
Quantification of Gut Microbiota Dysbiosis-Related Organic Acids in Human Urine Using LC-MS/MS


Abstract
:
1. Introduction
2. Results and Discussion
2.1. LC-MS/MS Modifier Optimization
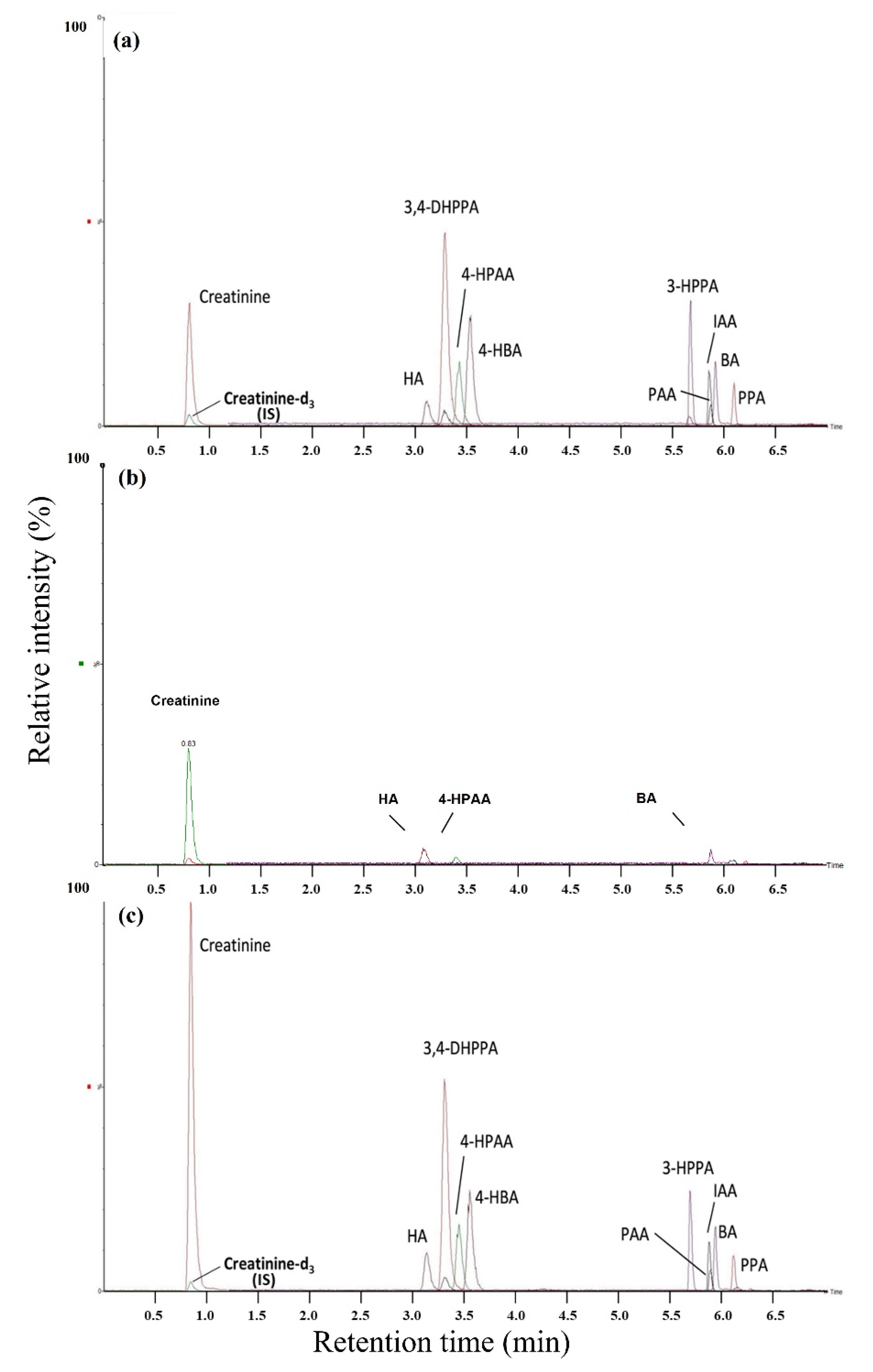
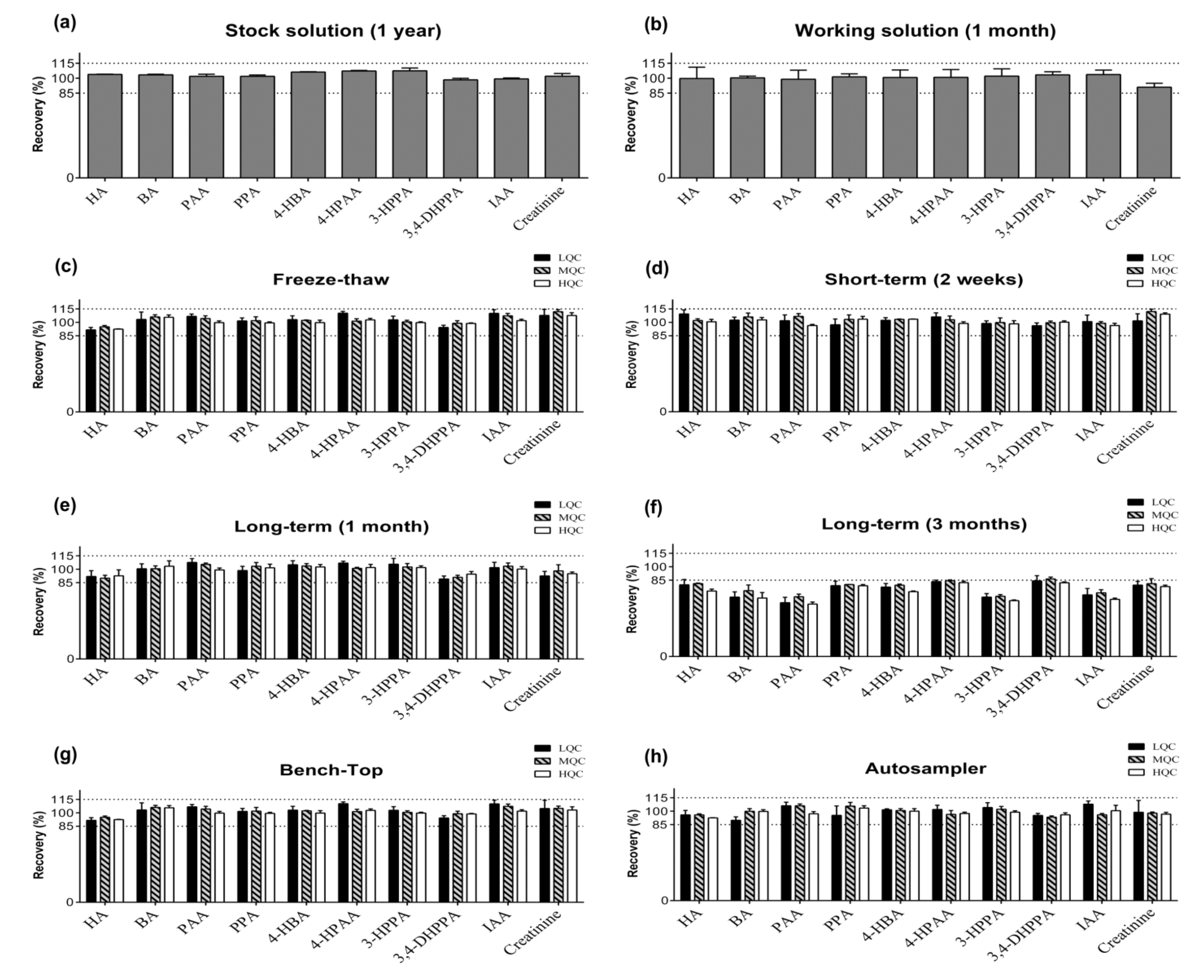
2.2. Method Validation
3. Material and Methods
3.1. Chemicals and Reagents
3.2. Sample Preparation
3.3. Calibration Curves
3.4. Instrumental and Analytic Conditions
3.5. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P. The human urine metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar] [CrossRef] [Green Version]
- Lord, R.S.; Bralley, J.A. Clinical applications of urinary organic acids. Part 2. Dysbiosis markers. Altern. Med. Rev. 2008, 13, 292–306. [Google Scholar] [PubMed]
- Guerra, A.; Folesani, G.; Mena, P.; Ticinesi, A.; Allegri, F.; Nouvenne, A.; Pinelli, S.; Del Rio, D.; Borghi, L.; Meschi, T. Hippuric acid in 24 h urine collections as a biomarker of fruits and vegetables intake in kidney stone formers. Int. J. Food Sci. Nutr. 2014, 65, 1033–1038. [Google Scholar] [CrossRef]
- Salmi, H.; Kuitunen, M.; Viljanen, M.; Lapatto, R. Cow’s milk allergy is associated with changes in urinary organic acid concentrations. Pediatr. Allergy Immunol. 2010, 21, e401–e406. [Google Scholar] [CrossRef]
- Salek, R.M.; Maguire, M.L.; Bentley, E.; Rubtsov, D.V.; Hough, T.; Cheeseman, M.; Nunez, D.; Sweatman, B.C.; Haselden, J.N.; Cox, R. A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol. Genom. 2007, 29, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvani, R.; Miccheli, A.; Capuani, G.; Miccheli, A.T.; Puccetti, C.; Delfini, M.; Iaconelli, A.; Nanni, G.; Mingrone, G. Gut microbiome-derived metabolites characterize a peculiar obese urinary metabotype. Int. J. Obes. 2010, 34, 1095–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Heiden, C.; Wauteks, E.; Ketting, D.; Duran, M.; Wadman, S. Gas chromatographic analysis of urinary tyrosine and phenylalanine metabolites in patients with gastrointestinal disorders. Clin. Chim. Acta 1971, 34, 289–296. [Google Scholar] [CrossRef]
