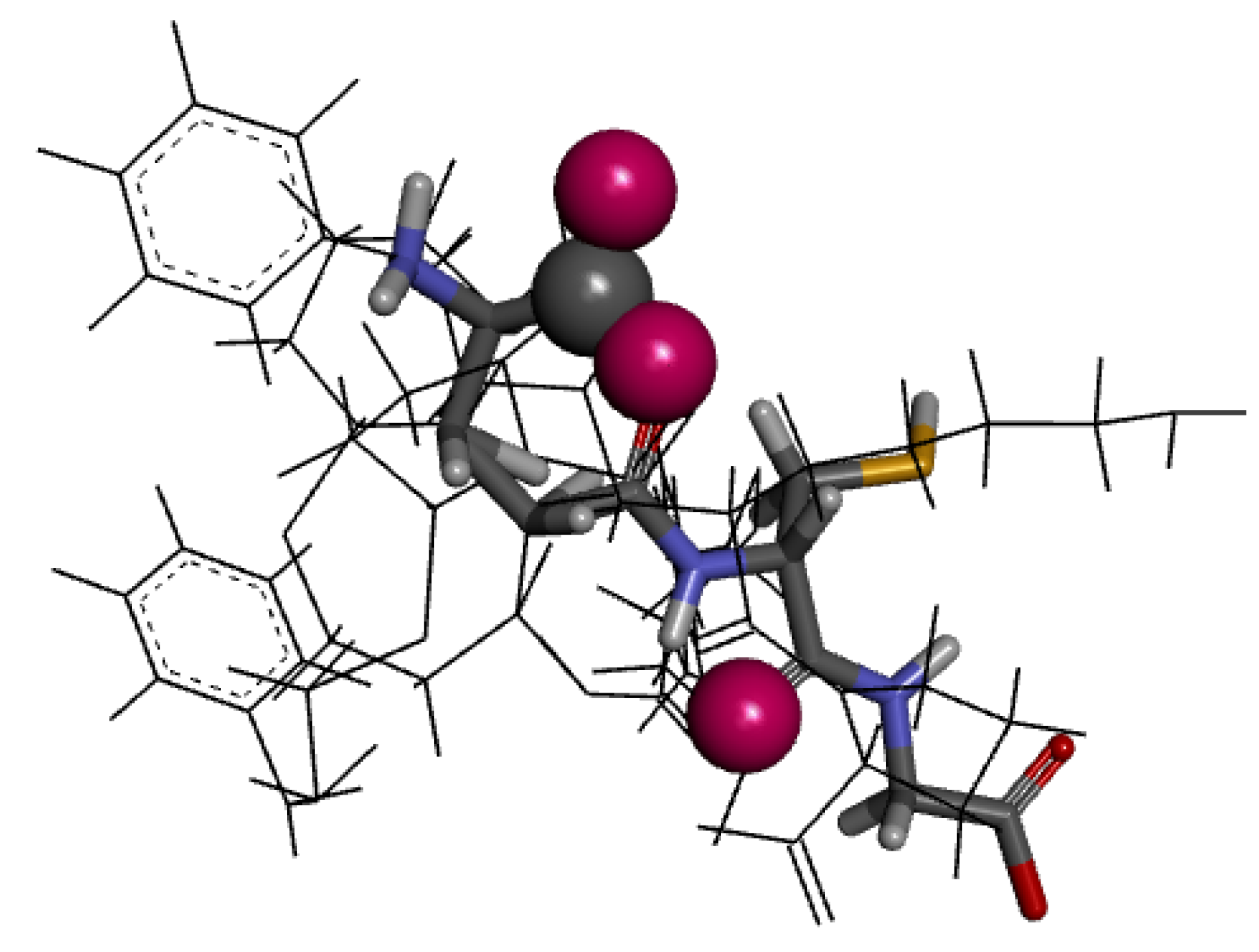
2.4. Molecular Docking Analysis
3. Computational Procedures
3.1. Computational Methods
3.2. Supramolecular Arrangement
3.3. Molecular Docking Analysis
3.4. Pharmacophore Design
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mc Namara, K.; Alzubaidi, H.; Jackson, J.K. Cardiovascular Disease as a Leading Cause of Death: How Are Pharmacists Getting Involved. Integr. Pharm. Res. Pract. 2019, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Birger, M.; Kaldjian, A.S.; Roth, G.A.; Moran, A.E.; Dieleman, J.L.; Bellows, B.K. Spending on Cardiovascular Disease and Cardiovascular Risk Factors in the United States: 1996 to 2016. Circulation 2021, 144, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Malinowski, B.; Zalewska, K.; Węsierska, A.; Sokołowska, M.M.; Socha, M.; Liczner, G.; Pawlak-Osińska, K.; Wiciński, M. Intermittent Fasting in Cardiovascular Disorders—An Overview. Nutrients 2019, 11, 673. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Taub, P.R.; Epstein, E.; Michos, E.D.; Ferraro, R.A.; Bailey, A.L.; Kelli, H.M.; Ferdinand, K.C.; Echols, M.R.; Weintraub, H.; et al. Ten Things to Know about Ten Cardiovascular Disease Risk Factors. Am. J. Prev. Cardiol. 2021, 5, 100149. [Google Scholar] [CrossRef] [PubMed]
- Flora, G.D.; Nayak, M.K. A Brief Review of Cardiovascular Diseases, Associated Risk Factors and Current Treatment Regimes. Curr. Pharm. Des. 2019, 25, 4063–4084. [Google Scholar] [CrossRef]
- Beevers, G. ABC of Hypertension: The Pathophysiology of Hypertension. BMJ 2001, 322, 912–916. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Touyz, R.M. Cell Signaling of Angiotensin II on Vascular Tone: Novel Mechanisms. Curr. Hypertens. Rep. 2011, 13, 122–128. [Google Scholar] [CrossRef]
- Wong, M.K.S. Angiotensin Converting Enzymes. In Handbook of Hormones; Elsevier: Amsterdam, The Netherlands, 2016; pp. 263–e29D-4. [Google Scholar]
- Chen, J.; Wang, Y.; Ye, R.; Wu, Y.; Xia, W. Comparison of Analytical Methods to Assay Inhibitors of Angiotensin I-Converting Enzyme. Food Chem. 2013, 141, 3329–3334. [Google Scholar] [CrossRef]
- Hawgood, B.J. Maurício Rocha E Silva MD: Snake Venom, Bradykinin and the Rise of Autopharmacology. Toxicon 1997, 35, 1569–1580. [Google Scholar] [CrossRef]
- Ferreira, S.H. Angiotensin Converting Enzyme: History and Relevance. Semin. Perinatol. 2000, 24, 7–10. [Google Scholar] [CrossRef]
- Acharya, K.R.; Sturrock, E.D.; Riordan, J.F.; Ehlers, M.R.W. Ace Revisited: A New Target for Structure-Based Drug Design. Nat. Rev. Drug Discov. 2003, 2, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, I.; Kim, S.-K. Angiotensin-I-Converting Enzyme (ACE) Inhibitors from Marine Resources: Prospects in the Pharmaceutical Industry. Mar. Drugs 2010, 8, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Roy, S. Angiotensin-Converting Enzyme Inhibitors from Plants: A Review of Their Diversity, Modes of Action, Prospects, and Concerns in the Management of Diabetes-Centric Complications. J. Integr. Med. 2021, 19, 478–492. [Google Scholar] [CrossRef]
