
Tetrahydrocurcumin-Related Vascular Protection: An Overview of the Findings from Animal Disease Models


 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
1.1. CUR- and THC-Related Vascular Protection in L-NAME-Induced Hypertension
1.2. THC-Induced Vascular Protection in the Context of NO Deficit
2. Protective Effects of THC in Cd2+-Induced Hypertension
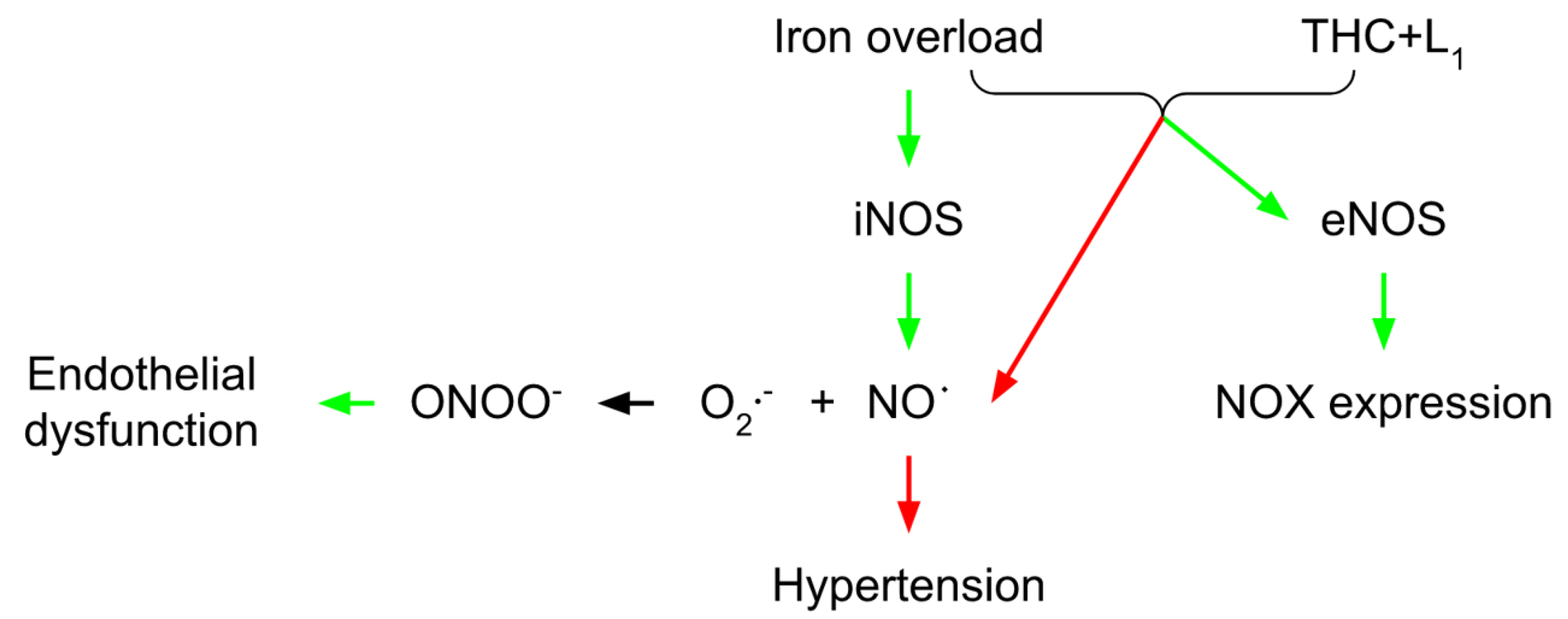
3. Vascular THC Protection in Conditions of Iron Overload
4. THC-Mediated Mitochondrial Impact in Brain Vasculature
5. THC-Induced Mitochondrial Remodeling in Brain Vascular Endothelial Cells
6. Antiangiogenic and Anti-Hypoxic Properties of THC
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ach | Acetylcholine |
| Ang II | Angiotensin |
| ARE | Antioxidant-response element |
| BBB | Blood–brain barrier |
| CAT | Catalase |
| CBS | Cystathionine-β-synthase |
| COO | Protein carbonyls |
| CSE | Cystathionine gamma lyase |
| CUR | Curcumin |
| DNMT | DNA methyltransferases |
| GCL | Glutamate-cysteine ligase |
| ECM | Extracellular matrix |
| eNOS | Endothelial nitric oxide synthase |
| GSH | Glutathione |
| Hcy | Homocysteine |
| HHcy | Hyperhomocysteinemia |
| HIF-1α | Hypoxia-inducible factor-1alpha |
| ICAM | Intracellular cell adhesion molecule-1 |
| iNOS | Inducible nitric oxide synthase |
| I/R | Ischemia/reperfusion |
| L-NAME | (ω)-nitro-L-arginine methyl ester |
| MDA | Malondialdehyde |
| MMP | Matrix metalloproteinase |
| MnSOD | Manganese superoxide dismutase |
| MPT | Mitochondrial permeability transition |
| MTHFR | Methylenetetrahydrofolate reductase |
| MVD | Microvascular density |
| Nrf2 | Nuclear factor-erythroid factor 2-related factor |
| NQO1 | NAD(P)H oxidoreductase |
| NTBI | Non-transferrin-bound iron |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SAHH | S-adenosyl-L-homocysteine hydrolase |
| SH | Sulfhydryl |
| SAH | S-adenosylhomocysteine |
| SAM | S-adenosylmethionine |
| SOD | Superoxide dismutase |
| STZ | Streptozotocin |
| THC | Tetrahydrocurcumin |
| TIMP | Tissue inhibitor of metalloproteinase |
| TJP | Tight junction protein |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
References
- Imran, M.; Nadeem, M.; Khan, M.A.; Ahmed, S.; Imran, A.; Amir, R.M.; Arshad, M.U.; Rauf, A.; Saeed, F.; Khan, S.; et al. Curcumin and its allied analogues: Epigenetic and health perspectives—A review. Czech J. Food Sci. 2017, 35, 285–310. [Google Scholar]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alamet, K.; et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Cox, F.F.; Misiou, A.; Vierkant, A.; Ale-Agha, N.; Grandoch, M.; Haendeler, J.; Altschmied, J. Protective effects of curcumin in cardiovascular diseases—impact on oxidative stress and mitochondria. Cells 2022, 11, 342. [Google Scholar] [CrossRef] [PubMed]
- Pourbagher-Shahri, A.M.; Farkhondeh, T.; Ashrafizadeh, M.; Talebi, M.; Samargahndian, S. Curcumin and cardiovascular diseases: Focus on cellular targets and cascades. Biomed. Pharmacother. 2021, 136, 111214. [Google Scholar] [CrossRef] [PubMed]