- Phipps, A.; Stewart, J.; Wright, B.; Wilson, I. Effect of diet on the urinary excretion of hippuric acid and other dietary-derived aromatics in rat. A complex interaction between diet, gut microflora and substrate specificity. Xenobiotica 1998, 28, 527–537. [Google Scholar] [CrossRef]
- De Luca, C.; Passi, S.; Quattrucci, E. Simultaneous determination of sorbic acid, benzoic acid and parabens in foods: A new gas chromatography-mass spectrometry technique adopted in a survey on Italian foods and beverages. Food Addit. Contam. 1995, 12, 1–7. [Google Scholar] [CrossRef]
- Akira, K.; Negishi, E.; Yamamoto, C.; Baba, S. Evaluation of liver function by co-administration methodology using 13C-labelled benzoic acid and hippuric acid coupled with nuclear magnetic resonance spectroscopy. J. Pharm. Pharmacol. 1997, 49, 1242–1247. [Google Scholar] [CrossRef]
- Hasegawa, K.; Shiojima, S.; Koizmui, A.; Ikeda, M. Hippuric acid and o-cresol in the urine of workers exposed to toluene. Int. Arch. Occup. Environ. Health 1983, 52, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.A.; Macfarlane, G.T. Formation of phenolic and indolic compounds by anaerobic bacteria in the human large intestine. MicEc 1997, 33, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Van der Heiden, C.; Wadman, S.; Ketting, D.; De Bree, P. Urinary and faecal excretion of metabolizes of tyrosine and phenylalanine in a patient with cystic fibrosis and severely impaired amino acid absorption. Clin. Chim. Acta 1971, 31, 133–141. [Google Scholar] [CrossRef]
- Pollitt, R. Phenylpropionic acid in the urine of patients with phenylketonuria and normals. Clin. Chim. Acta 1974, 55, 317–322. [Google Scholar] [CrossRef]
- Sabelli, H.; Fawcett, J.; Gusovsky, F.; Javaid, J.; Edwards, J.; Jeffriess, H. Urinary phenyl acetate: A diagnostic test for depression? Science 1983, 220, 1187–1188. [Google Scholar] [CrossRef] [PubMed]
- Pietta, P.; Simonetti, P.; Gardana, C.; Brusamolino, A.; Morazzoni, P.; Bombardelli, E. Catechin metabolites after intake of green tea infusions. BioFactors 1998, 8, 111–118. [Google Scholar] [CrossRef]
- Chalmers, R.; Valman, H.; Liberman, M. Measurement of 4-hydroxyphenylacetic aciduria as a screening test for small-bowel disease. Clin. Chem. 1979, 25, 1791–1794. [Google Scholar] [CrossRef]
