
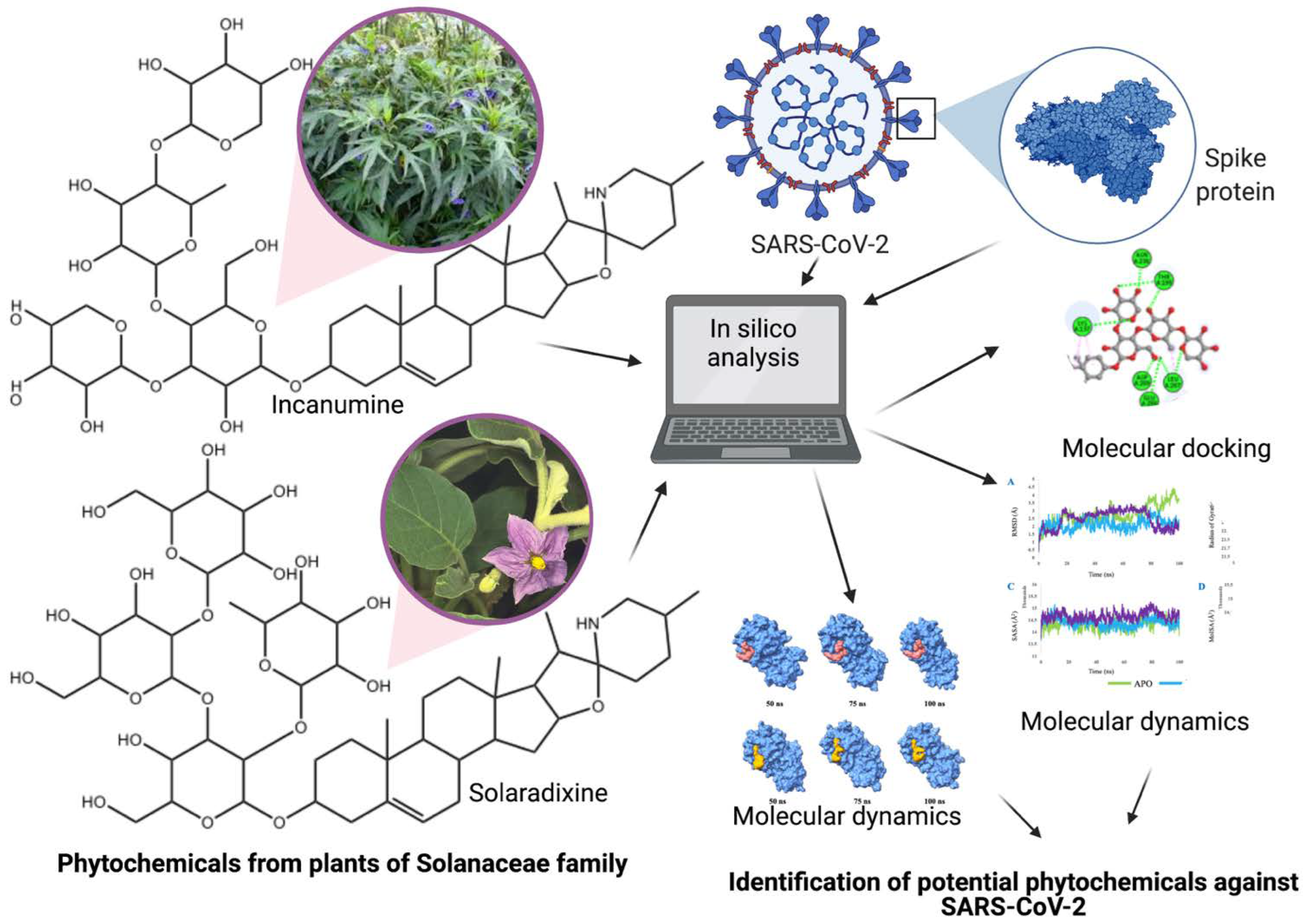
Solanaceae Family Phytochemicals as Inhibitors of 3C-Like Protease of SARS-CoV-2: An In Silico Analysis

 , , , ,
, , , , 
 , ,
, , 
Abstract
:1. Introduction
2. Methods
2.1. Molecular Docking
2.2. Lipinski’s Rule of Five
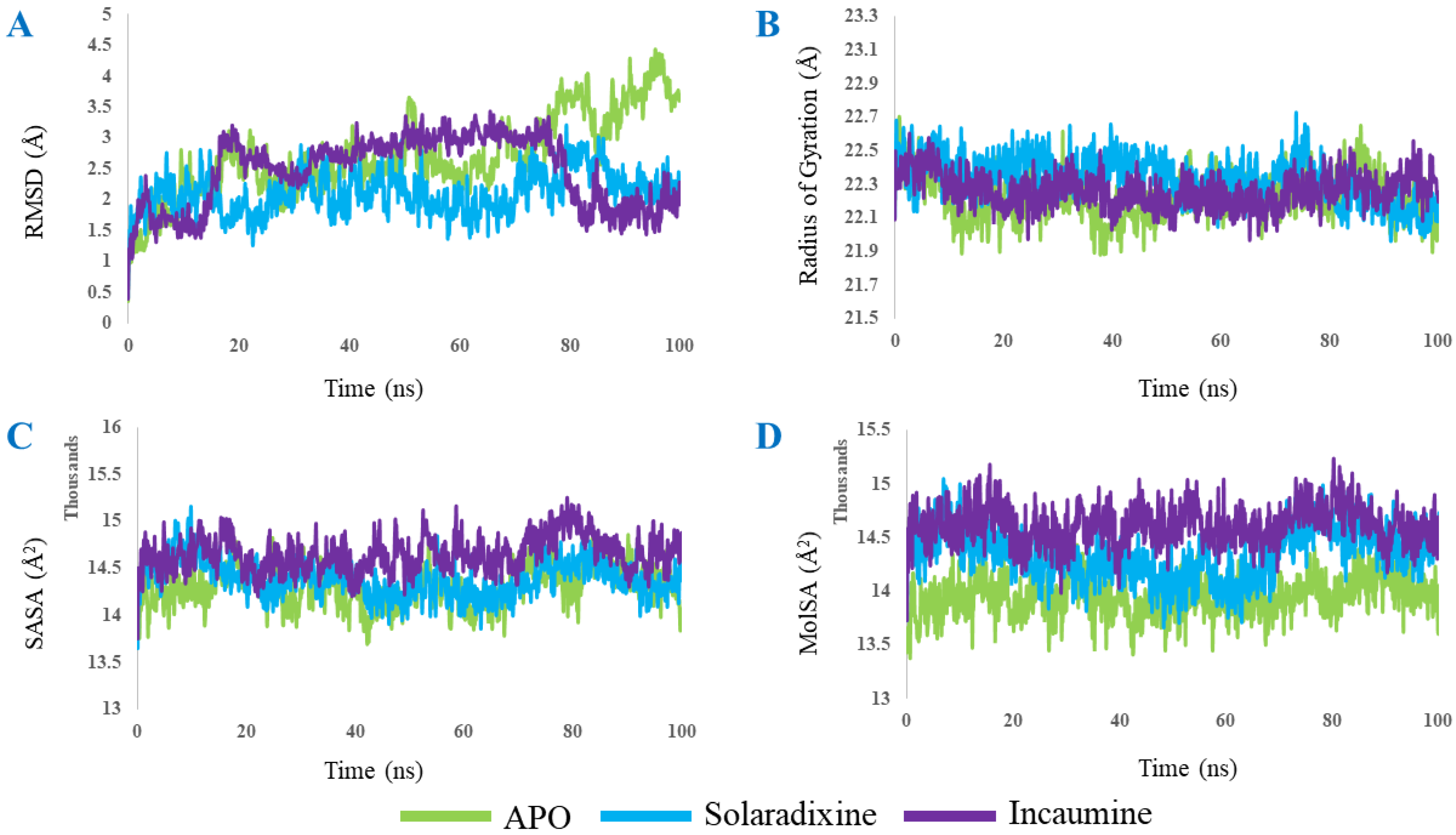
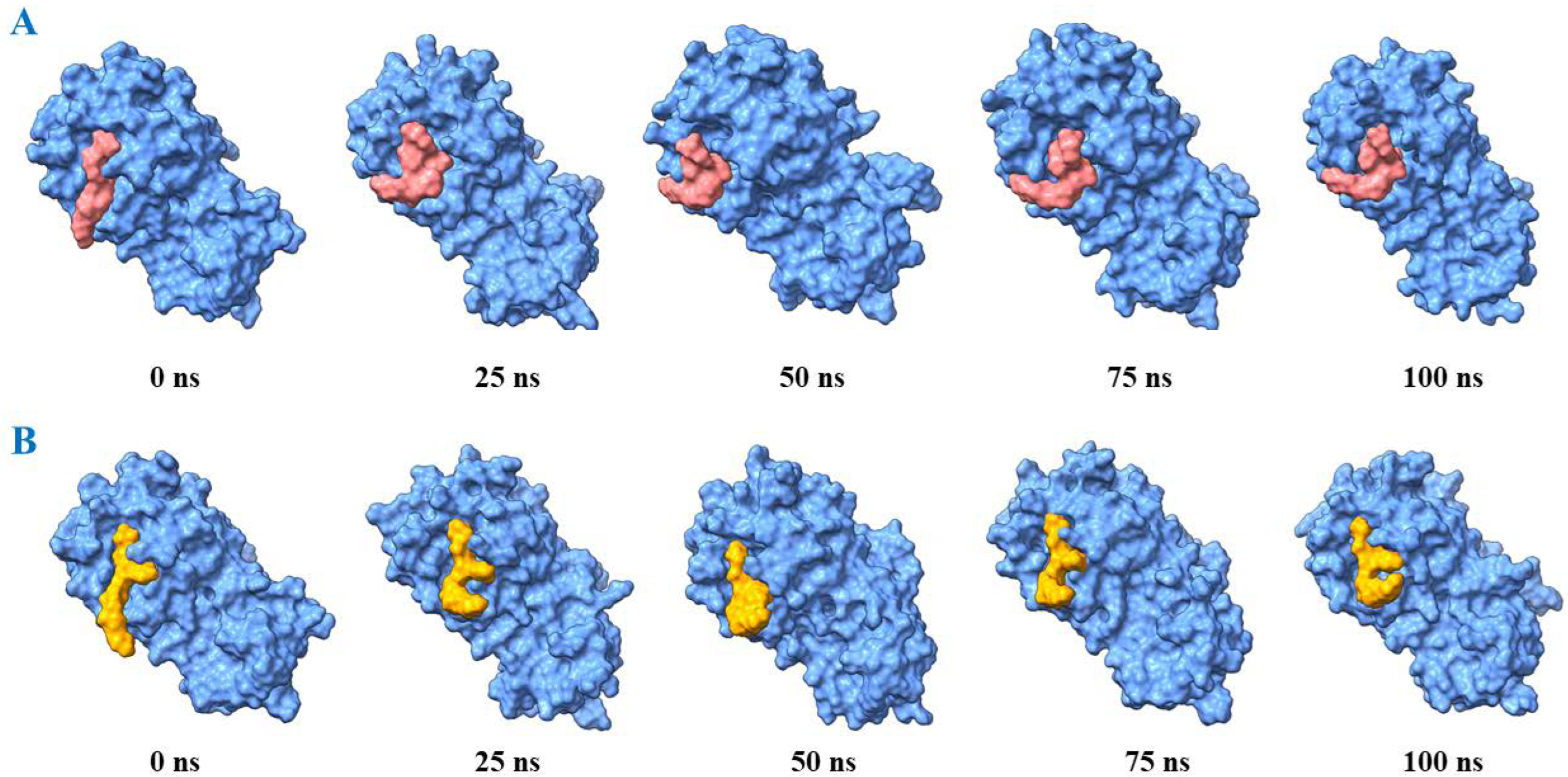
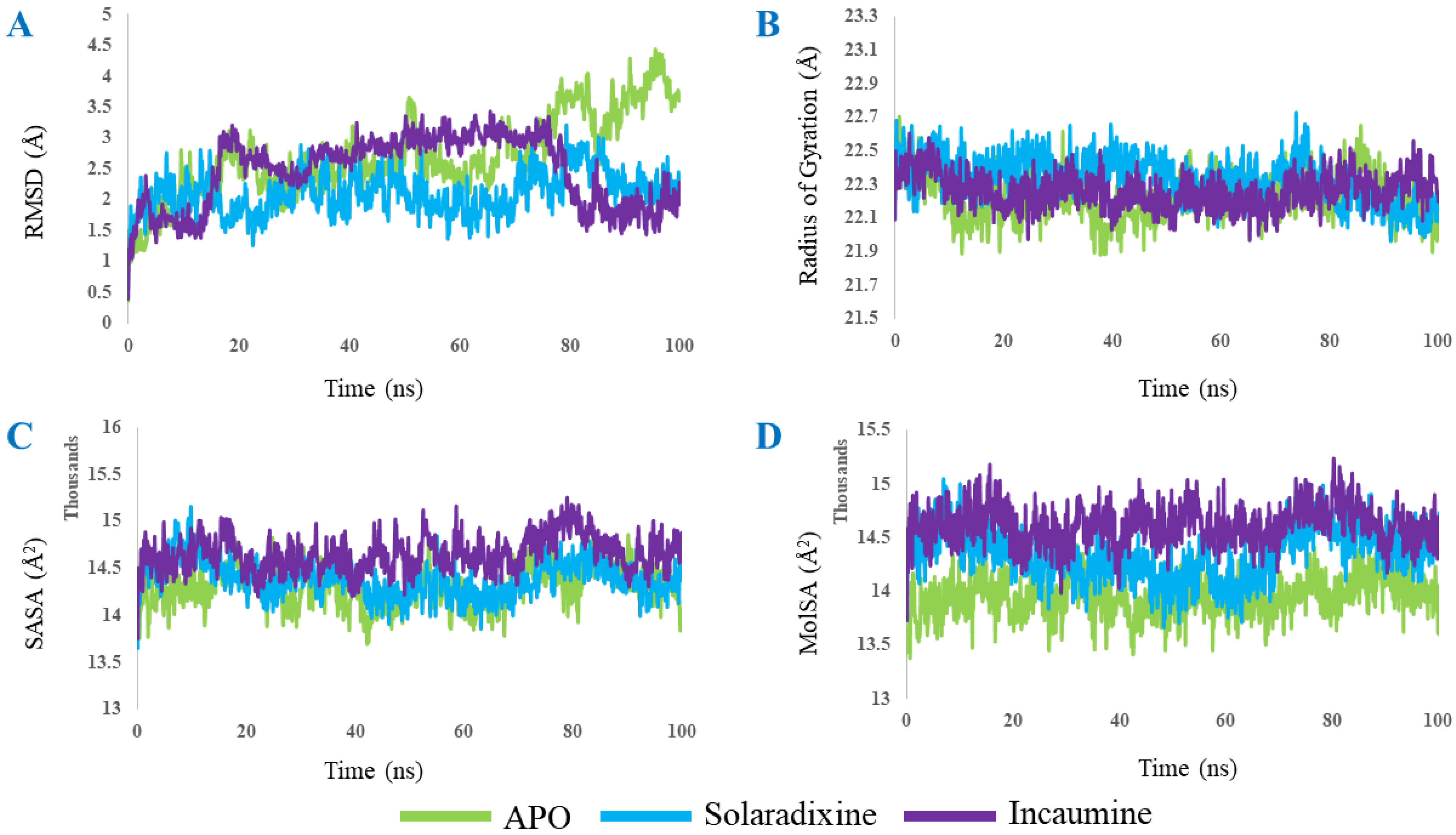
2.3. Molecular Dynamics
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tyrrell, D.A.J.; Bynoe, M.L. Cultivation of novel type of common-cold virus in organ cultures. BMJ 1965, 1, 1467–1470. [Google Scholar] [CrossRef] [Green Version]
- Hamre, D.; Procknow, J.J. A new virus isolated from the human respiratory tract. Proc. Soc. Exp. Biol. Med. 1966, 121, 190–193. [Google Scholar] [CrossRef]
- Bradburne, A.F.; Bynoe, M.L.; Tyrrell, D.A. Effects of a “new” human respiratory virus in volunteers. BMJ 1967, 3, 767–769. [Google Scholar] [CrossRef] [Green Version]
- van der Hoek, L. Human coronaviruses: What do they cause? Antivir. Ther. 2007, 12, 651–658. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-CoV: A comparative overview. Le Infez. Med. 2020, 2, 174–184. [Google Scholar]
- Ozili, P.; Arun, T. Spillover of COVID-19: Impact on the Global Economy. 2020. Available online: https://ssrn.com/abstract=3562570 (accessed on 22 July 2022).