- Turner, J.M.; Kodali, R. Should Angiotensin-Converting Enzyme Inhibitors Ever Be Used for the Management of Hypertension? Curr. Cardiol. Rep. 2020, 22, 95. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.H.; Imig, J.D. Antihypertensive Drugs. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Alderman, C.P. Adverse Effects of the Angiotensin-Converting Enzyme Inhibitors. Ann. Pharmacother. 1996, 30, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Nchinda, A.T.; Chibale, K.; Redelinghuys, P.; Sturrock, E.D. Synthesis and Molecular Modeling of a Lisinopril–Tryptophan Analogue Inhibitor of Angiotensin I-Converting Enzyme. Bioorganic Med. Chem. Lett. 2006, 16, 4616–4619. [Google Scholar] [CrossRef] [PubMed]
- Kharazmi-Khorassani, J.; Asoodeh, A.; Tanzadehpanah, H. Antioxidant and Angiotensin-Converting Enzyme (ACE) Inhibitory Activity of Thymosin Alpha-1 (Thα1) Peptide. Bioorg. Chem. 2019, 87, 743–752. [Google Scholar] [CrossRef]
- Papakyriakou, A.; Spyroulias, G.A.; Sturrock, E.D.; Manessi-Zoupa, E.; Cordopatis, P. Simulated Interactions between Angiotensin-Converting Enzyme and Substrate Gonadotropin-Releasing Hormone: Novel Insights into Domain Selectivity. Biochemistry 2007, 46, 8753–8765. [Google Scholar] [CrossRef]
- Corradi, H.R.; Chitapi, I.; Sewell, B.T.; Georgiadis, D.; Dive, V.; Sturrock, E.D.; Acharya, K.R. The Structure of Testis Angiotensin-Converting Enzyme in Complex with the C Domain-Specific Inhibitor RXPA380. Biochemistry 2007, 46, 5473–5478. [Google Scholar] [CrossRef]
- Alvarenga, D.J.; Matias, L.M.F.; Cordeiro, C.F.; de Souza, T.B.; Lavorato, S.N.; Pereira, M.G.A.G.; Dias, D.F.; Carvalho, D.T. Synthesis of Eugenol-Derived Glucosides and Evaluation of Their Ability in Inhibiting the Angiotensin Converting Enzyme. Nat. Prod. Res. 2022, 36, 2246–2253. [Google Scholar] [CrossRef]
- Basi, Z.; Turkoglu, V. In Vitro Effect of Oxidized and Reduced Glutathione Peptides on Angiotensin Converting Enzyme Purified from Human Plasma. J. Chromatogr. B 2019, 1104, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Bas, Z. Inhibition Effect of Nicotinamide (Vitamin B3) and Reduced Glutathione (GSH) Peptide on Angiotensin-Converting Enzyme Activity Purified from Sheep Kidney. Int. J. Biol. Macromol. 