- Ward, N.C.; Croft, K.D.; Puddey, I.B.; Hodgson, J.M. Supplementation with grape seed polyphenols results in increased urinary excretion of 3-hydroxyphenylpropionic acid, an important metabolite of proanthocyanidins in humans. J. Agric. Food Chem. 2004, 52, 5545–5549. [Google Scholar] [CrossRef]
- Rechner, A.R.; Smith, M.A.; Kuhnle, G.; Gibson, G.R.; Debnam, E.S.; Srai, S.K.S.; Moore, K.P.; Rice-Evans, C.A. Colonic metabolism of dietary polyphenols: Influence of structure on microbial fermentation products. Free Radic. Biol. Med. 2004, 36, 212–225. [Google Scholar] [CrossRef]
- Chung, K.; Anderson, G.; Fulk, G. Formation of indoleacetic acid by intestinal anaerobes. J. Bacteriol. 1975, 124, 573–575. [Google Scholar] [CrossRef] [Green Version]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, K.N.; Michael, A.F.; Good, R.A. Tryptophan metabolism in a patient with phenylketonuria and scleroderma: A proposed explanation of the indole defect in phenylketonuria. Can. Med. Assoc. J. 1966, 94, 834. [Google Scholar] [PubMed]
- Tůma, P.; Samcová, E.; Štulík, K. Determination of the spectrum of low molecular mass organic acids in urine by capillary electrophoresis with contactless conductivity and ultraviolet photometric detection—An efficient tool for monitoring of inborn metabolic disorders. Anal. Chim. Acta 2011, 685, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-P.; Liao, C.-S. Comparison of ion-pair chromatography and capillary zone electrophoresis for the assay of organic acids as markers of abnormal metabolism. J. Chromatogr. 2004, 1051, 213–219. [Google Scholar] [CrossRef]
- Fernández-Bravo, J.; de Andrés, F.; Zougagh, M.; Ríos, Á. Selective screening of glutaric acid acidurias by capillary electrophoresis-mass spectrometry. J. Pharm. Biomed. Anal. 2017, 145, 40–45. [Google Scholar] [CrossRef]
- Wajner, M.; de Moura Coelho, D.; Ingrassia, R.; de Oliveira, A.B.; Busanello, E.N.B.; Raymond, K.; Pires, R.F.; de Souza, C.F.M.; Giugliani, R.; Vargas, C.R. Selective screening for organic acidemias by urine organic acid GC–MS analysis in Brazil: Fifteen-year experience. Clin. Chim. Acta 2009, 400, 77–81. [Google Scholar] [CrossRef]