- Li, K.X.; Wang, Z.C.; Machuki, J.O.; Li, M.Z.; Wu, Y.J.; Niu, M.K.; Yu, K.Y.; Lu, Q.B.; Sun, H.J. Benefits of curcumin in the vasculature: A therapeutic candidate for vascular remodeling in arterial hypertension and pulmonary arterial hypertension? Front. Physiol. 2022, 13, 848867. [Google Scholar] [CrossRef] [PubMed]
- Alidadi, M.; Liberale, L.; Montecucco, F.; Majeed, M.; Al-Rasadi, K.; Banach, M.; Jamialahmadi, T.; Sahebkar, A. Protective effects of curcumin on endothelium: An updated review. Adv. Exp. Med. Biol. 2021, 1291, 103–119. [Google Scholar] [PubMed]
- Zhang, Z.B.; Luo, D.D.; Xie, J.H.; Xian, Y.F.; Lai, Z.Q.; Liu, Y.H.; Liu, W.H.; Chen, J.N.; Lai, X.P.; Lin, Z.X.; et al. Curcumin’s metabolites, tetrahydrocurcumin and octahydrocurcumin, possess superior anti-inflammatory effects in vivo through suppression of tak1-nf-κb pathway. Front. Pharmacol. 2018, 9, 1181. [Google Scholar] [CrossRef]
- Rege, S.A.; Varshneya, M.A.; Momin, S.A. A Mini-Review: Comparison between curcumin and tetrahydrocurcumin based on their activities. Croat. J. Food Sci. Technol. 2021, 13, 128–132. [Google Scholar] [CrossRef]
- Lai, C.S.; Ho, C.T.; Pan, M.H. The cancer chemopreventive and therapeutic potential of tetrahydrocurcumin. Biomolecules 2020, 10, 831. [Google Scholar] [CrossRef]
- Kukongviriyapan, U.; Apaijit, K.; Kukongviriyapan, V. Oxidative stress and cardiovascular dysfunction associated with cadmium exposure: Beneficial effects of curcumin and tetrahydrocurcumin. Tohoku. J. Exp. Med. 2016, 239, 25–38. [Google Scholar] [CrossRef]
- Nakmareong, S.; Kukongviriyapan, U.; Pakdeechote, P.; Donpunha, W.; Kukongviriyapan, V.; Kongyingyoes, B.; Sompamit, K.; Phisalaphong, C. Antioxidant and vascular protective effects of curcumin and tetrahydrocurcumin in rats with L-NAME-induced hypertension. Naunyn Schmiedeberg’s Arch. Pharmacol. 2011, 383, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Chai, H.; Yao, Q.; Lin, P.H.; Lumsden, A.B.; Chen, C. Curcumin blocks homocysteine-induced endothelial dysfunction in porcine coronary arteries. J. Vasc. Surg. 2004, 40, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- De Gennaro, C.V.; Rigamonti, A.; Fioretti, S.; Bonomo, S.; Manfredi, B.; Ferrario, P.; Bianchi, M.; Berti, F.; Muller, E.E.; Rossoni, G. Angiotensin-converting enzyme inhibition, and angiotensin AT1- receptor antagonism equally improve endothelial vasodilator function in L-NAME-induced hypertensive rats. Eur. J. Pharmacol. 2005, 516, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, V.; Sachdeva, J.; Golechha, M.; Kumari, S.; Arya, D.S. Curcumin protects rat myocardium against isoproterenol-induced ischemic injury: Attenuation of ventricular dysfunction through increased expression of Hsp27 along with strengthening antioxidant defense system. J. Cardiovasc. Pharmacol. 2010, 55, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovska, M.; Hadzi-Petrushev, N.; Nikodinovski, A.; Gagov, H.; Atanasova Panchevska, N.; Mitrokhin, V.; Kamkin, A.; Mladenov, M. Application of curcumin and its derivatives in the treatment of cardiovascular diseases: A review. Int. J. Food Prop. 2021, 24, 1510–1528. [Google Scholar] [CrossRef]
- Hadzi-Petrushev, N.; Angelovski, M.; Rebok, K.; Kamkin, A.; Mladenov, M. Antioxidant and anti-inflammatory effects of the monocarbonyl curcumin analogs B2BRBC and C66 in monocrotaline-induced right ventricular hypertrophy. J. Biochem. Mol. Toxicol. 2019, 33, e22353. [Google Scholar] [CrossRef]