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020, 30, 12. [Google Scholar] [CrossRef]
- Ang, L.; Lee, H.W.; Choi, J.Y.; Zhang, J.; Lee, M.S. Herbal medicine and pattern identification for treating COVID-19: A rapid review of guidelines. Integr. Med. Res. 2020, 9, 100407. [Google Scholar] [CrossRef]
- World Health Organization. Therapeutics and COVID-19. Living Guideline. 2022. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2022.4 (accessed on 15 July 2022).
- World Health Organization. Traditional 0407, Complementary and Integrative Medicine. In Proceedings of the WHO Expert Meeting on Evaluation of Traditional Chinese Medicine in the Treatment of COVID-19, Virtual meeting, 28 February–2 March 2022. [Google Scholar]
- Nugraha, R.V.; Ridwansyah, H.; Ghozali, M.; Khairani, A.F.; Atik, N. Traditional herbal medicine candidates as complementary treatments for COVID-19: A review of their mechanisms, pros and cons. Evid. -Based Complementary Altern. Med. 2020, 2020, 2560645. [Google Scholar] [CrossRef]
- Soleymani, S.; Naghizadeh, A.; Karimi, M.; Zarei, A.; Mardi, R.; Kordafshari, G.; Esmaelzadeh, N.; Zargaran, A. COVID-19: General strategies for herbal therapies. J. Evid. -Based Integr. Med. 2022, 27, 1–18. [Google Scholar] [CrossRef]
- Majnooni, M.B.; Fakhri, S.; Bahrami, G.; Naseri, M.; Farzaei, M.H.; Echeverría, J. Alkaloids as potential phytochemicals against SARS-CoV-2: Approaches to the associated pivotal mechanisms. Evid. -Based Complementary Integr. Med. 2021, 2021, 6632623. [Google Scholar] [CrossRef]
- Paul, A.K.; Hossain, M.K.; Mahboob, T.; Nissapatorn, V.; Wilairatana, P.; Jahan, R.; Jannat, K.; Bondhon, T.A.; Hasan, A.; de Lourdes Pereira, M.; et al. Does oxidative stress management help alleviation of COVID-19 symptoms in patients experiencing diabetes? Nutrients 2022, 14, 321. [Google Scholar] [CrossRef]
- Jannat, K.; Paul, A.K.; Bondhon, T.A.; Hasan, A.; Nawaz, M.; Jahan, R.; Mahboob, T.; Nissapatorn, V.; Wilairatana, P.; Pereira, M.d.L.; et al. Nanotechnology applications of flavonoids for viral diseases. Pharmaceutics 2021, 13, 1895. [Google Scholar] [CrossRef]
- Paul, A.K.; Jahan, R.; Bondhon, T.A.; Jannat, K.; Hasan, A.; Rahmatullah, M.; Nissapatorn, V.; Pereira, M.L.; Wiart, C. Potential role of flavonoids against SARS-CoV-2 induced diarrhea. Trop. Biomed. 2021, 38, 360–365. [Google Scholar] [CrossRef]
- Solnier, J.; Fladerer, J.-P. Flavonoids: A complementary approach to conventional therapy of COVID-19? Phytochem. Rev. 2021, 20, 773–795. [Google Scholar] [CrossRef]
- Kaul, R.; Paul, P.; Kumar, S.; Büsselberg, D.; Dwivedi, V.D.; Chaari, A. Promising antiviral activities of natural flavonoids against SARS-CoV-2 targets: Systematic review. Int. J. Mol. Sci. 2021, 22, 11069. [Google Scholar] [CrossRef]
- Tabari, M.A.K.; Iranpanah, A.; Bahramsoltani, R.; Rahimi, R. Flavonoids as promising antiviral agents against SARS-CoV-2 infection: A mechanistic review. Molecules 2021, 26, 3900. [Google Scholar] [CrossRef]
- Hasan, A.; Nahar, N.; Jannat, K.; Bondhon, T.A.; Jahan, R.; Rahmatullah, M. In silico studies on phytochemicals of Pimpinella anisum L. (Apiaceae) as potential inhibitors of SARS-CoV-2 3C-like protease. Int. J. Ayurveda 2020, 5, 8–14. [Google Scholar]