- Jones, P.M.; Bennett, M.J. Urine organic acid analysis for inherited metabolic disease using gas chromatography-mass spectrometry. In Clinical Applications of Mass Spectrometry; Springer: Berlin/Heidelberg, Germany, 2010; pp. 423–431. [Google Scholar] [CrossRef]
- Griffin, J.L.; Atherton, H.; Shockcor, J.; Atzori, L. Metabolomics as a tool for cardiac research. Nat. Rev. Cardiol. 2011, 8, 630–643. [Google Scholar] [CrossRef]
- Zhou, L.; Yu, D.; Zheng, S.; Ouyang, R.; Wang, Y.; Xu, G. Gut microbiota-related metabolome analysis based on chromatography-mass spectrometry. TrAC Trends Anal. Chem. 2021, 143, 116375. [Google Scholar] [CrossRef]
- Höcker, O.; Flottmann, D.; Schmidt, T.C.; Neusüß, C. Non-targeted LC-MS and CE-MS for biomarker discovery in bioreactors: Influence of separation, mass spectrometry and data processing tools. Sci. Total Environ. 2021, 798, 149012. [Google Scholar] [CrossRef]
- Ramautar, R.; Nevedomskaya, E.; Mayboroda, O.A.; Deelder, A.M.; Wilson, I.D.; Gika, H.G.; Theodoridis, G.A.; Somsen, G.W.; de Jong, G.J. Metabolic profiling of human urine by CE-MS using a positively charged capillary coating and comparison with UPLC-MS. Mol. Biosyst. 2011, 7, 194–199. [Google Scholar] [CrossRef]
- Obrenovich, M.E.; Tima, M.; Polinkovsky, A.; Zhang, R.; Emancipator, S.N.; Donskey, C.J. Targeted metabolomics analysis identifies intestinal microbiota-derived urinary biomarkers of colonization resistance in antibiotic-treated mice. Antimicrob. Agents Chemother. 2017, 61, e00477-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Lin, Y.; Li, L.; Li, Y.; Zhang, X.; Wang, M.; Chen, Y.; Luo, L.; Lu, B.; Xie, Z. Targeted metabolomics for the quantitative measurement of 9 gut microbiota–host co-metabolites in rat serum, urine and feces by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2019, 1110, 133–143. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on Bioanalytical Method Validation. 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 3 June 2021).
- European Medicines Agency. ICH Guideline M10 on Bioanalytical Method Validation. 2019. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-guideline-m10-bioanalytical-method-validation-step-2b_en.pdf (accessed on 3 June 2021).