- Hadzi-Petrushev, N.; Bogdanov, J.; Krajoska, J.; Ilievska, J.; Bogdanova-Popov, B.; Gjorgievska, E.; Mitrokhin, V.; Sopi, R.; Gagov, H.; Kamkin, A.; et al. Comparative study of the antioxidant properties of monocarbonyl curcumin analogs C66 and B2BrBC in isoproterenol-induced cardiac damage. Life Sci. 2018, 197, 10–18. [Google Scholar] [CrossRef]
- Bengmark, S. Curcumin, an atoxic antioxidant, and natural NFkappaB, cyclooxygenase-2, lipooxygenase, and inducible nitric oxide synthase inhibitor: A shield against acute and chronic diseases. J. Parenter. Enteral. Nutr. 2006, 30, 45–51. [Google Scholar] [CrossRef]
- Pae, H.O.; Jeong, G.S.; Jeong, S.O.; Kim, H.S.; Kim, S.A.; Kim, Y.C.; Yoo, S.J.; Kim, H.D.; Chung, H.T. Roles of heme oxygenase-1 in curcumin-induced growth inhibition in rat smooth muscle cells. Exp. Mol. Med. 2007, 39, 267–277. [Google Scholar] [CrossRef]
- Farombi, E.O.; Shrotriya, S.; Na, H.K.; Kim, S.H.; Surh, Y.J. Curcumin attenuates dimethylnitrosamine-induced liver injury in rats through Nrf2-mediated induction of heme oxygenase-1. Food Chem. Toxicol. 2008, 46, 1279–1287. [Google Scholar] [CrossRef]
- Okada, K.; Wangpoengtrakul, C.; Tanaka, T.; Toyokuni, S.; Uchida, K.; Osawa, T. Curcumin and especially tetrahydrocurcumin ameliorate oxidative stress-induced renal injury in mice. J. Nutr. 2001, 131, 2090–2095. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, Y.; Kawakishi, S.; Osawa, T. Involvement of the beta-diketone moiety in the antioxidative mechanism of tetrahydrocurcumin. Biochem. Pharmacol. 1996, 52, 519–525. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.H.; Lin-Shiau, S.Y.; Lin, J.K. Comparative studies on the suppression of nitric oxide synthase by curcumin and its hydrogenated metabolites through down-regulation of IkappaB kinase and NFkappaB activation in macrophages. Biochem. Pharmacol. 2000, 60, 1665–1676. [Google Scholar] [CrossRef]
- Kitamoto, S.; Egashira, K.; Kataoka, C.; Koyanagi, M.; Katoh, M.; Shimokawa, H.; Morishita, R.; Kaneda, Y.; Sueishi, K.; Takeshita, A. Increased activity of nuclear factor-kappaB participates in cardiovascular remodeling induced by chronic inhibition of nitric oxide synthesis in rats. Circulation 2000, 102, 806–812. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gauss, K.A.; Nelson-Overton, L.K.; Siemsen, D.W.; Gao, Y.; DeLeo, F.R.; Quinn, M.T. Role of NF-kappaB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-alpha. J. Leukoc. Biol. 2007, 82, 729–741. [Google Scholar] [CrossRef]
- Anrather, J.; Racchumi, G.; Iadecola, C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 2006, 281, 5657–5667. [Google Scholar] [CrossRef]
- Nakmareong, S.; Kukongviriyapan, U.; Pakdeechote, P.; Kukongviriyapan, V.; Kongyingyoes, B.; Donpunha, W.; Prachaney, P.; Phisalaphong, C. Tetrahydrocurcumin alleviates hypertension, aortic stiffening and oxidative stress in rats with nitric oxide deficiency. Hypertens. Res. 2012, 35, 418–425. [Google Scholar] [CrossRef]
- Murugan, P.; Pari, L. Antioxidant effect of tetrahydrocurcumin in streptozotocin-nicotinamide induced diabetic rats. Life Sci. 2006, 79, 1720–1728. [Google Scholar] [CrossRef]