2021, 189, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Yesiltepe, O.; Güler Çelik, E.; Geyik, C.; Gümüs, Z.P.; Odaci Demirkol, D.; Coşkunol, H.; Timur, S. Preparation of Glutathione Loaded Nanoemulsions and Testing of Hepatoprotective Activity on THLE-2 Cells. Turk. J. Chem. 2021, 45, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New Roles in Redox Signaling for an Old Antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, G.; Chen, J. Glutathione: A Review on Biotechnological Production. Appl. Microbiol. Biotechnol. 2004, 66, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Glutathione. Monograph; European Pharmacopoeia: Strasbourg, France, 2018. [Google Scholar]
- Pál, P.; Caridad, N.P.; Hamilton, B.N.; Hawsar, O.M. Physico-Chemical Characterization Pf Glutathione (GSH). In Glutathione. Biosynthesis, Functions and Biological Implications; Nova Science Publishers: Hauppauge, NY, USA, 2019; pp. 3–53. [Google Scholar]
- Jefferies, H.; Coster, J.; Khalil, A.; Bot, J.; McCauley, R.D.; Hall, J.C. Glutathione. ANZ J. Surg. 2003, 73, 517–522. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Exchange-Correlation Functional with Broad Accuracy for Metallic and Nonmetallic Compounds, Kinetics, and Noncovalent Interactions. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical Hybrid Density Functional with Perturbative Second-Order Correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef]
- Heitmann, P. A Model for Sulfhydryl Groups in Proteins. Hydrophobic Interactions of the Cysteine Side Chain in Micelles. Eur. J. Biochem. 1968, 3, 346–350. [Google Scholar] [CrossRef]
- LoPachin, R.M.; Barber, D.S. Synaptic Cysteine Sulfhydryl Groups as Targets of Electrophilic Neurotoxicants. Toxicol. Sci. 2006, 94, 240–255. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aurell, M.J.; Contreras, R. Quantitative Characterization of the Global Electrophilicity Pattern of Some Reagents Involved in 1,3-Dipolar Cycloaddition Reactions. Tetrahedron 2003, 59, 3117–3125. [Google Scholar] [CrossRef]
- Fukui, K. The Role of Frontier Orbitals in Chemical Reactions. Angew. Chem.-Int. Ed. 1982, 21, 801–876. [Google Scholar] [CrossRef]
- Fukui, K. Role of Frontier Orbitals in Chemical Reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting Intermolecular Interactions in Molecular Crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Çakmak, Ş.; Demircioğlu, Z.; Uzun, S.; Veyisoğlu, A.; Yakan, H.; Ersanli, C.C. Synthesis, X-Ray Structure, Antimicrobial Activity, DFT and Molecular Docking Studies of N-(Thiophen-2-Ylmethyl)Thiophene-2-Carboxamide. Acta Crystallogr. C Struct. Chem. 2022, 78, 390–397. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Matta, C.F.M.; Boyd, R.J. The Quantum Theory of Atoms in Molecules; Wiley-VCH: Weinheim, Germany, 2007; ISBN 9783527307487. [Google Scholar]
- March, N.H. Electron Density Theory of Atoms and Molecules. J. Phys. Chem. 1982, 86, 2262–2267. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Clarendon Press Publication: Oxford, UK, 1994; ISBN 9780198558651. [Google Scholar]