- Bondhon, T.A.; Mahamud, R.A.; Jannat, K.; Hasan, A.; Jahan, R.; Rahmatullah, M. In silico binding studies with b-sitosterol and some of its fatty acid esters to 3C-like protease of SARS-CoV-2. J. Med. Plants Stud. 2020, 8, 86–90. [Google Scholar] [CrossRef]
- Bondhon, T.A.; Rana, M.A.H.; Hasan, A.; Jahan, R.; Jannat, K.; Rahmatullah, M. Evaluation of phytochemicals of Cassia occidentalis L. for their binding affinities to SARS-CoV-2 3C-like protease: An in-silico approach. Asian J. Res. Infect. Dis. 2020, 4, 8–14. [Google Scholar] [CrossRef]
- Jannat, K.; Hasan, A.; Mahamud, R.A.; Jahan, R.; Bondhon, T.A.; Farzana, B.; Rahmatullah, M. In silico screening of Vigna radiata and Vigna mungo phytochemicals for their binding affinity to SARS-CoV-2 (COVID-19) main protease (3CLpro). J. Med. Plants Stud. 2020, 8, 89–95. [Google Scholar]
- Hasan, A.; Mahamud, R.A.; Bondhon, T.A.; Jannat, K.; Farzana, B.; Jahan, R.; Rahmatullah, M. Molecular Docking of Quassinoid Compounds Javanicolides A-F and H with C3-Like Protease (or 3CLpro) of SARS and SARS-CoV-2 (COVID-19) and VP8* Domain of the Outer Capsid Protein VP4 of Rotavirus A. J. Med. Plants Stud. 2020, 8, 14–19. [Google Scholar]
- Hasan, A.; Mahamud, R.A.; Bondhon, T.A.; Jannat, K.; Jahan, R.; Rahmatullah, M. Can javanicins be potential inhibitors of SARS-CoV-2 C-3 like protease? An evaluation through molecular docking studies. J. Nat. Ayurvedic Med. 2020, 4, 250. [Google Scholar] [CrossRef]
- Hossain, S.; Jahan, R.; Hasan, A.; Jannat, K.; Bondhon, T.A.; Rahmatullah, M. Spices and plants as home remedies for COVID-19: A survey in Rajbari district, Bangladesh. J. Nat. Ayurvedic Med. 2020, 4, 268. [Google Scholar] [CrossRef]
- Hasan, A.; Biswas, P.; Bondhon, T.A.; Jannat, K.; Paul, T.K.; Paul, A.K.; Jahan, R.; Nissapatorn, V.; Mahboob, T.; Wilairatana, P.; et al. Can Artemisia herba-alba be useful for managing COVID-19 and comorbidities? Molecules 2022, 27, 492. [Google Scholar] [CrossRef]
- Jannat, K.; Hasan, A.; Bondhon, T.A.; Mahboob, T.; Paul, A.K.; Jahan, R.; Nissapatorn, V.; Pereira, M.L.; Wiart, C.; Rahmatullah, M. Can Costus afer be used for co-treatment of COVID-19, its symptoms and comorbidities? A novel approach for combating the pandemic and implications for sub-Saharan Africa. Trop. Biomed. 2021, 38, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Mahamud, R.A.; Jannat, K.; Bondhon, T.A.; Farzana, B.; Fariba, M.H.; Jahan, R.; Rahmatullah, M. Phytochemicals from Solanum surattense Burm.f. have high binding affinities for C-3 like main protease of COVID-19 (SARS-CoV-2). J. Med. Plants Stud. 2020, 8, 20–26. [Google Scholar]
- Mazza, G.; Brouillard, R. Recent developments in the stabilization of anthocyanins in food products. Food Chem. 1987, 25, 207–225. [Google Scholar] [CrossRef]
- Lapidot, T.; Harel, S.; Akiri, B.; Granit, R.; Kanner, J. pH-dependent forms of red wine anthocyanins as antioxidants. J. Agric. Food Chem. 1999, 47, 67–70. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, B.; Jin, Z.; Yang, H.; Rao, Z. The Crystal Structure of COVID-19 Main Protease in Complex with an Inhibitor N3. Available online: https://www.rcsb.org/structure/6LU7 (accessed on 16 December 2020).