- Pari, L.; Amali, D.R. Protective role of tetrahydrocurcumin (THC) an active principle of turmeric on chloroquine induced hepatotoxicity in rats. J. Pharm. Pharm. Sci. 2005, 8, 115–123. [Google Scholar]
- Wallerath, T.; Deckert, G.; Ternes, T.; Anderson, H.; Li, H.; Witte, K.; Forstermann, U. Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation 2002, 106, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- De Gennaro Colonna, V.; Rossoni, G.; Rigamonti, A.; Bonomo, S.; Manfredi, B.; Berti, F.; Muller, E. Enalapril and quinapril improve endothelial vasodilator function and aortic eNOS gene expression in L-NAME-treated rats. Eur. J. Pharmacol. 2002, 450, 61–66. [Google Scholar] [CrossRef]
- Sangartit, W.; Kukongviriyapan, U.; Donpunha, W.; Pakdeechote, P.; Kukongviriyapan, V.; Surawattanawan, P.; Greenwald, S.E. Tetrahydrocurcumin protects against cadmium-induced hypertension, raised arterial stiffness and vascular remodeling in mice. PLoS ONE 2014, 9, e114908. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, S.; Lemarie, C.A.; Esposito, B.; Lijnen, H.R.; Tedgui, A. Pressure-induced matrix metalloproteinase-9 contributes to early hypertensive remodeling. Circulation 2004, 109, 1041–1047. [Google Scholar] [CrossRef]
- Watts, S.W.; Rondelli, C.; Thakali, K.; Li, X.; Uhal, B.; Pervaiz, H.M.; Watson, E.R.; Fink, D.G. Morphological and biochemical characterization of remodeling in aorta and vena cava of DOCA-salt hypertensive rats. Am. J. Physiol. Heart. Circ. Physiol. 2007, 292, 2438–2448. [Google Scholar] [CrossRef]
- Grote, K.; Flach, I.; Luchtefeld, M.; Akin, E.; Holland, S.M.; Drexler, H.; Schieffer, B. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase-derived reactive oxygen species. Circ. Res. 2003, 92, 80–86. [Google Scholar] [CrossRef]
- Kirschvink, N.; Vincke, G.; Fievez, L.; Onclinx, C.; Wirth, D.; Belleflamme, M.; Louis, R.; Cataldo, D.; Peck, M.J.; Gustin, P. Repeated cadmium nebulizations induce pulmonary MMP-2 and MMP-9 production and emphysema in rats. Toxicology 2005, 211, 36–48. [Google Scholar] [CrossRef]
- Kundu, S.; Sengupta, S.; Chatterjee, S.; Mitra, S.; Bhattacharyya, A. Cadmium induces lung inflammation independent of lung cell proliferation: A molecular approach. J. Inflamm. 2009, 6, 19. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Huang, P.L. Disruption of the endothelial nitric oxide synthase gene: Effect on vascular response to injury. Am. J. Cardiol. 1998, 82, 57S–59S. [Google Scholar] [CrossRef]
- Javanmard, S.H.; Nematbakhsh, M.; Mahmoodi, F.; Mohajeri, M.R. L-Arginine supplementation enhances eNOS expression in experimental model of hypercholesterolemic rabbits’ aorta. Pathophysiology 2009, 16, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Yoopan, N.; Watcharasit, P.; Wongsawatkul, O.; Piyachaturawat, P.; Satayavivad, J. Attenuation of eNOS expression in cadmium-induced hypertensive rats. Toxicol. Lett. 2008, 176, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Alissa, E.M.; Ferns, G.A. Heavy metal poisoning and cardiovascular disease. J. Toxicol. 