- Natesh, R.; Schwager, S.L.U.; Sturrock, E.D.; Acharya, K.R. Crystal Structure of the Human Angiotensin-Converting Enzyme–Lisinopril Complex. Nature 2003, 421, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Tzakos, A.G.; Gerothanassis, I.P. Domain-Selective Ligand-Binding Modes and Atomic Level Pharmacophore Refinement in Angiotensin I Converting Enzyme (ACE) Inhibitors. ChemBioChem 2005, 6, 1089–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhou, B. Insight into Structural Requirements of ACE Inhibitory Dipeptides: QSAR and Molecular Docking Studies. Mol. Divers. 2020, 24, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Lin, G.; Zhang, R.; Wu, W. Studies on the Bioactivities of ACE-Inhibitory Peptides with Phenylalanine C-Terminus Using 3D-QSAR, Molecular Docking and in Vitro Evaluation. Mol. Inform. 2017, 36, 1600157. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Amaya, J.A.; Cabrera, D.Z.; Matallana, A.M.; Arevalo, K.G.; Guevara-Pulido, J. In-Silico Design of New Enalapril Analogs (ACE Inhibitors) Using QSAR and Molecular Docking Models. Inform. Med. Unlocked 2020, 19, 100336. [Google Scholar] [CrossRef]
- Gu, Y.; Wu, J. LC–MS/MS Coupled with QSAR Modeling in Characterising of Angiotensin I-Converting Enzyme Inhibitory Peptides from Soybean Proteins. Food Chem. 2013, 141, 2682–2690. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, X.; Ma, Q.; Yi, J.; Cai, S. Phytochemical Bioaccessibility and in Vitro Antidiabetic Effects of Chinese Sumac (Rhus Chinensis Mill.) Fruits after a Simulated Digestion: Insights into the Mechanisms with Molecular Docking Analysis. Int. J. Food Sci. Technol. 2022, 57, 2656–2669. [Google Scholar] [CrossRef]
- Oke, A.M.; Adelakun, A.O.; Akintelu, S.A.; Soetan, E.A.; Oyebamiji, A.K.; Ewemoje, T.A. Inhibition of Angiotensin Converting Enzyme by Phytochemicals in Cucurbita Pepo L.: In Silico Approach. Pharmacol. Res.-Mod. Chin. Med. 2022, 4, 100142. [Google Scholar] [CrossRef]
- Juárez Olguín, H.; Calderón Guzmán, D.; Hernández García, E.; Barragán Mejía, G. The Role of Dopamine and Its Dysfunction as a Consequence of Oxidative Stress. Oxidative Med. Cell. Longev. 2016, 2016, 9730467. [Google Scholar] [CrossRef]
- Kanazawa, K.; Sakakibara, H. High Content of Dopamine, a Strong Antioxidant, in Cavendish Banana. J. Agric. Food Chem. 2000, 48, 844–848. [Google Scholar] [CrossRef]
- Gumieniczek, A. Modification of Oxidative Stress by Pioglitazone in the Heart of Alloxan-Induced Diabetic Rabbits. J. Biomed. Sci. 2005, 12, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Villa, P.; Saccani, A.; Sica, A.; Ghezzi, P. Glutathione Protects Mice from Lethal Sepsis by Limiting Inflammation and Potentiating Host Defense. J. Infect. Dis. 2002, 185, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Delles, C.; Miller, W.H.; Dominiczak, A.F. Targeting Reactive Oxygen Species in Hypertension. Antioxid. Redox Signal. 2008, 10, 1061–1078. [Google Scholar] [CrossRef]