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Zin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Ihlenfeldt, W.D. PubChem. In Applied Chemoinformatics: Achievements and Future Opportunities; Wiley-VCH: Weinheim, Germany, 2018; pp. 245–258. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. Autodock Vina: Improving the speed and accuracy of docking. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Studio, D. Dassault Systemes BIOVIA, discovery studio modelling environment; release 4.5; Accelrys Softw. Inc.: San Diego, CA, USA, 2015; pp. 98–104. [Google Scholar]
- Giménez, B.G.; Santos, M.S.; Ferrarini, M.; Fernandes, J.P.S. Evaluation of blockbuster drugs under the rule-of-five. Pharmazie 2010, 65, 148–152. [Google Scholar]
- Rakib, A.; Paul, A.; Chy, M.N.U.; Sami, S.A.; Baral, S.K.; Majumder, M.; Tareq, A.M.; Amin, M.N.; Shahriar, A.; Uddin, M.Z.; et al. Biochemical and computational approach of selected phytocompounds from Tinospora crispa in the management of COVID-19. Molecules 2020, 25, 3936. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins 2004, 57, 678–683. [Google Scholar] [CrossRef]
- Cherrak, S.A.; Merzouk, H.; Mokhtari-Soulimane, N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking study. PLoS ONE 2020, 15, e0240653. [Google Scholar] [CrossRef]
- Niño-Medina, G.; Urías-Orona, V.; Muy-Rangel, M.D.; Heredia, J.B. Structure and content of phenolics in eggplant (Solanum melongena)—A review. South Afr. J. Bot. 2017, 111, 161–169. [Google Scholar] [CrossRef]
- Cao, G.; Sofic, E.; Prior, R.L. Antioxidant capacity of tea and common vegetables. J. Agric. Food Chem. 1996, 44, 3426–3431. [Google Scholar] [CrossRef]
- Soto, M.E.; Guarner-Lans, V.; Soria-Castro, E.; Pech, L.M.; Pérez-Torres, I. Is antioxidant therapy a useful complementary measure for COVID-19 treatment? An algorithm for its application. Medicina 2020, 56, 386. [Google Scholar] [CrossRef]
- Fakhar, Z.; Faramarzi, B.; Pacifico, S.; Faramarzi, S. Anthocyanin derivatives as potent inhibitors of SARS-CoV-2 main protease: An in-silico perspective of therapeutic targets against COVID-19 pandemic. J. Biomol. Struct. Dyn. 2020, 39, 6171–6183. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Song, J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of Coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Li, Q.; Kang, C.B. Progress in developing inhibitors of SARS-CoV-2 3C-like protease. Microorganisms 2020, 8, 1250. [Google Scholar] [CrossRef]
- Goyal, B.; Goyal, D. Targeting the dimerization of main protease of Coronaviruses: A potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 2020, 22, 297–305. [Google Scholar] [CrossRef]
- Muramatsu, T.; Takemoto, C.; Kim, Y.-T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.-F.; Kuo, C.-J.; Chang, K.-T.; Chang, H.-C.; Chou, C.-C.; Ko, T.-P.; Shr, H.-L.; Chang, G.-G.; Wang, A.H.-J.; Liang, P.-H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005, 280, 31257–31266. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.W.; Yiu, C.-P.B.; Wong, K.-Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000 Res. 2020, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Mittal, L.; Kumari, A.; Srivastava, M.; Singh, M.; Asthana, S. Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. J. Biomol. Struct. Dyn. 2020, 39, 3662–3680. [Google Scholar] [CrossRef]