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N. HMDB 4.0: The human metabolome database for 2018. NAR 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Kostiainen, R.; Kauppila, T.J. Effect of eluent on the ionization process in liquid chromatography–mass spectrometry. J. Chromatogr. 2009, 1216, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Gao, W.; Phelps, M.A.; Wu, D.; Miller, D.D.; Dalton, J.T. Favorable effects of weak acids on negative-ion electrospray ionization mass spectrometry. AnaCh 2004, 76, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Rabaneda, F.; Jáuregui, O.; Casals, I.; Andrés-Lacueva, C.; Izquierdo-Pulido, M.; Lamuela-Raventós, R.M. Liquid chromatographic/electrospray ionization tandem mass spectrometric study of the phenolic composition of cocoa (Theobroma cacao). J. Mass Spectrom. 2003, 38, 35–42. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness metric approach and software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]
- Zou, B.; Sun, Y.; Xu, Z.; Chen, Y.; Li, L.; Lin, L.; Zhang, S.; Liao, Q.; Xie, Z. Rapid simultaneous determination of gut microbial phenylalanine, tyrosine, and tryptophan metabolites in rat serum, urine, and faeces using LC–MS/MS and its application to a type 2 diabetes mellitus study. Biomed. Chromatogr. 2021, 35, e4985. [Google Scholar] [CrossRef]
- Gasperotti, M.; Masuero, D.; Guella, G.; Mattivi, F.; Vrhovsek, U. Development of a targeted method for twenty-three metabolites related to polyphenol gut microbial metabolism in biological samples, using SPE and UHPLC–ESI-MS/MS. Talanta 2014, 128, 221–230. [Google Scholar] [CrossRef]
- Chiu, C.-H.; Chen, C.-T.; Cheng, M.-H.; Pao, L.-H.; Wang, C.; Wan, G.-H. Use of urinary hippuric acid and o-/p-/m-methyl hippuric acid to evaluate surgical smoke exposure in operating room healthcare personnel. Ecotoxicol. Environ. Saf. 2021, 217, 112231. [Google Scholar] [CrossRef]
- Jones, M.G.; Cooper, E.; Amjad, S.; Goodwin, C.S.; Barron, J.L.; Chalmers, R.A. Urinary and plasma organic acids and amino acids in chronic fatigue syndrome. Clin. Chim. Acta 2005, 361, 150–158. [Google Scholar] [CrossRef]
- Loke, W.M.; Jenner, A.M.; Proudfoot, J.M.; McKinley, A.J.; Hodgson, J.M.; Halliwell, B.; Croft, K.D. A metabolite profiling approach to identify biomarkers of flavonoid intake in humans. J. Nutr. 2009, 139, 2309–2314. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Analyte | Responses | |||
|---|---|---|---|---|
| 0.025% FA (pH = 2.95) | 0.025% AA (pH = 3.45) | 0.05% AA (pH = 3.29) | 0.1% AA (pH = 3.14) | |
| HA | 1.27 × 107 | 2.01 × 106 | 1.69 × 106 | 1.64 × 106 |
| BA | 3.92 × 105 | 1.85 × 107 | 1.5 × 107 | 1.09 × 107 |
| PAA | 8.21 × 104 | 9.53 × 105 | 9.11 × 105 | 6.94 × 105 |