- Paravicini, T.M.; Touyz, R.M. NADPH Oxidases, Reactive Oxygen Species, and Hypertension: Clinical Implications and Therapeutic Possibilities. Diabetes Care 2008, 31, S170–S180. [Google Scholar] [CrossRef] [PubMed]
- Bessa, S.S.; Ali, E.M.M.; Hamdy, S.M. The Role of Glutathione S- Transferase M1 and T1 Gene Polymorphisms and Oxidative Stress-Related Parameters in Egyptian Patients with Essential Hypertension. Eur. J. Intern. Med. 2009, 20, 625–630. [Google Scholar] [CrossRef]
- Allen, F.H.; Bellard, S.; Brice, M.D.; Cartwright, B.A.; Doubleday, A.; Higgs, H.; Hummelink, T.; Hummelink-Peters, B.G.; Kennard, O.; Motherwell, W.D.S.; et al. The Cambridge Crystallographic Data Centre: Computer-Based Search, Retrieval, Analysis and Display of Information. Acta Crystallogr. B 1979, 35, 2331–2339. [Google Scholar] [CrossRef]
- Zhang, G.; Musgrave, C.B. Comparison of DFT Methods for Molecular Orbital Eigenvalue Calculations. J. Phys. Chem. A 2007, 111, 1554–1561. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Sjoberg, P.; Politzer, P. Use of the Electrostatic Potential at the Molecular Surface to Interpret and Predict Nucleophilic Processes. J. Phys. Chem. 1990, 94, 3959–3961. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The Fundamental Nature and Role of the Electrostatic Potential in Atoms and Molecules. Theor. Chem. Acc. 2002, 108, 134–142. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W.; Hill, C.; Carolina, N. Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6 2016; Semichem Inc.: Shawnee, KS, USA, 2016. [Google Scholar]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer, Version 21.5; University of Western Australia: Perth, Australia, 2021. [Google Scholar]
- Lu, T. Multiwfn. Software Manual, Version 2014; Beijing Kein Research Center for Natural Sciences: Beijing, China, 2014; Volume 3. [Google Scholar]
- Weinhold, F.; Landis, C.R. Natural Bond Orbitals and Extensions of Localized Bonding Concepts. Chem. Educ. Res. Pract. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking 1 1Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A Webserver for Ligand-Based Pharmacophore Detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Deterministic Pharmacophore Detection via Multiple Flexible Alignment of Drug-Like Molecules. J. Comput. Biol. 2008, 15, 737–754. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystallographic Data | GSHA | GSHB |
|---|---|---|
| Empirical formula | C10H17N3O6S | C10H17N3O6S |
| Formula weight | 307.33 | 307.33 |