- Vázquez-Calvo, Á.; Jiménez de Oya, N.; Martin-Acebes, M.A.; Garcia-Moruno, E.; Saiz, J.-C. Antiviral properties of the natural polyphenols delphinidin and epigallocatechin gallate against the Flaviviruses West Nile virus, Zika virus, and Dengue virus. Front. Microbiol. 2017, 8, 1314. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phytochemical | Source | Binding Energy (ΔG = kcal/mol) | ||
|---|---|---|---|---|
| 6LU7 | 6LZE | 7BRO | ||
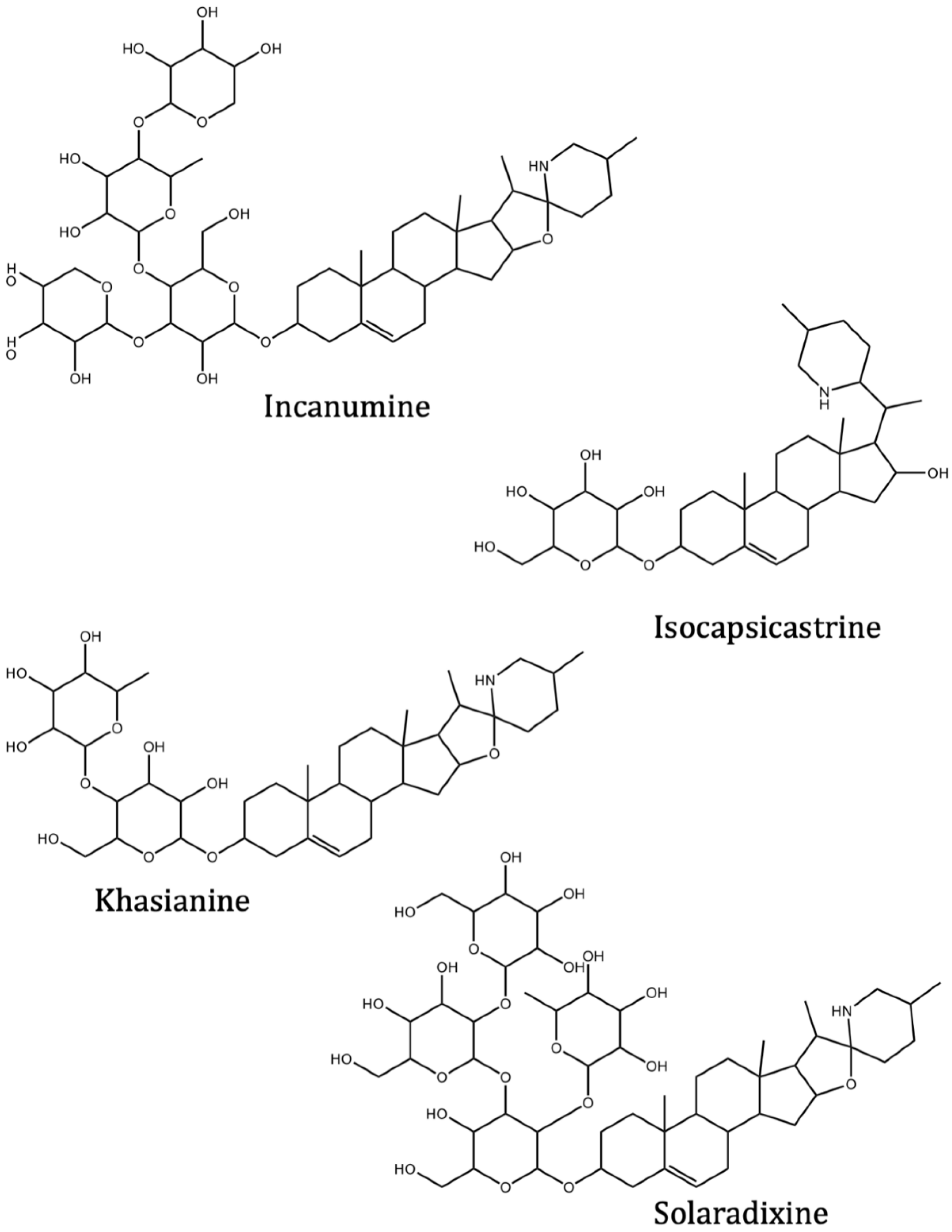
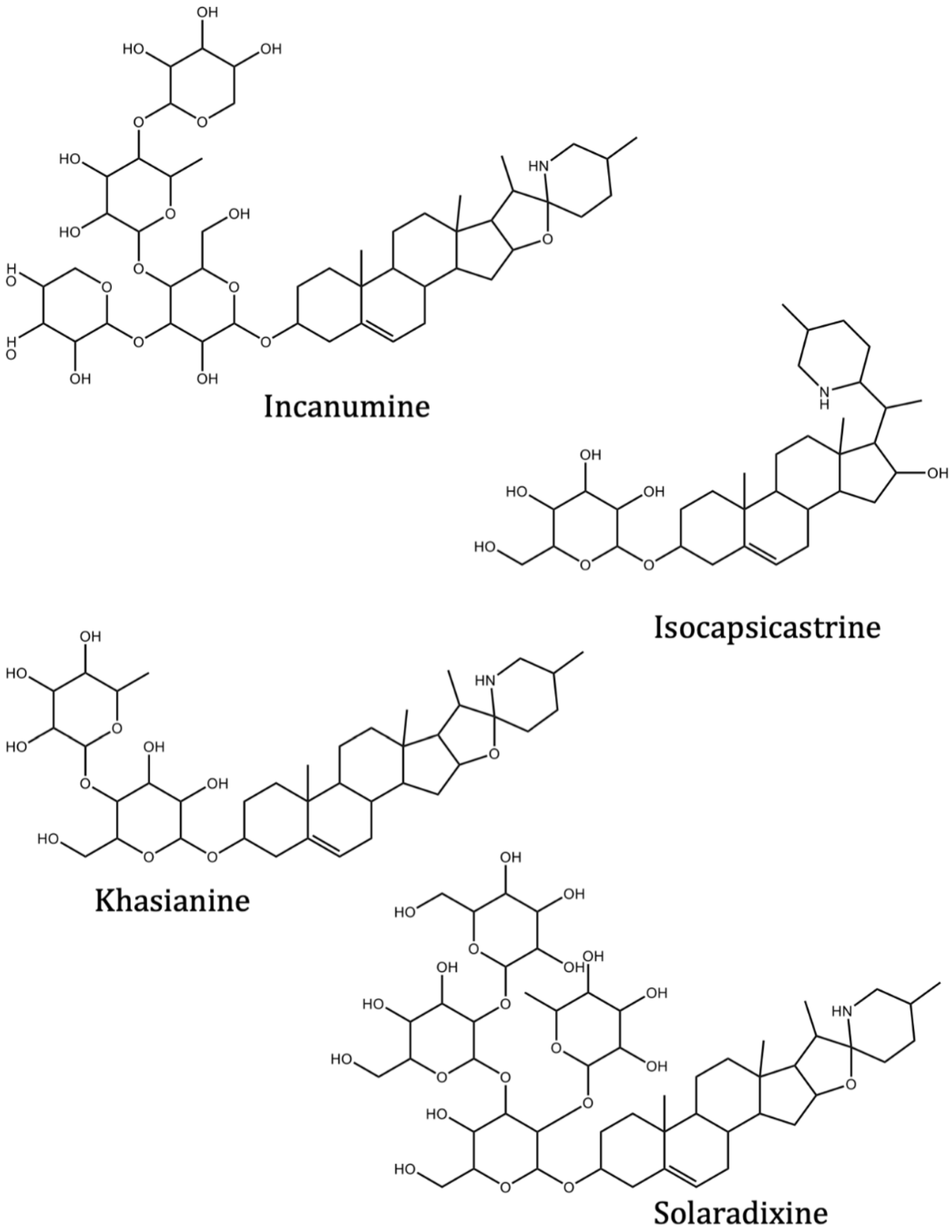
| Incanumine | Solanum incanum L. | −9.8 | −9.3 | |
| Isocapsicastrine | Solanum capsicastrum Link ex Schauer. | −8.4 | ||
| Khasianine | Solanum xanthocarpum Schrad. and Wendl. | −9.2 | −8.8 | |
| Solaradixine | Solanum laciniatum Aiton. | −9.4 | −8.3 | |
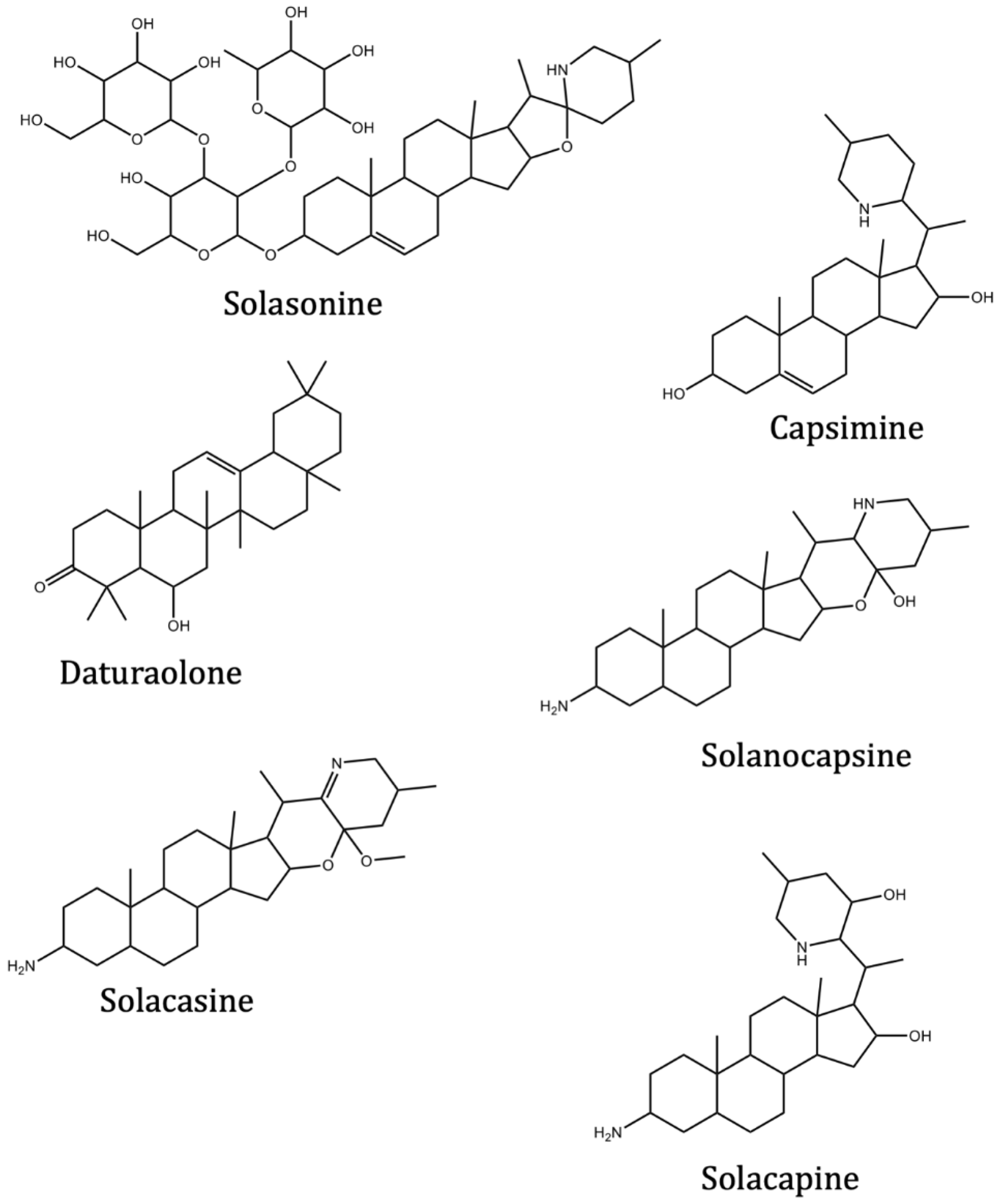
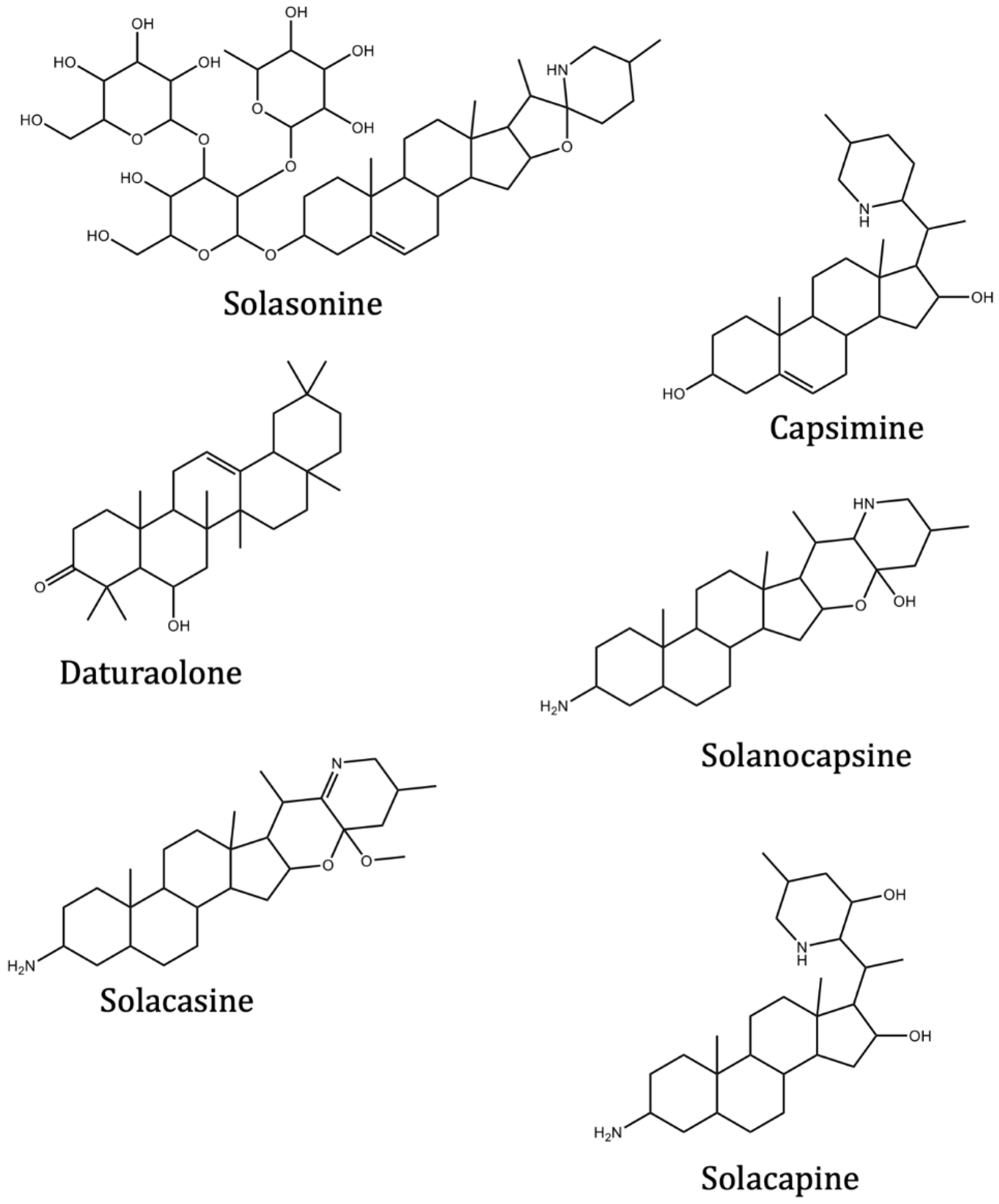
| Solasonine | Solanum asperum Rich. | −9.2 | −9.3 | |
| Capsimine | Solanum capsicastrum Link ex Schauer. | −7.5 | ||
| Daturaolone | Solanum arundo Mattei | −8.1 | −8.3 | |
| Solanocapsine | Solanum capsicastrum Link ex Schauer. | −8.3 | ||
| Solacasine | Solanum capsicastrum Link ex Schauer. | −8.1 | ||
| Solacapine | Solanum capsicastrum Link ex Schauer. | −7.6 | −7.7 | |
| Episolacapine | Solanum capsicastrum Link ex Schauer. | −7.9 | ||
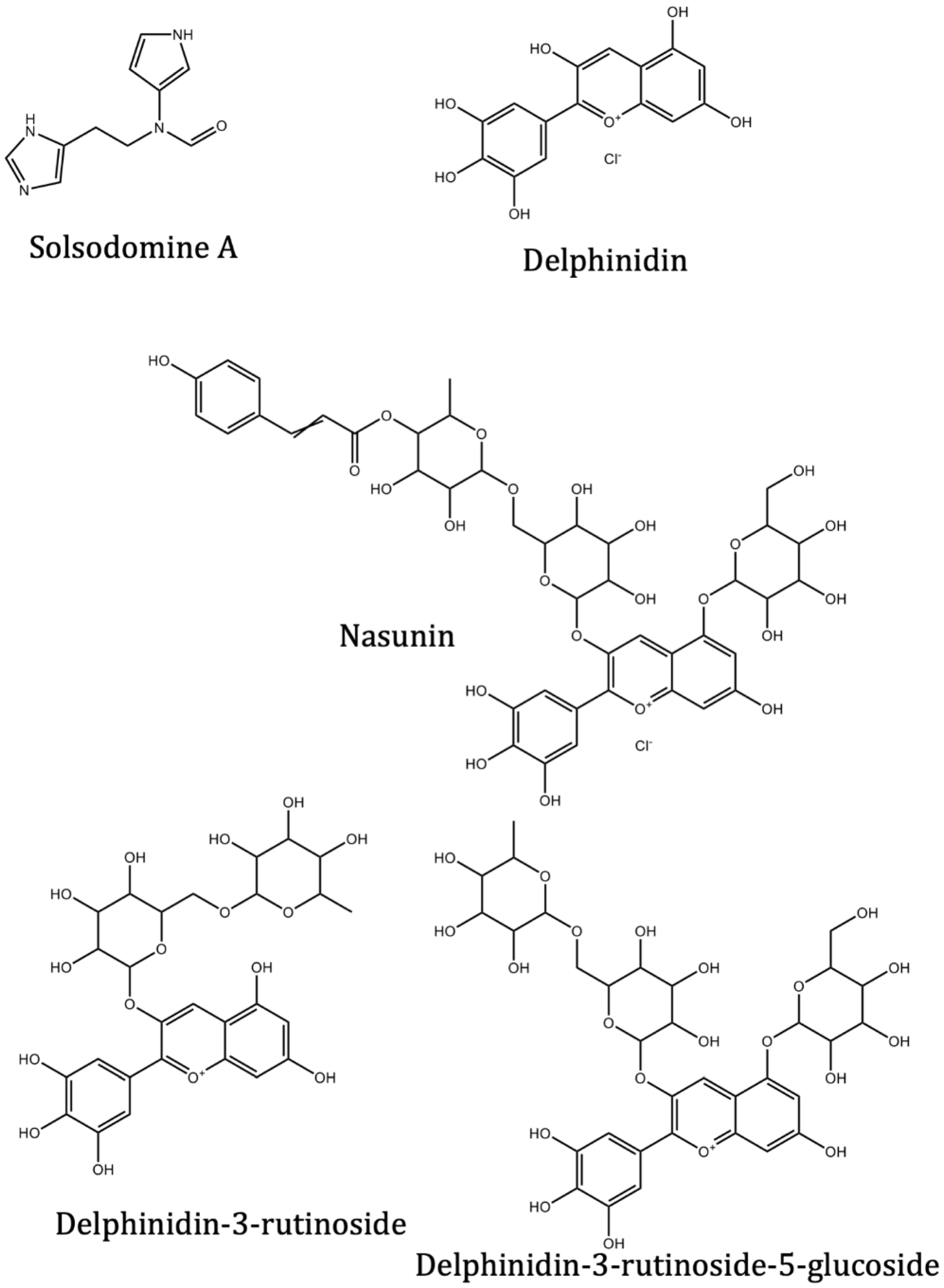
| Solsodomine A | Solanum sodomeum L. | −5.1 | −5.1 | |
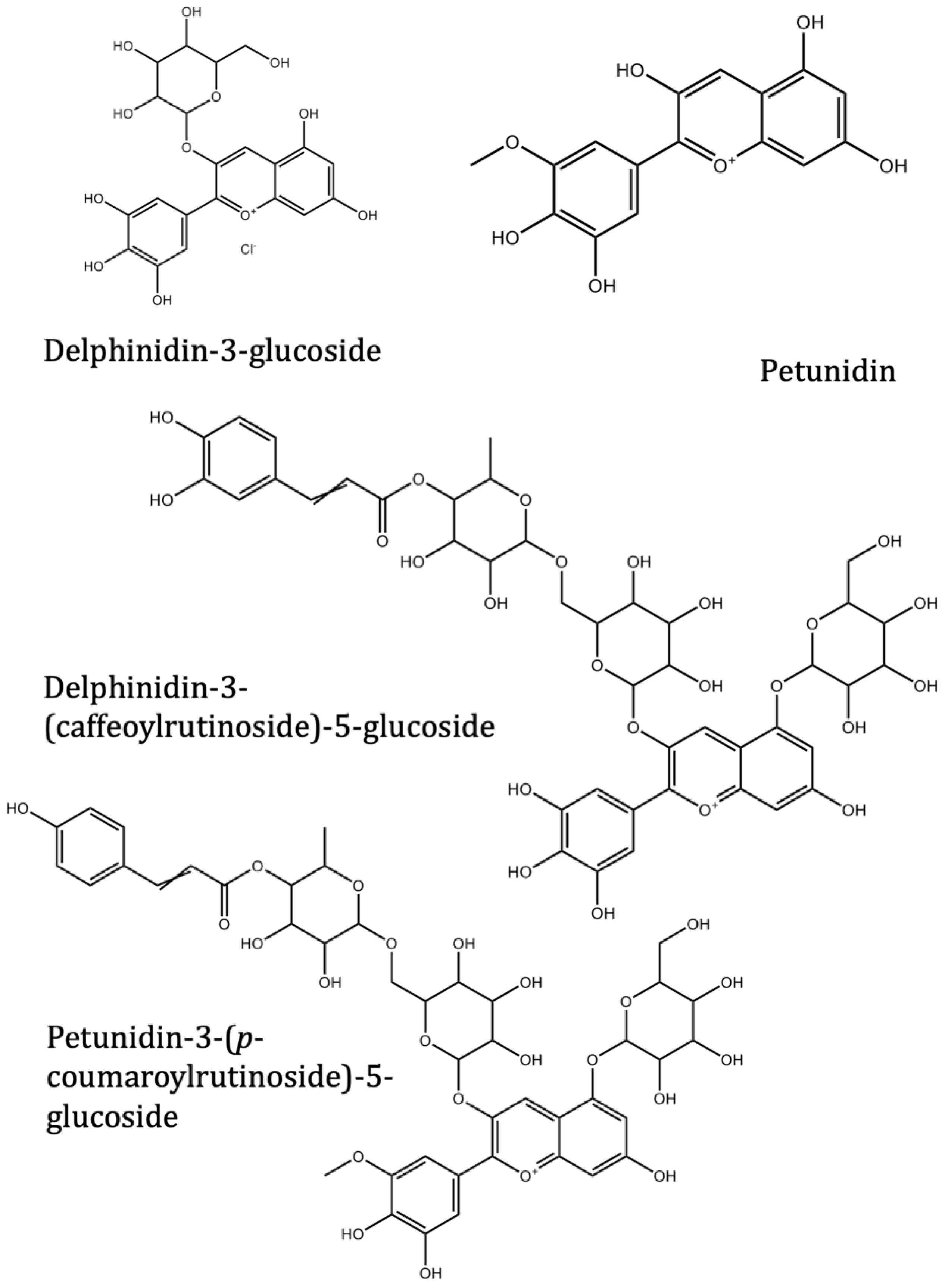
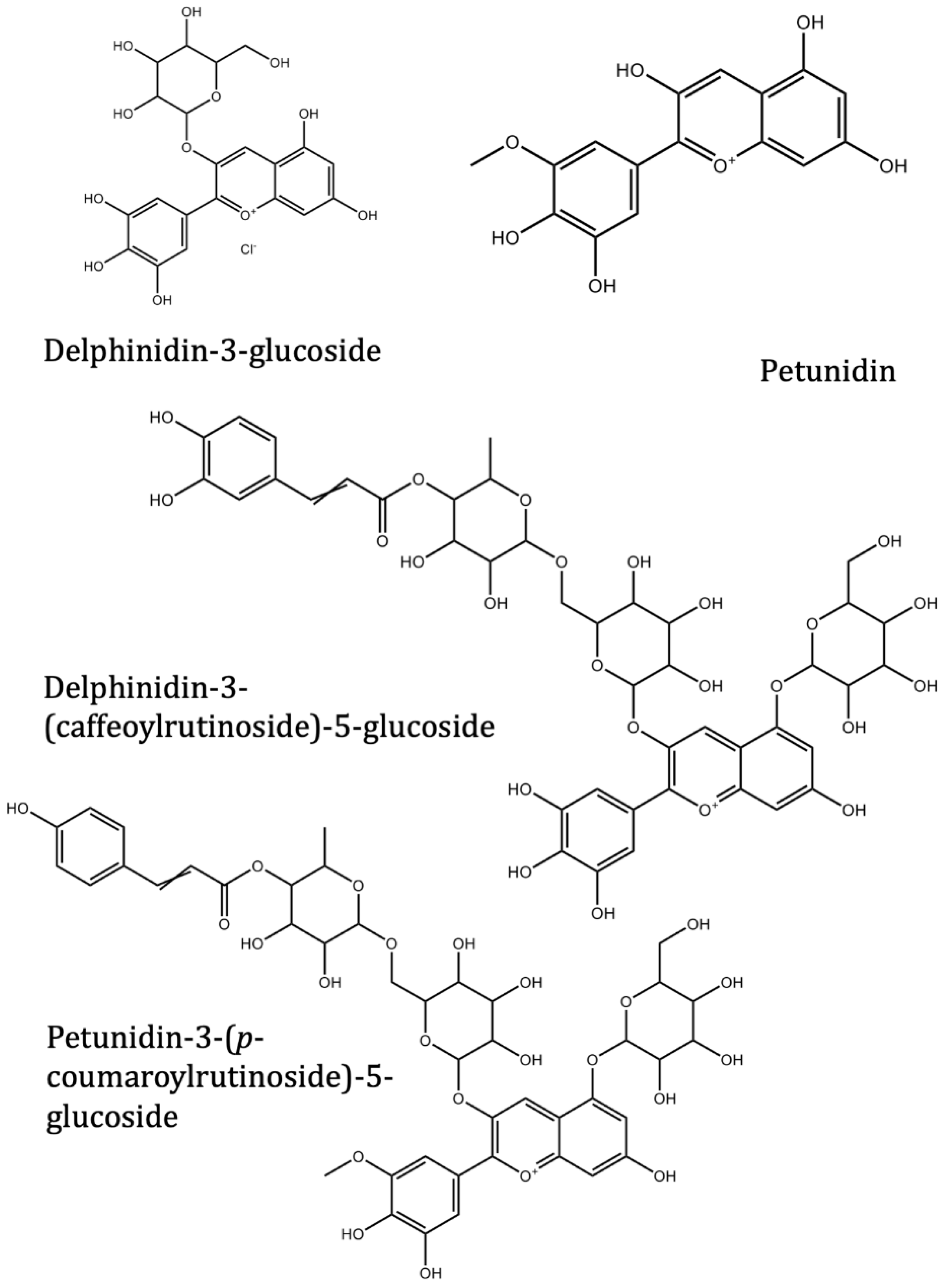
| Delphinidin | Solanum melongena L. | −7.4 | −7.3 | |
| Nasunin (delphinidin-3-p-coumaroylrutinoside-5-glucoside) | Solanum melongena L. | −8.5 | ||
| Delphinidin-3-rutinoside (Tulipanin) | Solanum melongena L. | −8.6 | ||
| Delphinidin-3-rutinoside-5-glucoside | Solanum melongena L. | −8.5 | ||
| Delphinidin-3-glucoside (Myrtillin/Mirtillin) | Solanum melongena L. | −8.4 | ||
| Delphinidin-3-(caffeoyl-rutinoside)-5-glucoside | Solanum melongena L. | −8.2 | ||
| Petunidin-3-(p-coumaroylrutinoside)-5-glucoside | Solanum melongena L. | −8.6 | ||
| Petunidin | Solanum melongena L. | −7.5 | −7.3 | |
| Lopinavir | Antiviral control drug | −8.2 | ||
| Baricitinib | Antiviral control drug | −6.6 | −7.2 | |
| Capsaicin | Alkaloid in Capsicum annuum L. | −5.9 | −5.8 | |
| Ivermectin | Anti-parasitic control drug | −9.1 | −8.8 | |
| Molnupiravir | Antiviral control drug | −6.6 | −6.4 | |
| Quercetin | Flavonoid found in grapes | −6.6 | −7.3 | |
| Luteolin | Reported Mpro inhibitor | −7.5 | −7.5 | |
| Remdesivir | Antiviral control drug | −7.4 | −7.8 | |
| Nirmatrelvir | Antiviral control drug | −7.5 | −7.0 | |
| Bedaquiline | Mpro inhibitor | −6.9 | −6.8 | |
| Boceprevir | Mpro inhibitor | −6.4 | −6.9 | |
| Efonidipine | Mpro inhibitor | −7.5 | −7.3 | |
| Lercanidipine | Mpro inhibitor | −7.8 | −7.3 | |
| Manidipine | Mpro inhibitor | −7.2 | −6.5 |
| Name of the Chemicals | Chemical Structure | ΔG = kcal/mol |
|---|---|---|
| Delphinidin (PubChem) | 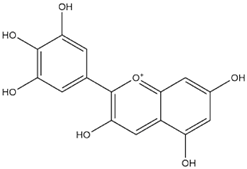 | −7.4 |
| Delphinidin-E-Chalcone |  | −6.9 |
| Delphinidin Hemiketal | 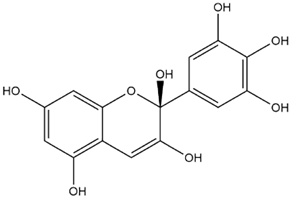 | −7.2 |
| Delphinidin Hemiacetal | 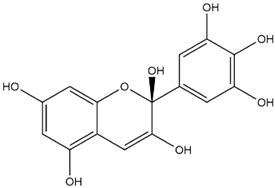 | −7.7 |
| Delphinidin Quinonoid-1 |  | −7.3 |
| Delphinidin Quinonoid-2 |  | −6.9 |
| Delphinidin-3-glucoside (PubChem) |  | −8.4 |
| Mirtillin-E-Chalcone (Delphinidin-3-glucoside chalcone form) | 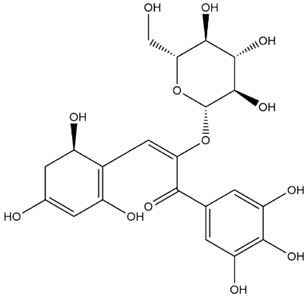 | −7.9 |
| Mirtillin Quinonoid-1 |  | −8.3 |
| Mirtillin Quinonoid-2 |  | −7.6 |