2011, 2011, 870125. [Google Scholar] [CrossRef]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.D.; Seggara, G.; Vo, P.A.; Macallister, R.J.; Hobbs, A.J.; Ahluwalia, A. Protection against lipopolysaccharide-induced endothelial dysfunction in resistance and conduit vasculature of iNOS knockout mice. FASEB J. 2003, 17, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Gaubin, Y.; Vaissade, F.; Croute, F.; Beau, B.; Soleilhavoup, J.; Murat, J. Implication of free radicals and glutathione in the mechanism of cadmium-induced expression of stress proteins in the A549 human lung cell-line. Biochim. Biophys. Acta. 2000, 1495, 4–13. [Google Scholar] [CrossRef]
- Tandon, S.K.; Singh, S.; Prasad, S.; Khandekar, K.; Dwivedi, V.K.; Chatterjee, M.; Mathur, N. Reversal of cadmium induced oxidative stress by chelating agent, antioxidant or their combination in rat. Toxicol. Lett. 2003, 145, 211–217. [Google Scholar] [CrossRef]
- Cuypers, A.; Plusquin, M.; Remans, T.; Jozefczak, M.; Keunen, E.; Gielen, H.; Opdenakker, K.; Nair, R.A.; Munters, E.; Artois, J.T.; et al. Cadmium stress: An oxidative challenge. Biometals 2010, 23, 927–940. [Google Scholar] [CrossRef]
- Lambertucci, R.H.; Leandro, C.G.; Vinolo, M.A.; Nachbar, R.T.; Dos Reis Silveira, L.; Hirabara, S.M.; Curi, R.; Pithon-Curi, T.C. The effects of palmitic acid on nitric oxide production by rat skeletal muscle: Mechanism via superoxide and iNOS activation. Cell. Physiol. Biochem. 2012, 30, 1169–1180. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity, and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef]
- Murugan, P.; Pari, L. Effect of tetrahydrocurcumin on lipid peroxidation and lipids in streptozotocin nicotinamide-induced diabetic rats. Basic. Clin. Pharmacol. Toxicol. 2006, 99, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.S.; Mudagal, M.P.; Goli, D. Cardioprotective effect of tetrahydrocurcumin and rutin on lipid peroxides and antioxidants in experimentally induced myocardial infarction in rats. Die Pharm. Int. J. Pharm. Sci. 2009, 64, 132–136. [Google Scholar]
- Yeh, T.K.; Wu, J.P.; Chang, L.W.; Tsai, M.H.; Chang, W.H.; Tsai, H.T.; Yang, C.S.; Lin, P. Comparative tissue distributions of cadmium chloride and cadmium-based quantum dot 705 in mice: Safety implications and applications. Nanotoxicology 2011, 5, 91–97. [Google Scholar] [CrossRef]
- Eybl, V.; Kotyzova, D.; Koutensky, J. Comparative study of natural antioxidants—Curcumin, resveratrol, and melatonin—In cadmium-induced oxidative damage in mice. Toxicology 2006, 225, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.L.; Hung, S.C.; Lee, T.S.; Tarng, D.C. Iron sucrose accelerates early atherogenesis by increasing superoxide production and upregulating adhesion molecules in CKD. J. Am. Soc. Nephrol. 2014, 25, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Cornejo, P.; Varela, P.; Videla, L.A.; Fernandez, V. Chronic iron overload enhances inducible nitric oxide synthase expression in rat liver. Nitric Oxide 2005, 13, 54–61. [Google Scholar] [CrossRef]
- Wu, F.; Tyml, K.; Wilson, J.X. iNOS expression requires NADPH oxidase-dependent redox signaling in microvascular endothelial cells. J. Cell. Physiol. 2008, 217, 207–214. [Google Scholar] [CrossRef]