| Crystal system | orthorhombic | orthorhombic |
| Space group | P212121 | P212121 |
| a (Å) | 5.2748(2) | 5.6131(11) |
| b (Å) | 8.3459(3) | 8.720(2) |
| c (Å) | 25.496(3) | 27.940(5) |
| α (°) | 90 | 90 |
| β (°) | 90 | 90 |
| γ (°) | 90 | 90 |
| Volume (Å3) | 1122.39(14) | 1367.6(5) |
| Z | 4 | 4 |
| ρcalc (g/cm3) | 1.819 | 1.492 |
| μ (mm−1) | 0.077 | 0.146 |
| F(000) | 648.0 | 648.0 |
| Crystal size/mm3 | 0.2 × 0.2 × 0.1 | 0.19 × 0.14 × 0.11 |
| Radiation | synchrotron (λ = 0.47670) | synchrotron (λ = 0.5636) |
| 2Θ range for data collection/° | 3.444 to 34.714 | 2.312 to 55.978 |
| Index ranges | −6 ≤ h ≤ 6 −10 ≤ k ≤ 10 −14 ≤ l ≤ 14 | −9 ≤ h ≤ 9 −14 ≤ k ≤ 14 −46 ≤ l ≤ 44 |
| Reflections collected | 5512 | 36,183 |
| Goodness of fit on F2 | 1.115 | 1.268 |
| Final R indexes [all data] | R1 = 0.0483, wR2 = 0.0449 | R1 = 0.0340, wR2 = 0.0780 |
| Largest diff. peak/hole (eÅ−3) | 0.24/−0.29 | 0.50/−0.53 |
| Flack parameter | −0.2(3) | −0.01(4) |
| Thermochemical Property | Neutral | Zwitterion | |
|---|---|---|---|
| Electronic Energy (kcal/mol) | −881,611.16 | −881,477.12 | −134.03 |
| Zero-Point Energy (kcal/mol) | −881,430.11 | −881,287.93 | −142.18 |
| Internal Energy (kcal/mol) | −881,419.18 | −881,280.20 | −138.98 |
| Enthalpy (kcal/mol) | −881,418.59 | −881,279.61 | −138.98 |
| Free Energy (kcal/mol) | −881,458.22 | −881,313.73 | −144.48 |
| Entropy (cal/molK) | 132.92 | 114.45 | 18,47 |
| Heat Capacity (cal/molK) | 69.59 | 50.57 | - |
| Polarizability (a.u.) | 174.96 | 162.49 | - |
| Dipole Moment (Debye) | 6.46 | 13.45 | - |
| Descriptors | (kcal/mol) |
|---|---|
| −212.678 | |
| 29.969 | |
| 242.647 | |
| Ionization Energy () | 212.678 |
| Electronic Affinity () | −29.969 |
| Electronegativity () | 91.355 |
| Chemical Potential () | −91.355 |
| Chemical Hardness () | 242.647 |
| Chemical Softness () | 0.004 |
| Electrophilicity Index () | 17.197 |
| D–H⋯A | D–H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) | (a) (a.u.) | (b) (a.u.) | (c) (a.u.) | (d) (a.u.) | (e) (a.u.) | |
|---|---|---|---|---|---|---|---|---|---|---|
| GSHA | ||||||||||
| S–H⋯O1 I | 1.2000 | 2.5900 | 3.691(5) | 152 | 0.0175 | 0.0655 | 0.0140 | −0.0116 | 0.0024 | 0.8 |
| S–H⋯O2 I | 1.2000 | 2.1500 | 3.261(8) | 153′ | 0.0174 | 0.0660 | 0.0140 | −0.0115 | 0.0025 | 0.8 |
| O6–H⋯O3 II | 0.8300 | 1.6600 | 2.465(4) | 164 | 0.0469 | 0.2126 | 0.0543 | −0.0554 | −0.0011 | 1.0 |
| N1–H⋯O1 III | 0.8900 | 1.8200 | 2.669(10) | 157′ | 0.0134 | 0.0512 | 0.0110 | −0.0091 | 0.0018 | 0.8 |
| N1–H⋯O2 II | 0.8900 | 2.2900 | 2.947(4) | 130 | 0.0302 | 0.1483 | 0.0331 | −0.0291 | 0.0040 | 0.9 |
| N1–H⋯O4 IV | 0.8900 | 2.4800 | 3.122(6) | 130 | 0.0085 | 0.0319 | 0.0067 | −0.0055 | 0.0012 | 0.8 |
| N2–H⋯O2 II | 0.8600 | 1.9900 | 2.675(7) | 136′ | 0.0244 | 0.1175 | 0.0251 | −0.0208 | 0.0043 | 0.8 |