| PPA | 5.26 × 105 | 1.6 × 107 | 1.24 × 107 | 8.77 × 106 |
| 4-HBA | 2.47 × 107 | 3.61 × 106 | 2.92 × 106 | 3.18 × 106 |
| 4-HPAA | 2.67 × 106 | 7.87 × 106 | 9.15 × 106 | 6.18 × 106 |
| 3-HPPA | 1.46 × 107 | 3.69 × 107 | 3.25 × 107 | 2.84 × 107 |
| 3,4-DHPPA | 1.77 × 107 | 3.01 × 106 | 8.57 × 106 | 1.42 × 107 |
| IAA | 9.65 × 106 | 1.02 × 107 | 1.46 × 107 | 1.47 × 107 |
| Creatinine | 1.69 × 106 | 6.8 × 106 | 6.21 × 106 | 5.98 × 106 |
| Analyte | Nominal Concentration (ng/mL) | Within-Run (n = 6) | Between-Run (n = 9) | |||||
|---|---|---|---|---|---|---|---|---|
| Concentration (ng/mL) | Accuracy (%) | Precision (CV, %) | Concentration (ng/mL) | Accuracy (%) | Precision (CV, %) | |||
| HA | LLOQ | 40 | 40.9 ± 5.5 | 102.3 | 13.3 | 38.5 ± 4.8 | 96.2 | 12.4 |
| LQC | 120 | 126.9 ± 9.7 | 105.8 | 7.7 | 118.8 ± 11.7 | 99.0 | 9.8 | |
| MQC | 360 | 381.4 ± 18.9 | 105.9 | 5.0 | 377.4 ± 14.4 | 104.8 | 3.8 | |
| HQC | 640 | 662.5 ± 38.7 | 103.5 | 5.8 | 667.7 ± 29.0 | 104.3 | 4.3 | |
| BA | LLOQ | 10 | 9.5 ± 0.9 | 94.6 | 9.9 | 9.4 ± 0.7 | 93.6 | 7.5 |
| LQC | 30 | 29.5 ± 1.4 | 98.2 | 4.6 | 28.1 ± 1.7 | 93.6 | 6.2 | |
| MQC | 90 | 88.9 ± 2.8 | 98.8 | 3.1 | 88.5 ± 2.3 | 98.4 | 2.6 | |
| HQC | 160 | 159.7 ± 4.4 | 99.8 | 2.7 | 161.4 ± 4.7 | 100.9 | 2.9 | |
| PAA | LLOQ | 40 | 34.4 ± 3.8 | 85.9 | 11.0 | 34.3 ± 3.0 | 85.8 | 8.7 |
| LQC | 120 | 122.7 ± 6.9 | 102.3 | 5.7 | 124.4 ± 7.0 | 103.6 | 5.7 | |
| MQC | 360 | 372.3 ± 13.9 | 103.4 | 3.7 | 392.6 ± 26.9 | 109.1 | 6.8 | |
| HQC | 640 | 625.3 ± 22.2 | 97.7 | 3.6 | 634.0 ± 22.2 | 99.1 | 3.5 | |
| PPA | LLOQ | 10 | 10.1 ± 0.7 | 100.6 | 6.5 | 9.7 ± 0.8 | 97.0 | 7.8 |
| LQC | 30 | 29.7 ± 1.0 | 98.9 | 3.5 | 28.4 ± 1.8 | 94.6 | 6.3 | |
| MQC | 90 | 90.1 ± 3.2 | 100.1 | 3.5 | 88.2 ± 3.3 | 98.0 | 3.7 | |
| HQC | 160 | 161.6 ± 2.4 | 101.0 | 1.5 | 156.6 ± 6.1 | 97.9 | 3.9 | |
| 4-HBA | LLOQ | 10 | 9.7 ± 0.2 | 97.1 | 2.4 | 9.1 ± 0.7 | 91.0 | 7.9 |
| LQC | 30 | 28.8 ± 1.0 | 96.1 | 3.5 | 28.0 ± 1.3 | 93.2 | 4.7 | |
| MQC | 90 | 89.6 ± 1.2 | 99.6 | 1.4 | 88.0 ± 2.3 | 97.8 | 2.6 | |
| HQC | 160 | 158.1 ± 2.7 | 98.8 | 1.7 | 157.1 ± 4.1 | 98.2 | 2.6 | |
| 4-HPAA | LLOQ | 20 | 18.6 ± 1.2 | 92.8 | 6.7 | 18.6 ± 1.4 | 93.0 | 7.4 |
| LQC | 60 | 56.7 ± 2.3 | 94.5 | 4.1 | 55.0 ± 3.2 | 91.6 | 5.8 | |
| MQC | 180 | 179.3 ± 10.7 | 99.6 | 5.9 | 176.9 ± 8.3 | 98.3 | 4.7 | |
| HQC | 320 | 312.6 ± 18.4 | 97.7 | 5.9 | 317.7 ± 16.3 | 99.3 | 5.1 | |
| 3-HPPA | LLOQ | 10 | 9.7 ± 0.7 | 96.6 | 7.3 | 9.3 ± 0.7 | 92.9 | 7.8 |
| LQC | 30 | 29.1 ± 0.8 | 97.0 | 2.9 | 29.0 ± 0.6 | 96.7 | 2.2 | |
| MQC | 90 | 89.8 ± 2.0 | 99.8 | 2.2 | 89.8 ± 1.9 | 99.8 | 2.2 | |
| HQC | 160 | 158.5 ± 4.4 | 99.1 | 2.8 | 158.7 ± 5.3 | 99.2 | 3.3 | |