| Nasunin (PubChem) | 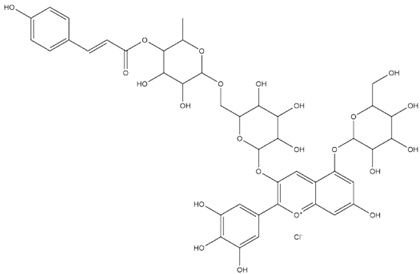 | −8.5 |
| Nasunin-E-Chalcone | 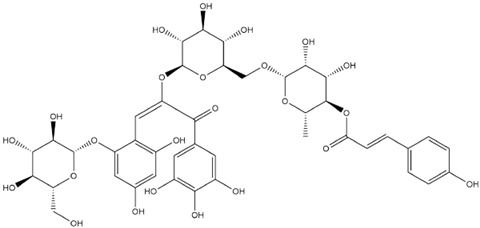 | −8.5 |
| Nasunin Hemiketal | 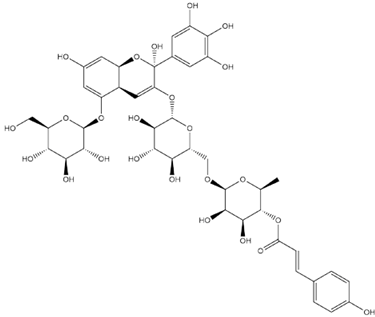 | −7.3 |
| Nasunin Quinonoid-1 |  | −8.3 |
| Nasunin Quinonoid-2 |  | −8.5 |
| Petunidin (PubChem) |  | −7.5 |
| Petunidin-E-Chalcone | 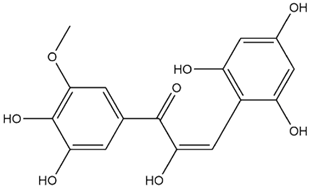 | −7.1 |
| Petunidin Hemiketal | 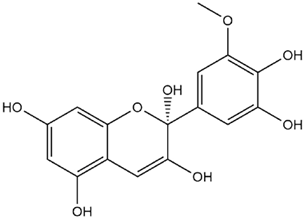 | −7.6 |
| Petunidin Quinonoid-1 |  | −7.3 |
| Petunidin Quinonoid-2 | 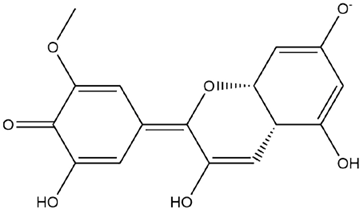 | −6.5 |
| Delphinidin-3-rutinoside (PubChem) |  | −8.6 |
| Tulipanin-E-Chalcone (Delphinidin-3-rutinoside chalcone form) |  | −7.9 |
| Tulipanin Hemiketal | 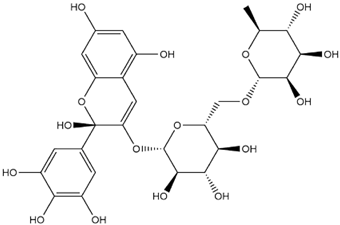 | −8.1 |
| Tulipanin Quinonoid-1 |  | −9.4 |
| Tulipanin Quinonoid-2 |  | −8.2 |
| Delphinidin-3-(Caffeoyl-Rutinoside)-5-Glucoside (PubChem) |  | −8.2 |
| Delphinidin-3-Caffeoyl-Rutinoside-5-Glucoside-E-Chalcone |  | −8.2 |
| Delphinidin-3-Caffeoyl-Rutinoside-5-Glucoside Hemiketal |  | −8.5 |
| Delphinidin-3-Caffeoyl-Rutinoside-5-Glucoside Quinonoid-1 | 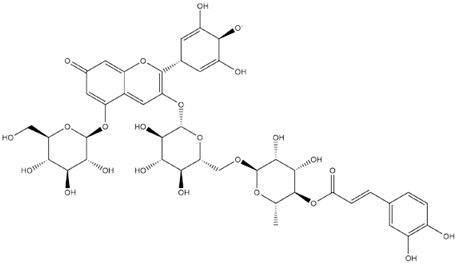 | −8.8 |
| Delphinidin-3-Caffeoyl-Rutinoside-5-Glucoside Quinonoid-2 |  | −8.0 |
| Delphinidin-3-Rutinoside-5-Glucoside (PubChem) | 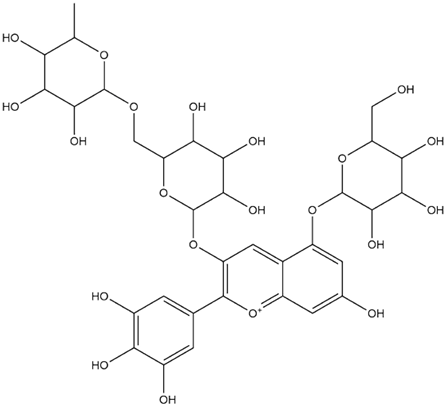 | −8.5 |
| Delphinidin-3-Rutinoside-5-Glucoside-E-Chalcone |  | −8.2 |
| Delphinidin-3-Rutinoside-5-Glucoside Hemiketal |  | −8.4 |
| Delphinidin-3-Rutinoside-5-Glucoside Quinonoid-1 |  | −7.8 |
| Delphinidin-3-Rutinoside-5-Glucoside Quinonoid-2 |  | −8.6 |
| Petunidin-3-(p-Coumaroyl-Rutinoside)-5-Glucoside |  | −8.6 |
| Petunidin-3-(p-Coumaroyl-Rutinoside)-5-Glucoside Chalcone |  | −8.3 |
| Petunidin-3-(p-Coumaroyl-Rutinoside)-5-Glucoside Hemiketal | 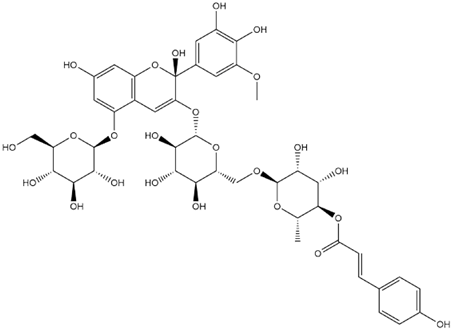 | −7.4 |
| Petunidin-3-(p-Coumaroyl-Rutinoside)-5-Glucoside Quinonoid-1 |  | −8.7 |
| Petunidin-3-(p-Coumaroyl-Rutinoside)-5-Glucoside Quinonoid-2 | 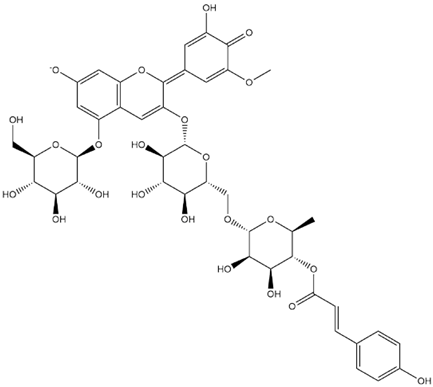 | −9.1 |
| Phytochemical (Binding Energy in kcal/mol) | Molecular Weight | Number of H-Bond Acceptors | Number of H-Bond Donors | Log P | Molar Refractivity | Number of Violations of Rule of Five |
|---|---|---|---|---|---|---|
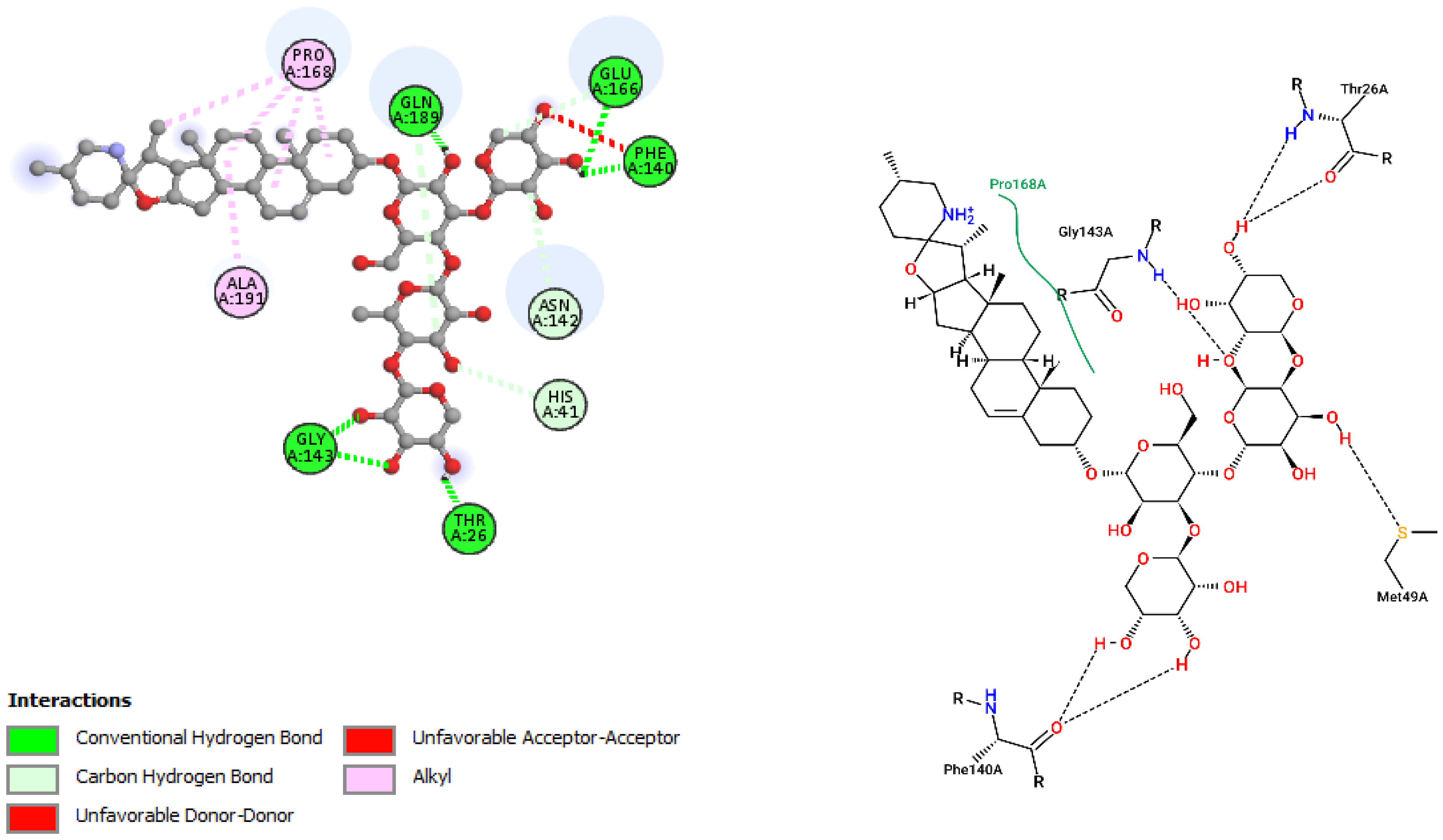
| Incanumine (−9.8) | 986.15 | 20 | 11 | 4.65 | 243.66 | 4 |
| Isocapsicastrine (−8.4) | 577.79 | 8 | 6 | 4.42 | 161.76 | 3 |
| Khasianine (−9.2) | 721.92 | 12 | 7 | 4.74 | 190.83 | 5 |
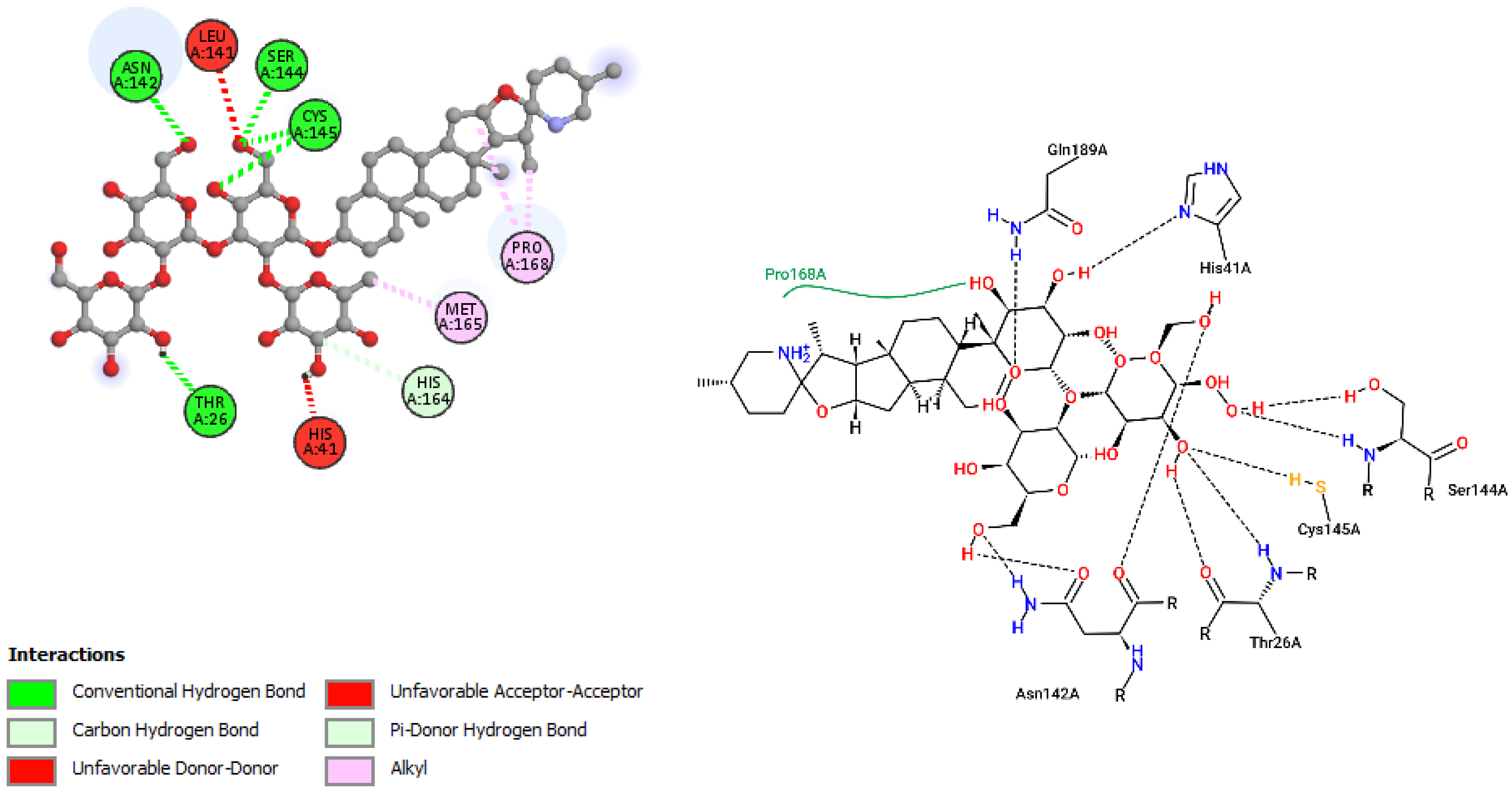
| Solaradixine (−9.4) | 1046.20 | 22 | 13 | 4.09 | 255.60 | 4 |
| Solasonine (−9.2) | 884.06 | 17 | 10 | 4.03 | 223.21 | 4 |
| Capsimine (−7.5) | 415.65 | 3 | 3 | 4.22 | 129.38 | 0 |
| Daturaolone (−8.1) | 440.70 | 2 | 1 | 4.37 | 135.08 | 0 |
| Solanocapsine (−8.3) | 430.67 | 4 | 3 | 4.14 | 130.41 | 0 |
| Solacasine (−8.1) | 442.68 | 4 | 1 | 4.53 | 135.43 | 0 |
| Solacapine (−7.6) | 432.68 | 4 | 4 | 3.85 | 132.56 | 0 |
| Episolacapine (7.9) | 442.68 | 4 | 1 | 4.53 | 135.43 | 0 |
| Solsodomine A (−5.1) | 204.23 | 2 | 2 | 0.91 | 57.03 | 0 |
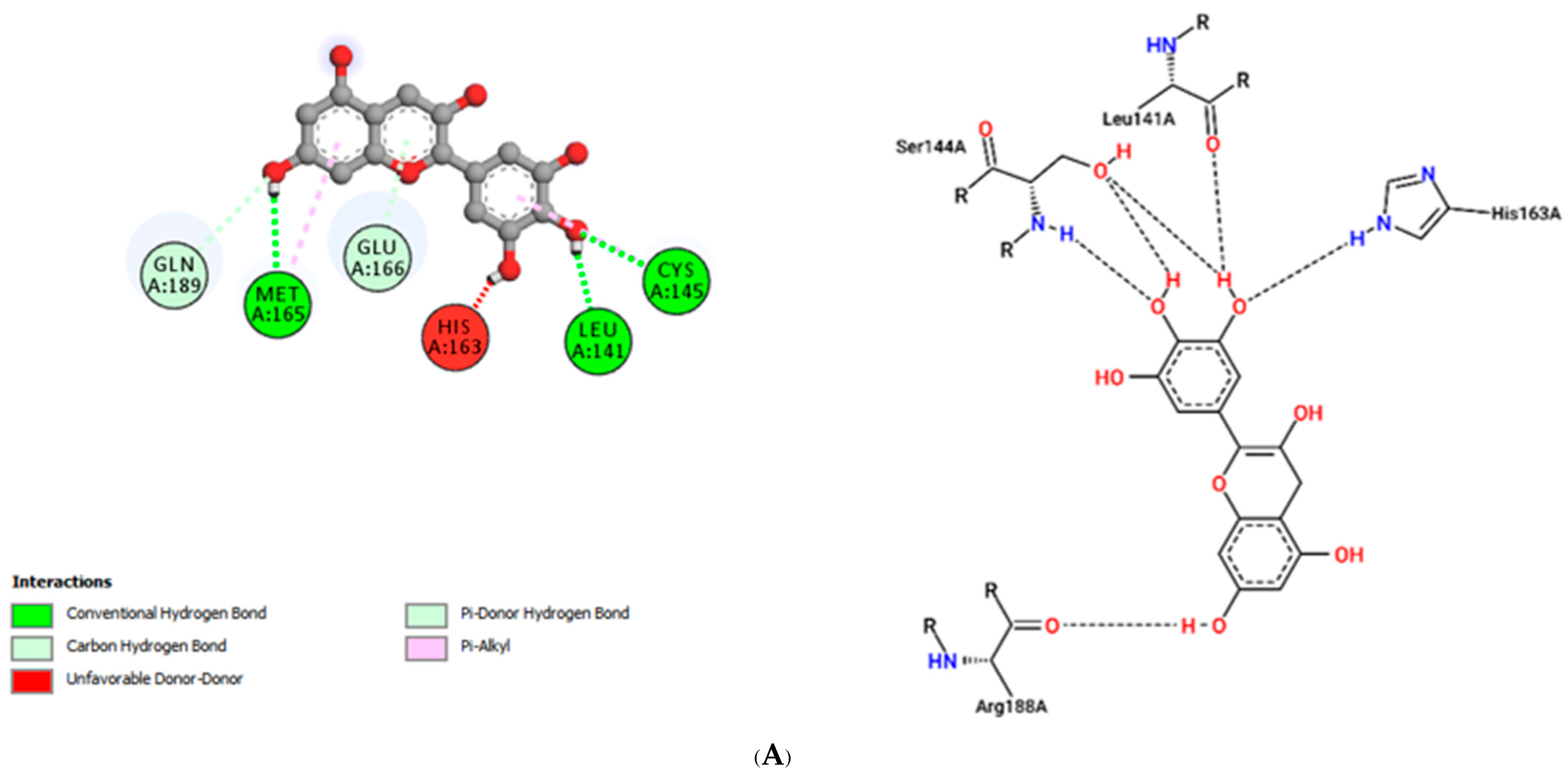
| Delphinidin (−7.4) | 303.24 | 7 | 6 | −3.10 | 78.20 | 1 |
| Nasunin (delphinidin-3-p-coumaroylrutinoside-5-glucoside (−8.5) | 955.26 | 23 | 14 | −5.04 | 220.89 | 4 |
| Delphinidin-3-rutinoside (−8.6) | 611.53 | 16 | 11 | −2.34 | 141.54 | 3 |