| N3–H⋯O5 V | 0.8600 | 2.0700 | 2.672(8) | 127′ | 0.0214 | 0.1034 | 0.0216 | −0.0173 | 0.0043 | 0.8 |
| C2–H⋯O4 IV | 0.9700 | 2.4200 | 3.097(5) | 127 | 0.0116 | 0.0416 | 0.0089 | −0.0075 | 0.0015 | 0.8 |
| GSHB | ||||||||||
| S–H⋯O1 VII | 1.3400 | 2.1800 | 3.4603(8) | 158 | 0.0154 | 0.0538 | 0.0114 | −0.0094 | 0.0020 | 0.8 |
| N1–H⋯O4 VIII | 1.0200 | 1.8400 | 2.8034(6) | 155 | 0.0310 | 0.1209 | 0.0285 | −0.0267 | 0.0018 | 0.9 |
| N1–H⋯O1 IX | 1.0200 | 1.9600 | 2.8383(7) | 142′ | 0.0222 | 0.0956 | 0.0208 | −0.0177 | 0.0031 | 0.9 |
| N1–H⋯O2 II | 1.0200 | 1.7200 | 2.6959(6) | 158′ | 0.0438 | 0,1527 | 0.0407 | −0.0431 | −0.0025 | 1.1 |
| N2–H⋯O2 II | 1.0100 | 1.9600 | 2.8984(7) | 154 | 0.0222 | 0.0993 | 0.0214 | −0.0179 | 0.0035 | 0.8 |
| N3–H⋯O5 X | 1.0100 | 2.0000 | 2.8712(7) | 144′ | 0.0214 | 0.0924 | 0.0199 | −0.0166 | 0.0032 | 0.8 |
| O6–H⋯O3 XI | 0.9600 | 1.6400 | 2.5987(6) | 172 | 0.0495 | 0.1628 | 0.0463 | −0.0519 | −0.0056 | 1.1 |
| C3–H⋯O2 II | 1.0900 | 2.5200 | 3.3842(8) | 135 | 0.0098 | 0.0297 | 0.0068 | −0.0062 | 0.0006 | 0.9 |
| Symmetry codes: | (I) | (V) | (IX) | |||||||
| (II) | (VI) | (X) | ||||||||
| (III) | (VII) | (XI) | ||||||||
| (IV) | (VIII) | |||||||||
| Topological Properties: | (a) Total electronic density on BCP | (c) Lagrangian Kinetic energy | (e) Total energy density | |||||||
| (b) Laplacian of electron density on BCP | (d) Potential energy density | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguiar, A.S.N.; Borges, I.D.; Borges, L.L.; Dias, L.D.; Camargo, A.J.; Perjesi, P.; Napolitano, H.B. New Insights on Glutathione’s Supramolecular Arrangement and Its In Silico Analysis as an Angiotensin-Converting Enzyme Inhibitor. Molecules 2022, 27, 7958. https://doi.org/10.3390/molecules27227958
Aguiar ASN, Borges ID, Borges LL, Dias LD, Camargo AJ, Perjesi P, Napolitano HB. New Insights on Glutathione’s Supramolecular Arrangement and Its In Silico Analysis as an Angiotensin-Converting Enzyme Inhibitor. Molecules. 2022; 27(22):7958. https://doi.org/10.3390/molecules27227958
Chicago/Turabian StyleAguiar, Antônio S. N., Igor D. Borges, Leonardo L. Borges, Lucas D. Dias, Ademir J. Camargo, Pál Perjesi, and Hamilton B. Napolitano. 2022. "New Insights on Glutathione’s Supramolecular Arrangement and Its In Silico Analysis as an Angiotensin-Converting Enzyme Inhibitor" Molecules 27, no. 22: 7958. https://doi.org/10.3390/molecules27227958
APA StyleAguiar, A. S. N., Borges, I. D., Borges, L. L., Dias, L. D., Camargo, A. J., Perjesi, P., & Napolitano, H. B. (2022). New Insights on Glutathione’s Supramolecular Arrangement and Its In Silico Analysis as an Angiotensin-Converting Enzyme Inhibitor. Molecules, 27(22), 7958. https://doi.org/10.3390/molecules27227958