| 3,4-DHPPA | LLOQ | 40 | 39.0 ± 3.4 | 97.6 | 8.7 | 36.3 ± 3.7 | 90.8 | 10.3 |
| LQC | 120 | 111.9 ± 4.1 | 93.2 | 3.7 | 114.4 ± 3.9 | 95.3 | 3.4 | |
| MQC | 360 | 353.7 ± 10.7 | 98.3 | 3.0 | 358.8 ± 9.6 | 99.7 | 2.7 | |
| HQC | 640 | 622.9 ± 11.1 | 97.3 | 1.8 | 629.2 ± 16.8 | 98.3 | 2.7 | |
| IAA | LLOQ | 10 | 9.9 ± 1.0 | 99.3 | 9.6 | 9.4 ± 0.9 | 93.8 | 10.1 |
| LQC | 30 | 30.1 ± 2.2 | 100.3 | 7.2 | 29.4 ± 1.9 | 98.2 | 6.3 | |
| MQC | 90 | 90.3 ± 4.2 | 100.4 | 4.6 | 90.0 ± 3.2 | 100.0 | 3.6 | |
| HQC | 160 | 159.6 ± 3.5 | 99.7 | 2.2 | 159.0 ± 4.0 | 99.4 | 2.5 | |
| Creatinine | LLOQ | 100 | 109.7 ± 9.8 | 109.7 | 9.8 | 109.3 ± 11.1 | 109.3 | 11.1 |
| LQC | 300 | 324.9 ± 21.9 | 108.3 | 7.3 | 324.6 ± 30.3 | 108.2 | 10.1 | |
| MQC | 900 | 893.7 ± 43.2 | 99.3 | 4.8 | 992.7 ± 27.9 | 110.3 | 3.1 | |
| HQC | 1600 | 1613 ± 78.4 | 100.8 | 4.9 | 1754 ± 41.6 | 109.6 | 2.6 | |
| Analyte | ESI Mode | Retention Time (min) | Q1 > Q3 (m/z) | Cone Voltage (V) | Collision Energy (eV) |
|---|---|---|---|---|---|
| HA | − | 3.1 | 178 > 134 | 10 | 12 |
| BA | − | 5.9 | 121 > 77 | 44 | 12 |
| PAA | − | 5.9 | 135 > 91 | 10 | 10 |
| PPA | − | 6.1 | 149 > 105 | 36 | 12 |
| 4-HBA | − | 3.6 | 137 > 93 | 10 | 14 |
| 4-HPAA | − | 3.5 | 151 > 107 | 14 | 12 |
| 3-HPPA | − | 5.7 | 165 > 121 | 10 | 12 |
| 3,4-DHPPA | − | 3.3 | 181 > 137 | 10 | 12 |
| IAA | − | 5.9 | 174 > 130 | 10 | 14 |
| Creatinine | + | 0.9 | 114 > 44 | 34 | 25 |
| Creatinine-d3 (IS) | + | 0.9 | 117 > 47 | 44 | 25 |
| Analyte | Nominal Concentration (ng/mL) | Matrix Factor, MF (%) | CV (%) | |
|---|---|---|---|---|
| HA | LQC | 120 | 105.8 | 7.7 |
| HQC | 640 | 103.5 | 5.8 | |
| BA | LQC | 30 | 99.6 | 5.7 |
| HQC | 160 | 98.9 | 4.6 | |
| PAA | LQC | 120 | 94.3 | 6.2 |
| HQC | 640 | 93.7 | 6.1 | |
| PPA | LQC | 30 | 107.8 | 4.7 |
| HQC | 160 | 105.3 | 3.7 | |
| 4-HBA | LQC | 30 | 105.8 | 4.3 |
| HQC | 160 | 103.3 | 4.0 | |
| 4-HPAA | LQC | 60 | 97.5 | 9.2 |
| HQC | 320 | 91.9 | 8.7 | |
| 3-HPPA | LQC | 30 | 105.8 | 4.7 |
| HQC | 160 | 104.0 | 5.0 | |
| 3,4-DHPPA | LQC | 120 | 93.2 | 3.7 |
| HQC | 640 | 97.3 | 1.8 | |
| IAA | LQC | 30 | 105.3 | 7.9 |
| HQC | 160 | 107.5 | 8.9 | |
| Creatinine | LQC | 300 | 27.7 | 8.1 |
| HQC | 1600 | 22.9 | 9.7 | |
| Creatinine | LQC | 300 | 108.3 a | 7.8 |
| HQC | 1600 | 100.8 a | 4.9 | |
| Analyte | Calibration a | Back-Calculated Concentration/Nominal Concentration b (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Range (ng/mL) | Slope | Intercept | r | 1st Level | 2nd Level | 3rd Level | 4th Level | 5th Level | 6th Level | 7th Level | 8th Level | |
| HA | 40–800 | 0.06 ± 0.01 | −0.72 ± 0.47 | 0.9973–0.9994 | 108.00 ± 0.04 | 101.46 ± 0.03 | 98.52 ± 0.07 | 96.24 ± 0.04 | 94.89 ± 0.01 | 95.63 ± 0.03 | 100.47 ± 0.03 | 104.75 ± 0.02 |