| Delphinidin-3-rutinoside-5-glucoside (−8.5) | 773.67 | 21 | 14 | −1.98 | 173.66 | 3 |
| Delphinidin-3-glucoside (−8.4) | 500.84 | 12 | 9 | −5.34 | 116.17 | 2 |
| Delphinidin-3-(caffeoyl-rutinoside)-5- Glucoside (−8.2) | 935.81 | 24 | 15 | −0.74 | 217.06 | 4 |
| Petunidin-3-(p-coumaroylrutinoside)-5- Glucoside (−8.6) | 933.84 | 23 | 13 | 0.49 | 219.50 | 4 |
| Petunidin (−7.5) | 317.27 | 7 | 5 | −1.72 | 85.66 | 0 |
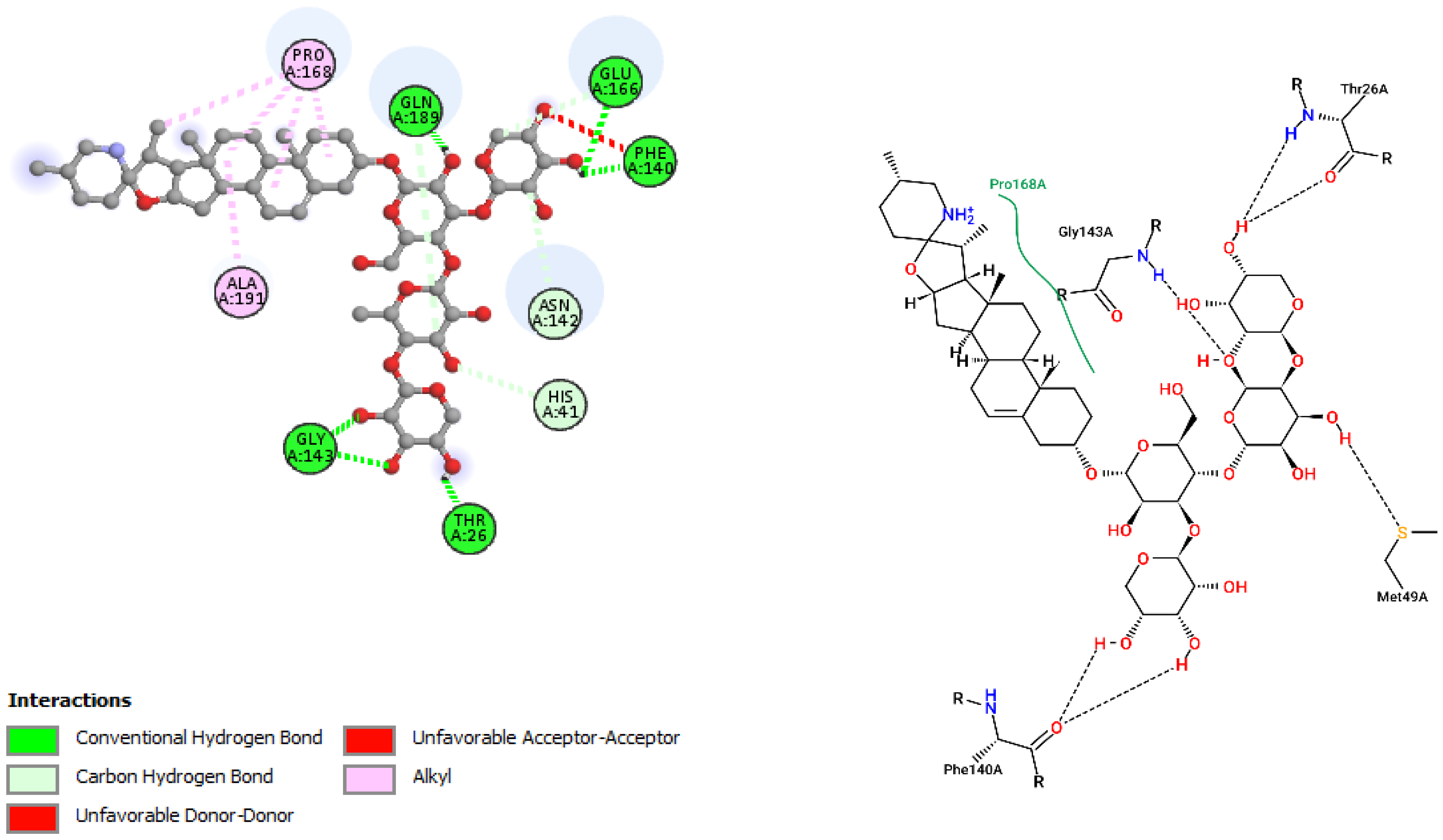
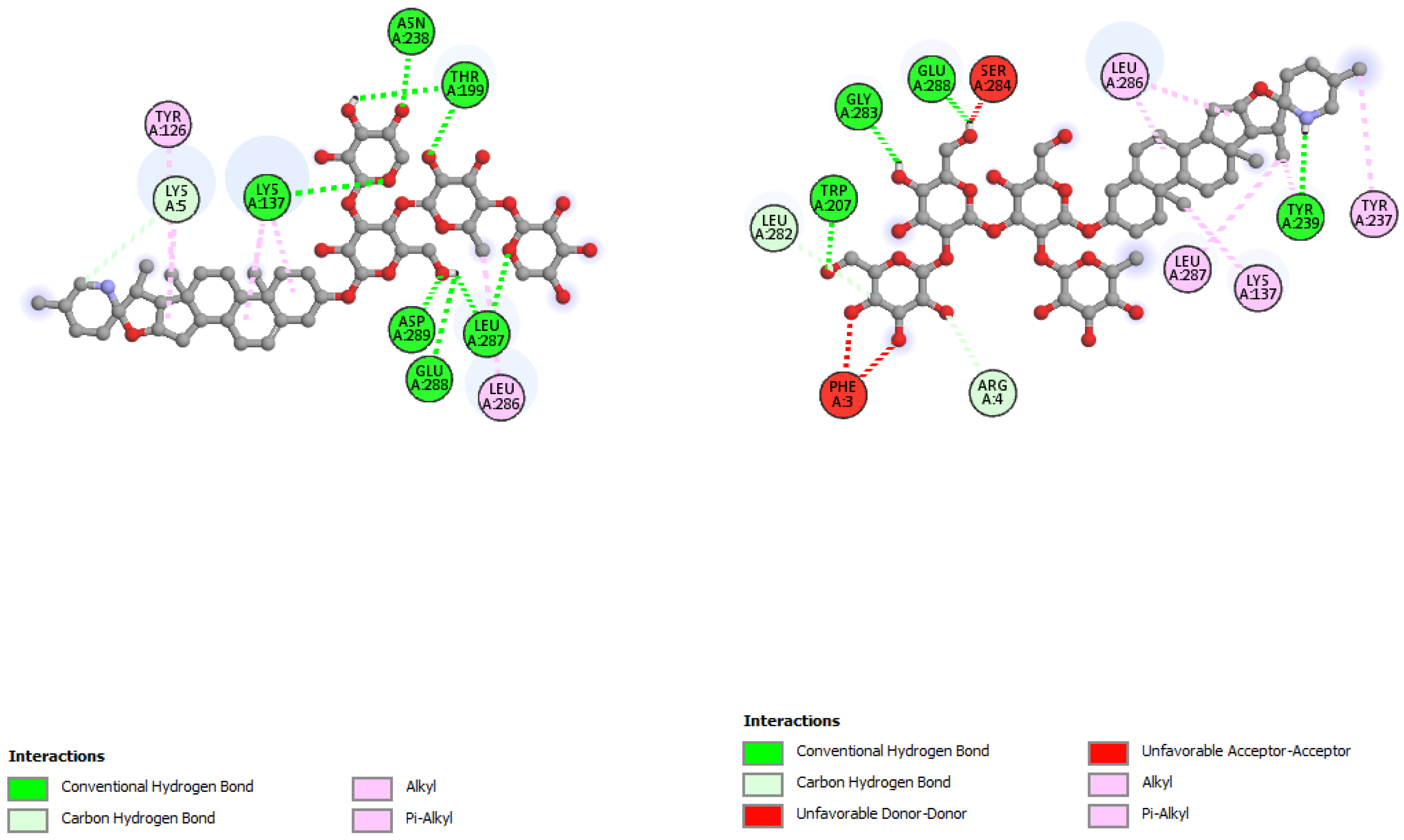
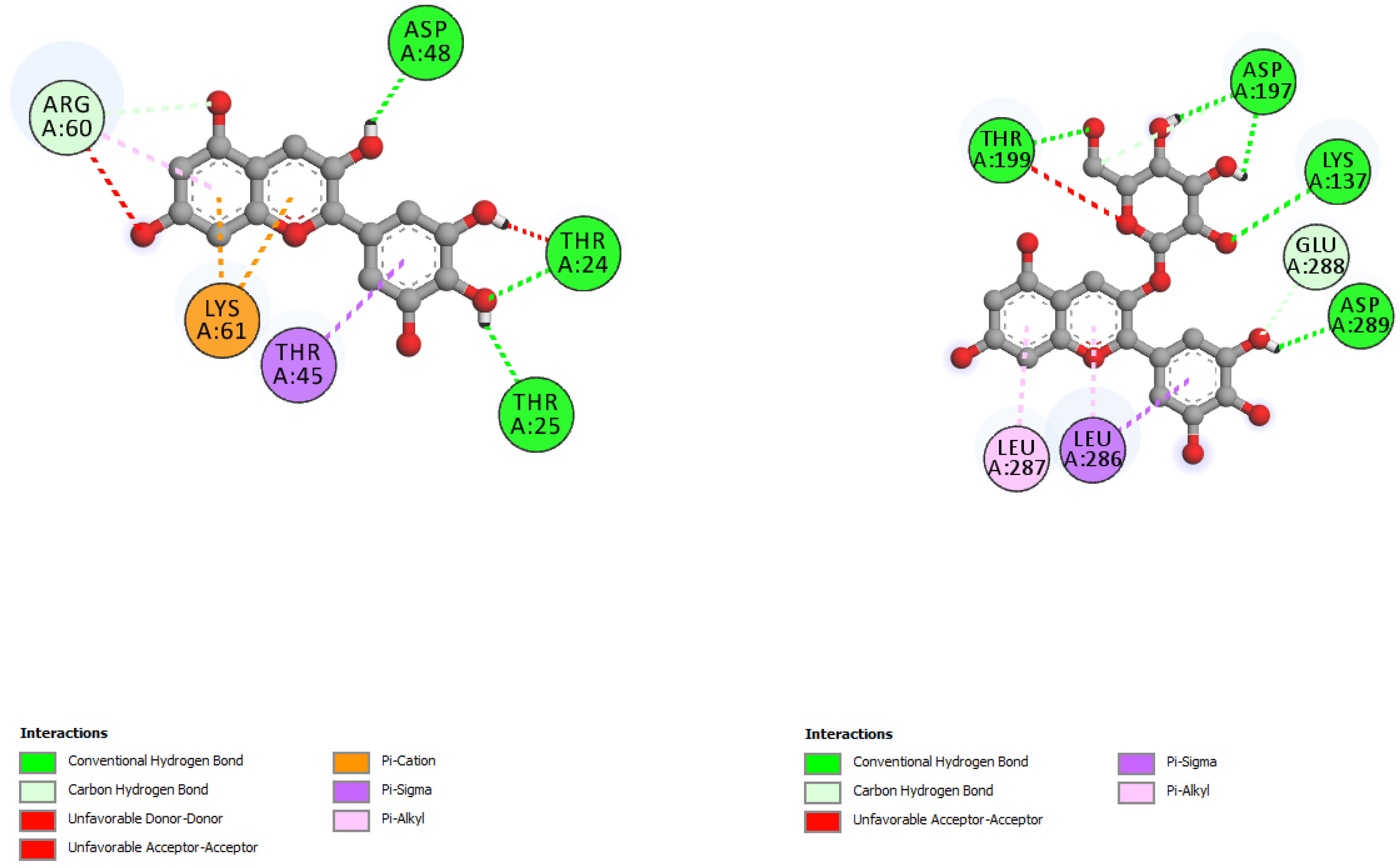
| Incanumine | |||
|---|---|---|---|
| Residues | Distance | Category | Type |
| GLY143 | 2.68 | Hydrogen Bond | CH |
| GLY143 | 2.14 | Hydrogen Bond | CH |
| GLN189 | 1.87 | Hydrogen Bond | CH |
| PHE140 | 2.09 | Hydrogen Bond | CH |
| GLU166 | 2.51 | Hydrogen Bond | CH |
| THR26 | 2.45 | Hydrogen Bond | CH |
| HIS41 | 3.75 | Hydrogen Bond | C |
| GLU166 | 3.25 | Hydrogen Bond | C |
| ASN142 | 3.23 | Hydrogen Bond | C |
| GLN189 | 3.40 | Hydrogen Bond | C |
| PRO168 | 4.84 | Hydrophobic | Alkyl |
| PRO168 | 4.67 | Hydrophobic | Alkyl |
| PRO168 | 4.65 | Hydrophobic | Alkyl |
| ALA191 | 4.80 | Hydrophobic | Alkyl |
| PRO168 | 4.33 | Hydrophobic | Alkyl |
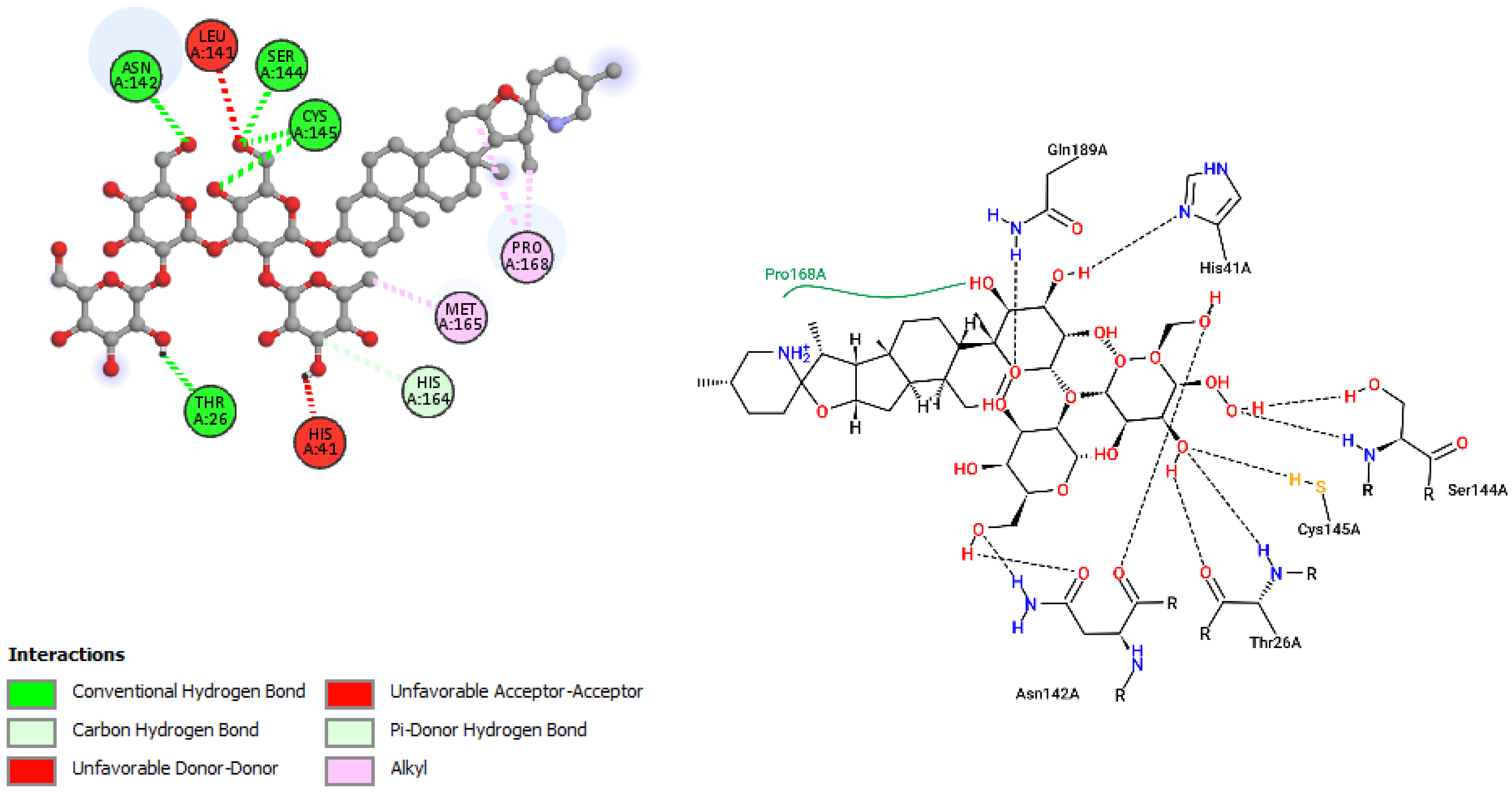
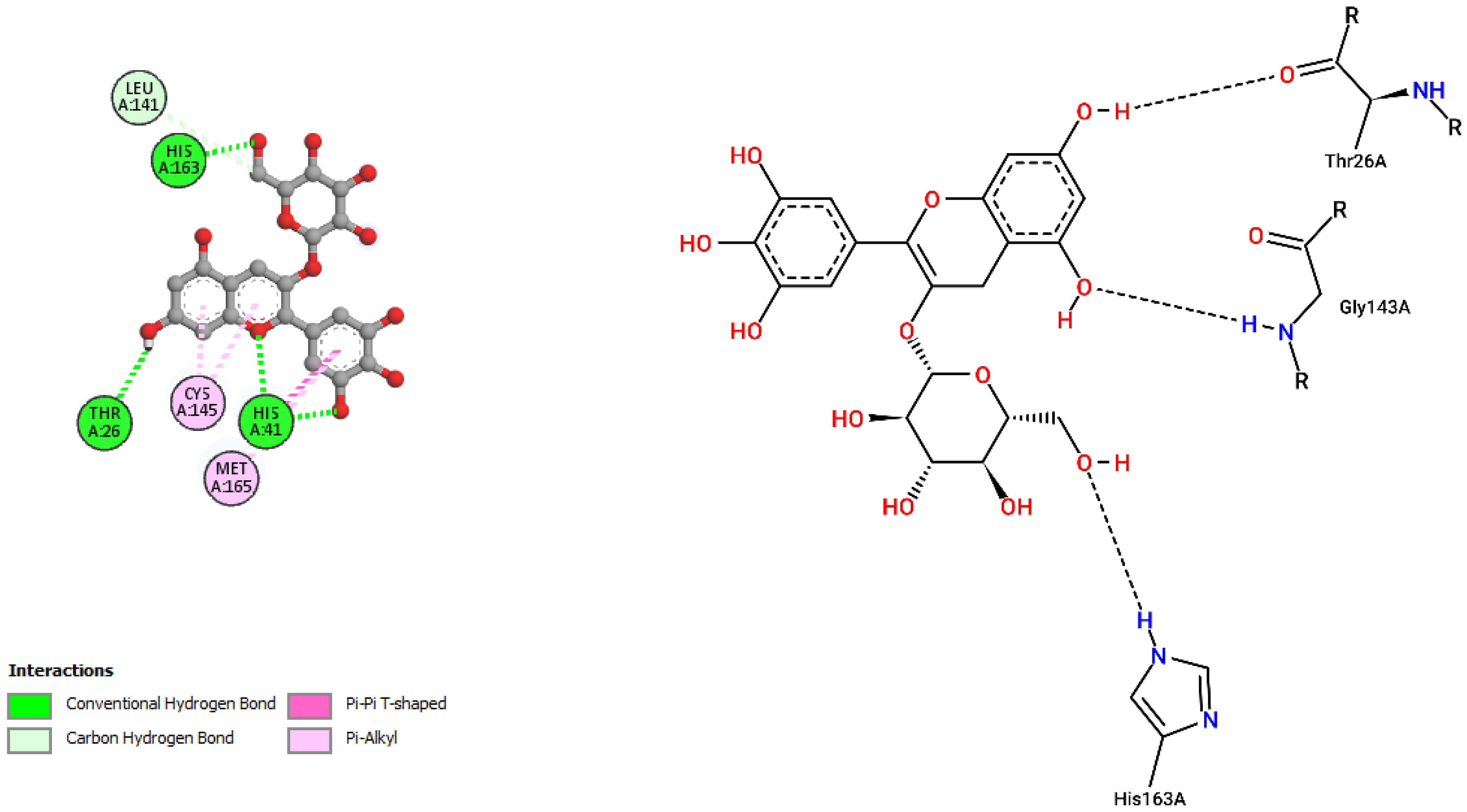
| Solaradixine | |||
| ASN142 | 2.26 | Hydrogen Bond | CH |
| SER144 | 2.38 | Hydrogen Bond | CH |
| CYS145 | 2.67 | Hydrogen Bond | CH |
| CYS145 | 2.76 | Hydrogen Bond | CH |
| THR26 | 1.97 | Hydrogen Bond | CH |
| HIS164 | 3.32 | Hydrogen Bond | C |
| HIS41 | 2.33 | Hydrogen Bond | Pi-Donor Hydrogen Bond |
| PRO168 | 4.52 | Hydrophobic | Alkyl |
| PRO168 | 4.31 | Hydrophobic | Alkyl |
| MET165 | 4.16 | Hydrophobic | Alkyl |
| Daturaolone | |||
| ASN142 | 3.63 | Hydrogen Bond | C |
| PRO168 | 4.79 | Hydrophobic | Alkyl |
| CYS145 | 4.64 | Hydrophobic | Alkyl |
| CYS145 | 4.09 | Hydrophobic | Alkyl |
| PRO168 | 4.47 | Hydrophobic | Alkyl |
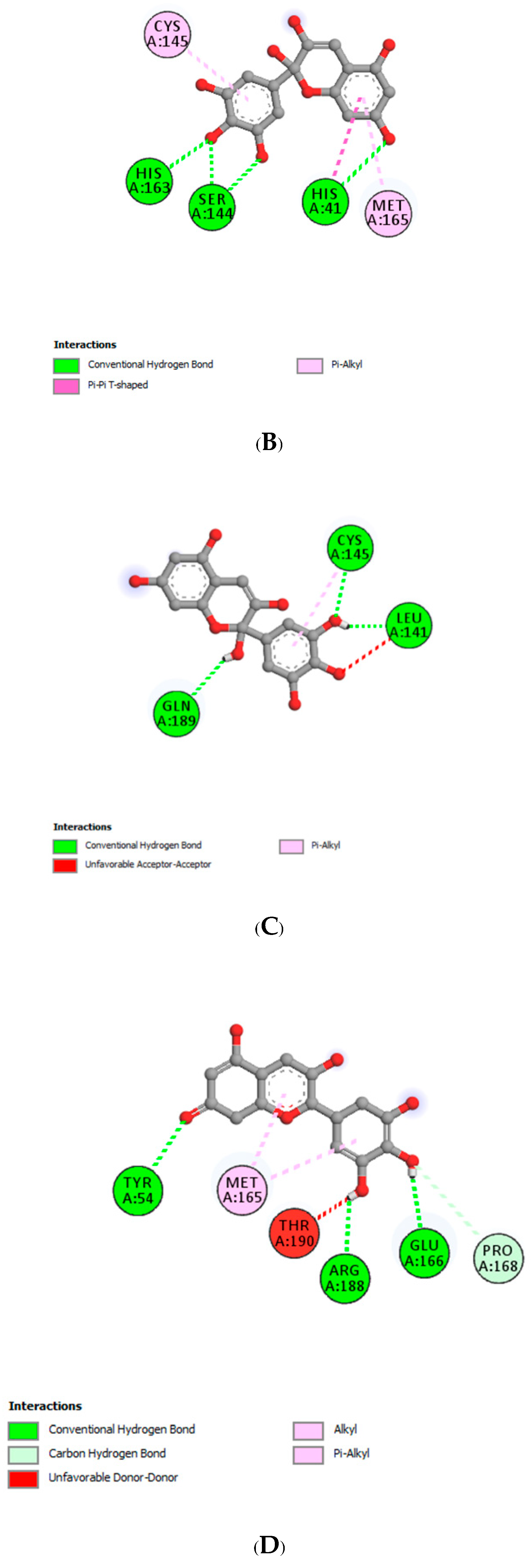
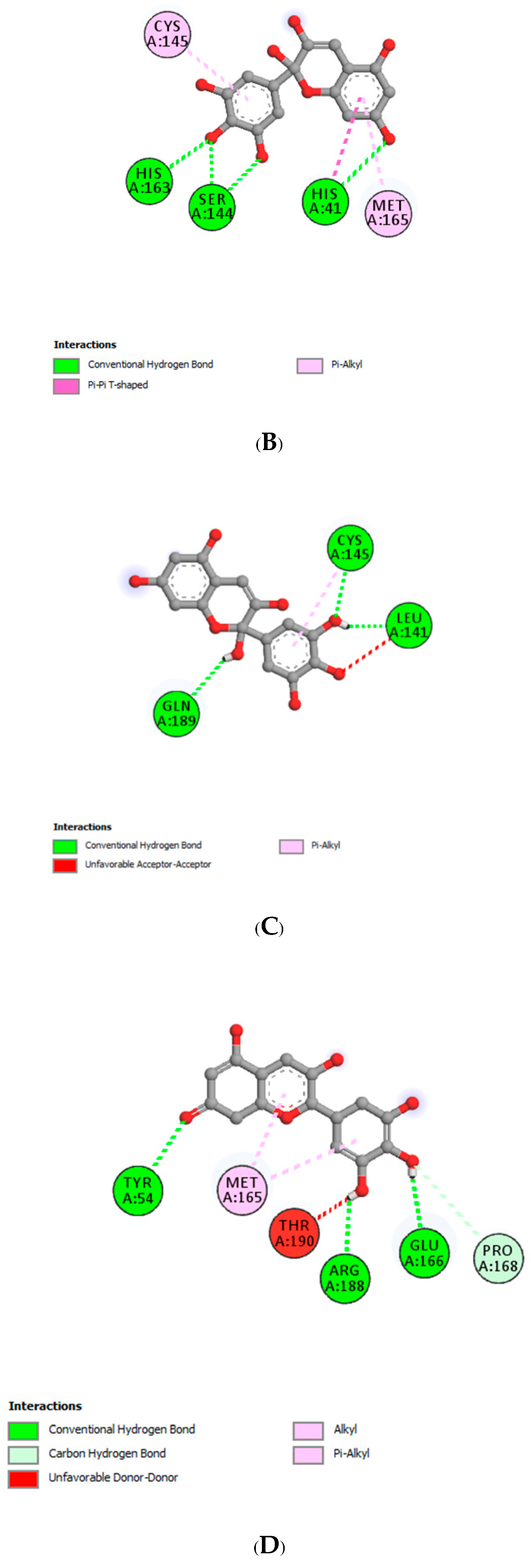
| Delphinidin | |||
| LEU141 | 1.79 | Hydrogen Bond | CH |
| MET165 | 2.76 | Hydrogen Bond | CH |
| GLN189 | 3.27 | Hydrogen Bond | CH |
| GLU166 | 3.07 | Hydrogen Bond | Pi-Donor Hydrogen Bond |
| MET165 | 5.31 | Hydrophobic | Pi-Alkyl |
| CYS145 | 4.86 | Hydrophobic | Pi-Alkyl |
| CYS145 | 2.63 | Hydrogen Bond | CH |
| Delphinidin-3-glucoside | |||
| HIS41 | 3.30 | Hydrogen Bond | CH |
| HIS41 | 2.96 | Hydrogen Bond | CH |
| HIS163 | 1.89 | Hydrogen Bond | CH |
| THR26 | 2.37 | Hydrogen Bond | CH |
| LEU141 | 3.43 | Hydrogen Bond | C |
| HIS41 | 4.78 | Hydrophobic | Pi-Pi T-shaped |
| CYS145 | 4.88 | Hydrophobic | Pi-Alkyl |
| CYS145 | 4.88 | Hydrophobic | Pi-Alkyl |
| MET165 | 5.10 | Hydrophobic | Pi-Alkyl |
| Delphinidin-3-rutinoside | |||
| CYS145 | 2.95 | Hydrogen Bond | CH |
| THR26 | 2.73 | Hydrogen Bond | CH |
| THR26 | 2.75 | Hydrogen Bond | CH |
| GLU166 | 2.58 | Hydrogen Bond | CH |
| CYS145 | 4.97 | Other | Pi-Sulfur |
| MET49 | 5.09 | Hydrophobic | Pi-Alkyl |
| MET49 | 4.66 | Hydrophobic | Pi-Alkyl |
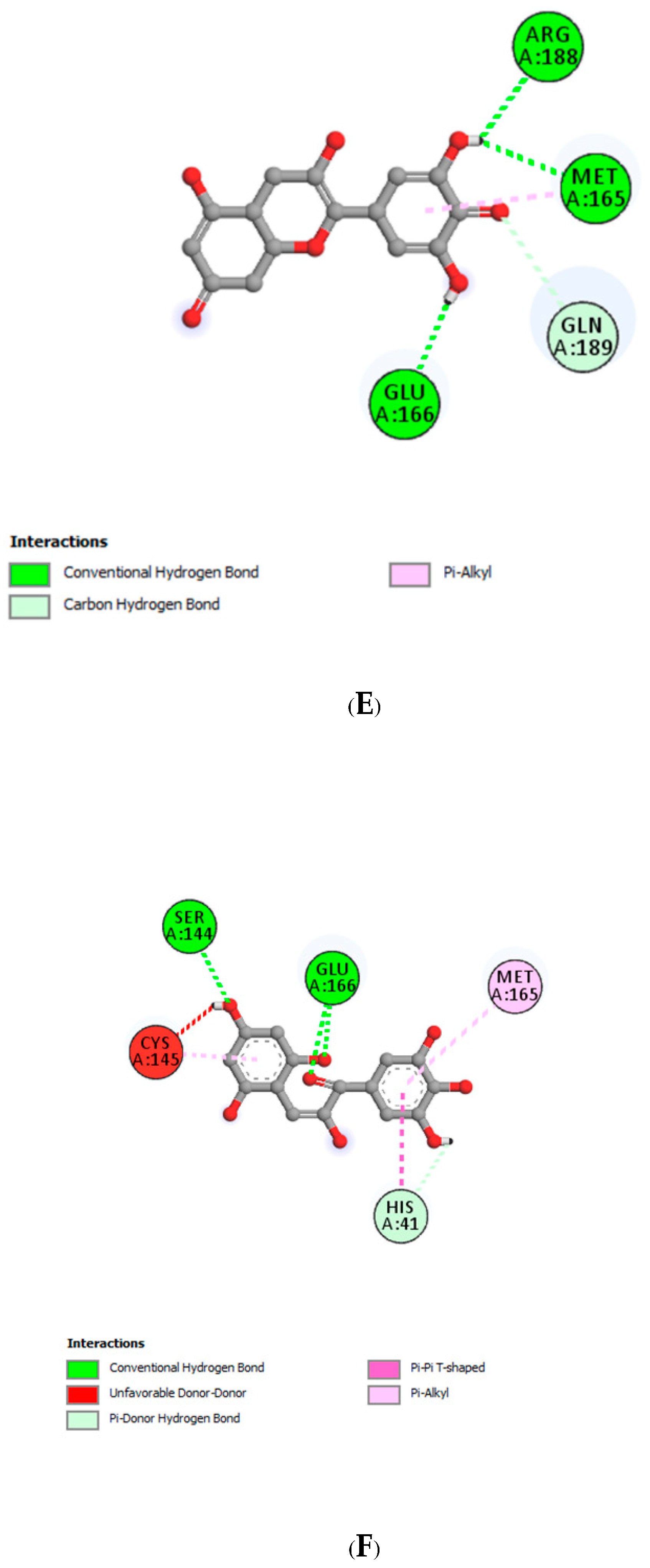
| Interacting Amino Acid Residues of Mpro with | |||||
|---|---|---|---|---|---|
| Delphinidin (PubChem) | Delphinidin Hemiacetal | Delphinidin Hemiketal | Delphinidin Quinonoid 1 | Delphinidin Quinonoid 2 | Delphinidin Chalcone |