- Oliveira-Paula, G.H.; Lacchini, R.; Tanus-Santos, J.E. Inducible nitric oxide synthase as a possible target in hypertension. Curr. Drug Targets 2014, 15, 164–174. [Google Scholar] [CrossRef]
- Kamkin, G.A.; Kamkina, V.O.; Shim, I.A.; Bilichenko, A.; Mitrokhin, M.V.; Kazansky, F.V.; Filatova, S.T.; Abramochkin, V.D.; Mladenov, I.M. The role of activation of two different sGC binding sites by NO-dependent and NO-independent mechanisms in the regulation of SACs in rat ventricular cardiomyocytes. Physiol. Rep. 2022, 10, e15246. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840844. [Google Scholar] [CrossRef]
- Sangartit, W.; Pakdeechote, P.; Kukongviriyapan, V.; Donpunha, W.; Shibahara, S.; Kukongviriyapan, U. Tetrahydrocurcumin in combination with deferiprone attenuates hypertension, vascular dysfunction, baroreflex dysfunction, and oxidative stress in iron-overloaded mice. Vascul. Pharmacol. 2016, 87, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Thephinlap, C.; Phisalaphong, C.; Lailerd, N.; Chattipakorn, N.; Winichagoon, P.; Vadolas, J.; Fucharoen, S.; Porter, J.B.; Srichairatanakool, S. Reversal of cardiac iron loading and dysfunction in thalassemic mice by curcuminoids. Med. Chem. 2011, 7, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Srichairatanakool, S.; Thephinlap, C.; Phisalaphong, C.; Porter, J.B.; Fucharoen, S. Curcumin contributes to in vitro removal of non-transferrin-bound iron by deferiprone and desferrioxamine in thalassemic plasma. Med. Chem. 2007, 3, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.Y.; Wilkinson, J.T.; Pietsch, E.C.; Buss, J.L.; Wang, W.; Planalp, R.; Torti, F.M.; Torti, S.V. Iron chelation in the biological activity of curcumin. Free Radic. Biol. Med. 2006, 40, 1152–1160. [Google Scholar] [CrossRef]
- Mondal, K.N.; Behera, J.; Kelly, E.K.; George, K.A.; Tyagi, K.P.; Tyagi, N. Tetrahydrocurcumin epigenetically mitigates mitochondrial dysfunction in brain vasculature during ischemic stroke. Neurochem. Int. 2019, 122, 120–138. [Google Scholar] [CrossRef]
- Veltkamp, R.; Siebing, D.A.; Sun, L.; Heiland, S.; Bieber, K.; Marti, H.H.; Nagel, S.; Schwab, S.; Schwaninger, M. Hyperbaric oxygen reduces blood-brain barrier damage and edema after transient focal cerebral ischemia. Stroke 2005, 36, 1679–1683. [Google Scholar] [CrossRef]
- Wald, D.S.; Law, M.; Morris, J.K. Homocysteine and cardiovascular disease: Evidence on causality from a meta-analysis. Br. Med. J. 2002, 325, 1202–1206. [Google Scholar] [CrossRef]
- Bostom, A.G.; Rosenberg, I.H.; Silbershatz, H.; Jacques, P.F.; Selhub, J.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Nonfasting plasma total homocysteine levels and stroke incidence in elderly persons: The Framingham Study. Ann. Intern. Med. 1999, 131, 352–355. [Google Scholar] [CrossRef]
- Iso, H.; Moriyama, Y.; Sato, S.; Kitamura, A.; Tanigawa, T.; Yamagishi, K.; Imano, H.; Ohira, T.; Okamura, T.; Naito, Y.; et al. Serum total homocysteine concentrations and risk of stroke and its subtypes in Japanese. Circulation 2004, 109, 2766–2772. [Google Scholar] [CrossRef]
- Nappo, F.; De, R.N.; Marfella, R.; De, L.D.; Ingrosso, D.; Perna, A.F.; Farzati, B.; Giugliano, D. Impairment of endothelial functions by acute hyperhomocysteinemia and reversal by antioxidant vitamins. JAMA 1999, 281, 2113–2118. [Google Scholar] [CrossRef]