| BA | 10–200 | 0.33 ± 0.06 | 0.56 ± 1.01 | 0.9979–0.9994 | 106.33 ± 0.03 | 98.00 ± 0.03 | 103.00 ± 0.04 | 94.39 ± 0.02 | 96.08 ± 0.04 | 99.00 ± 0.05 | 100.42 ± 0.03 | 102.72 ± 0.01 |
| PAA | 40–800 | 0.06 ± 0.02 | −1.24 ± 0.38 | 0.9969–0.9979 | 114.33 ± 0.02 | 97.33 ± 0.06 | 96.54 ± 0.08 | 95.00 ± 0.05 | 94.02 ± 0.03 | 96.34 ± 0.04 | 101.69 ± 0.02 | 104.69 ± 0.01 |
| PPA | 10–200 | 0.21 ± 0.08 | −0.57 ± 0.54 | 0.9956–0.9996 | 111.33 ± 0.07 | 95.83 ± 0.01 | 99.92 ± 0.02 | 95.56 ± 0.03 | 95.42 ± 0.03 | 96.97 ± 0.05 | 101.84 ± 0.05 | 103.13 ± 0.01 |
| 4-HBA | 10–200 | 0.85 ± 0.37 | −2.16 ± 1.22 | 0.9983–0.9993 | 108.00 ± 0.02 | 98.00 ± 0.02 | 99.92 ± 0.03 | 96.06 ± 0.02 | 96.08 ± 0.04 | 98.30 ± 0.03 | 100.67 ± 0.03 | 102.88 ± 0.00 |
| 4-HPAA | 20–400 | 0.22 ± 0.07 | −1.05 ± 0.72 | 0.9974–0.9991 | 109.17 ± 0.02 | 98.42 ± 0.02 | 100.46 ± 0.04 | 95.47 ± 0.03 | 94.52 ± 0.02 | 97.70 ± 0.04 | 100.61 ± 0.03 | 103.86 ± 0.00 |
| 3-HPPA | 10–200 | 0.54 ± 0.14 | −1.53 ± 0.59 | 0.9981–0.9995 | 108.33 ± 0.03 | 97.17 ± 0.02 | 98.67 ± 0.03 | 98.11 ± 0.04 | 96.67 ± 0.01 | 97.60 ± 0.03 | 100.47 ± 0.04 | 102.90 ± 0.03 |
| 3,4-DHPPA | 40–800 | 0.29 ± 0.26 | 2.54 ± 2.79 | 0.9975–0.9982 | 97.75 ± 0.15 | 98.25 ± 0.07 | 102.83 ± 0.08 | 99.93 ± 0.06 | 104.65 ± 0.08 | 97.94 ± 0.05 | 98.89 ± 0.03 | 99.74 ± 0.04 |
| IAA | 10–200 | 0.15 ± 0.09 | −0.09 ± 0.08 | 0.9992–0.9999 | 101.33 ± 0.05 | 98.33 ± 0.06 | 102.08 ± 0.01 | 96.72 ± 0.03 | 101.00 ± 0.02 | 101.70 ± 0.02 | 98.02 ± 0.02 | 100.88 ± 0.02 |
| Creatinine | 100–2000 | 0.26 ± 0.02 | 0.72 ± 2.15 | 0.9996–0.9998 | 98.50 ± 0.05 | 98.47 ± 0.03 | 101.59 ± 0.02 | 102.22 ± 0.02 | 101.20 ± 0.02 | 98.37 ± 0.02 | 100.34 ± 0.01 | 99.33 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-T.; Huang, S.-Q.; Lin, C.-H.; Pao, L.-H.; Chiu, C.-H. Quantification of Gut Microbiota Dysbiosis-Related Organic Acids in Human Urine Using LC-MS/MS. Molecules 2022, 27, 5363. https://doi.org/10.3390/molecules27175363
Lee Y-T, Huang S-Q, Lin C-H, Pao L-H, Chiu C-H. Quantification of Gut Microbiota Dysbiosis-Related Organic Acids in Human Urine Using LC-MS/MS. Molecules. 2022; 27(17):5363. https://doi.org/10.3390/molecules27175363
Chicago/Turabian StyleLee, Yu-Tsung, Sui-Qing Huang, Ching-Hao Lin, Li-Heng Pao, and Chun-Hui Chiu. 2022. "Quantification of Gut Microbiota Dysbiosis-Related Organic Acids in Human Urine Using LC-MS/MS" Molecules 27, no. 17: 5363. https://doi.org/10.3390/molecules27175363
APA StyleLee, Y.-T., Huang, S.-Q., Lin, C.-H., Pao, L.-H., & Chiu, C.-H. (2022). Quantification of Gut Microbiota Dysbiosis-Related Organic Acids in Human Urine Using LC-MS/MS. Molecules, 27(17), 5363. https://doi.org/10.3390/molecules27175363