- Rajeswari, A. Curcumin protects mouse brain from oxidative stress caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Eur. Rev. Med. Pharmacol. Sci. 2006, 10, 157–161. [Google Scholar] [PubMed]
- Stavrovskaya, I.G.; Kristal, B.S. The powerhouse takes control of the cell: Is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death. Free Radic. Biol. Med. 2005, 38, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Romanic, A.M.; White, R.F.; Arleth, A.J.; Ohlstein, E.H.; Barone, F.C. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: Inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 1998, 29, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Dollery, C.M.; Humphries, S.E.; McClelland, A.; Latchman, D.S.; McEwan, J.R. Expression of tissue inhibitor of matrix metalloproteinases 1 by use of an adenoviral vector inhibits smooth muscle cell migration and reduces neointimal hyperplasia in the rat model of vascular balloon injury. Circulation 1999, 99, 3199–3205. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Refsum, H.; Ueland, P.M.; Nygard, O.; Vollset, S.E. Homocysteine, and cardiovascular disease. Annu. Rev. Med. 1998, 49, 31–62. [Google Scholar] [CrossRef]
- Tyagi, N.; Ovechkin, A.V.; Lominadze, D.; Moshal, K.S.; Tyagi, S.C. Mitochondrial mechanism of microvascular endothelial cells apoptosis in hyperhomocysteinemia. J. Cell Biochem. 2006, 98, 1150–1162. [Google Scholar] [CrossRef]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox. Biol. 2014, 4, 6–13. [Google Scholar] [CrossRef]
- Reddy, P.H. Role of mitochondria in neurodegenerative diseases: Mitochondria as a therapeutic target in Alzheimer’s disease. CNS Spectrums 2009, 14, 8–13. [Google Scholar] [CrossRef]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009332. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-ros crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 2012, 329635. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Givvimani, S.; Sathnur, P.B.; Tyagi, S.C.; Tyagi, N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience 2013, 252, 302–319. [Google Scholar] [CrossRef] [PubMed]
- Kalani, A.; Kamat, P.K.; Givvimani, S.; Brown, K.; Metreveli, N.; Tyagi, S.C.; Tyagi, N. Nutri-epigenetics ameliorates blood-brain barrier damage and neurodegeneration in hyperhomocysteinemia: Role of folic acid. J. Mol. Neurosci. 2014, 52, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Vacek, C.J.; Behera, B.; George, K.A.; Kamat, K.P.; Kalani, A.; Tyagi, N. Tetrahydrocurcumin ameliorates homocysteine-mediated mitochondrial remodeling in brain endothelial cells. J. Cell. Physiol. 2018, 233, 3080–3092. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef]
- Tyagi, N.; Qipshidze, N.; Munjal, C.; Vacek, J.C.; Metreveli, N.; Givvimani, S.; Tyagi, S.C. Tetrahydrocurcumin ameliorates homocysteinylated cytochrome-c mediated autophagy in hyperhomocysteinemia mice after cerebral ischemia. J. Mol. Neurosci. 2012, 47, 128–138. [Google Scholar] [CrossRef]
- Yoysungnoen, B.; Bhattarakosol, P.; Patumraj, S.; Changtam, C. Effects of tetrahydrocurcumin on hypoxia-inducible factor-1α and vascular endothelial growth factor expression in cervical cancer cell-induced angiogenesis in nude mice. Biomed Res. Int. 2015, 2015, 391748. [Google Scholar] [CrossRef]
- Yoysungnoen-Chintana, P.; Bhattarakosol, P.; Patumraj, S. Antitumor and antiangiogenic activities of curcumin in cervical cancer xenografts in nude mice. Biomed Res. Int. 2014, 2014, 817972. [Google Scholar] [CrossRef]
- Pradeep, C.R.; Sunila, E.S.; Kuttan, G. Expression of vascular endothelial growth factor (VEGF) and VEGF receptors in tumor angiogenesis and malignancies. Integ. Cancer Ther. 2005, 4, 315–321. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Ferrara, N. Role of vascular endothelial growth factor in the regulation of physiological angiogenesis. Am. J. Physiol. Cell Physiol. 2001, 280, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: Therapeutic implications. Semin. Oncol. 2002, 29, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Kazanski, V.; Mitrokhin, V.M.; Mladenov, M.I.; Kamkin, A.G. Cytokine effects on mechano-induced electrical activity in atrial myocardium. Immunol. Investig. 2017, 46, 22–37. [Google Scholar] [CrossRef]
- Meyer, R.D.; Singh, A.; Majnoun, F.; Latz, C.; Lashkari, K.; Rahimi, N. Substitution of C-terminus of VEGFR-2 with VEGFR-1 promotes VEGFR-1 activation and endothelial cell proliferation. Oncogene 2004, 23, 5523–5531. [Google Scholar] [CrossRef]
- Shibuya, M. Tyrosine kinase receptor Flt/VEGFR family: Its characterization related to angiogenesis and cancer. Genes Cancer 2011, 1, 1119–1123. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signali ng in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Boehm, T.; Folkman, J.; Browder, T.; O’Reilly, M.S. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390, 404–407. [Google Scholar] [CrossRef]
- Yodkeeree, S.; Garbisa, S.; Limtrakul, P. Tetrahydrocurcumin inhibits HT1080 cell migration and invasion via downregulation of MMPs and uPA. Acta Pharmacol. Sin. 2008, 29, 853–860. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Li, C.; Wang, S.; Avtanski, D.; Hadzi-Petrushev, N.; Mitrokhin, V.; Mladenov, M.; Wang, F. Tetrahydrocurcumin-Related Vascular Protection: An Overview of the Findings from Animal Disease Models. Molecules 2022, 27, 5100. https://doi.org/10.3390/molecules27165100
Zhang L, Li C, Wang S, Avtanski D, Hadzi-Petrushev N, Mitrokhin V, Mladenov M, Wang F. Tetrahydrocurcumin-Related Vascular Protection: An Overview of the Findings from Animal Disease Models. Molecules. 2022; 27(16):5100. https://doi.org/10.3390/molecules27165100
Chicago/Turabian StyleZhang, Li, Changhu Li, Sicheng Wang, Dimiter Avtanski, Nikola Hadzi-Petrushev, Vadim Mitrokhin, Mitko Mladenov, and Feng Wang. 2022. "Tetrahydrocurcumin-Related Vascular Protection: An Overview of the Findings from Animal Disease Models" Molecules 27, no. 16: 5100. https://doi.org/10.3390/molecules27165100
APA StyleZhang, L., Li, C., Wang, S., Avtanski, D., Hadzi-Petrushev, N., Mitrokhin, V., Mladenov, M., & Wang, F. (2022). Tetrahydrocurcumin-Related Vascular Protection: An Overview of the Findings from Animal Disease Models. Molecules, 27(16), 5100. https://doi.org/10.3390/molecules27165100