| LEU141 | HIS41 | CYS145 | TYR54 | GLU166 | SER144 |
| MET165 | SER144 | LEU141 | ARG188 | MET165 | GLU166 |
| GLN189 | HIS163 | GLN189 | GLU166 | ARG188 | HIS41 |
| GLU166 | MET165 | PRO168 | GLN189 | MET165 | |
| MET165 | CYS145 | MET165 | MET165 | CYS145 | |
| CYS145 | |||||
| Phytochemicals | Interaction with Mpro | Binding Energy (ΔG = kcal/mol) | Interaction with N3-Mpro | Binding Energy (ΔG = kcal/mol) |
|---|---|---|---|---|
| N3 (irreversible inhibitor) | His41, Met49, Phe140, Leu141, Asn142, Gly143, His163, His164, Glu166, Leu167, Pro168, Gln189, Thr190, Ala191 | Not applicable | ||
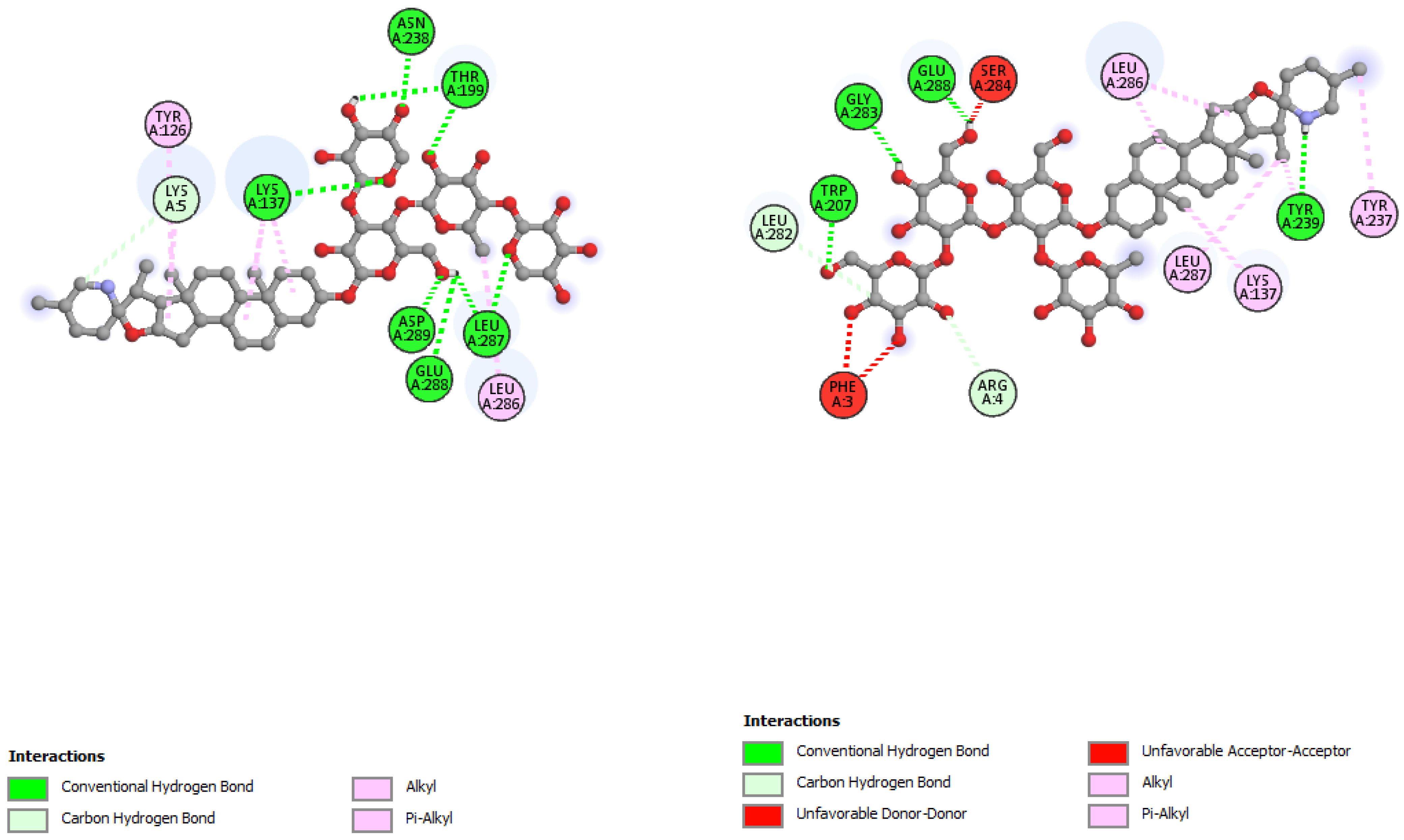
| Incanumine | Thr26, His41, Phe140, Asn142, Gly143, Glu166, Pro168, Gln189, Ala191 | −9.8 | Lys5, Tyr126, Lys137, Thr199, Asn238, Leu286, Leu287, Glu288, Asp289 | −8.9 |
| Solaradixine | Thr26, His 41, Leu141, Asn142, Ser144, Cys145, His164, Met165, Pro168 | −9.4 | Phe3, Arg4, Lys137, Trp207, Tyr237, Tyr239, Leu282, Gly283, Ser284, Leu286, Glu288 | −8.1 |
| Delphinidin | Leu141, Cys145, His163, Met165, Glu166, Gln189 | −7.4 | Thr24, Thr25, Thr45, Asp48, Arg60, Lys61 | −6.9 |
| Delphinidin−3-glucoside | Thr26, His41, Leu141, Cys145, His163, Met165 | −8.4 | Lys137, Asp197, Thr199, Leu286, Leu287, Glu288, Asp289 | −7.2 |
| Residues | Distance | Bond Category | Type |
|---|---|---|---|
| Non-bonded interactions of N3 with Mpro in the original pose. | |||
| GLY143 | 2.79 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLU166 | 2.97 | Hydrogen Bond | Conventional Hydrogen Bond |
| THR190 | 2.84 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLU166 | 2.83 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLN189 | 2.93 | Hydrogen Bond | Conventional Hydrogen Bond |
| PHE140 | 3.13 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLU166 | 3.38 | Hydrogen Bond | Conventional Hydrogen Bond |
| HIS164 | 3.07 | Hydrogen Bond | Conventional Hydrogen Bond |
| HIS172 | 3.32 | Hydrogen Bond | Carbon–Hydrogen Bond |
| MET49 | 4.66 | Hydrophobic | Alkyl |
| MET165 | 4.57 | Hydrophobic | Alkyl |
| LEU167 | 5.46 | Hydrophobic | Alkyl |
| HIS41 | 4.31 | Hydrophobic | Pi-Alkyl |
| PRO168 | 4.84 | Hydrophobic | Pi-Alkyl |
| ALA191 | 4.53 | Hydrophobic | Pi-Alkyl |
| Non-bonded interactions of N3 with Mpro in the re-docked pose. | |||
| HIS163 | 2.01 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLU166 | 2.32 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLN189 | 2.55 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLN189 | 2.27 | Hydrogen Bond | Conventional Hydrogen Bond |
| GLU166 | 3.73 | Hydrogen Bond | Carbon–Hydrogen Bond |
| GLU166 | 4.33 | Electrostatic | Pi-Anion |
| HIS41 | 3.92 | Hydrophobic | Pi-Sigma |
| MET49 | 3.89 | Hydrophobic | Alkyl |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmood, R.A.; Hasan, A.; Rahmatullah, M.; Paul, A.K.; Jahan, R.; Jannat, K.; Bondhon, T.A.; Mahboob, T.; Nissapatorn, V.; de Lourdes Pereira, M.; et al. Solanaceae Family Phytochemicals as Inhibitors of 3C-Like Protease of SARS-CoV-2: An In Silico Analysis. Molecules 2022, 27, 4739. https://doi.org/10.3390/molecules27154739
Mahmood RA, Hasan A, Rahmatullah M, Paul AK, Jahan R, Jannat K, Bondhon TA, Mahboob T, Nissapatorn V, de Lourdes Pereira M, et al. Solanaceae Family Phytochemicals as Inhibitors of 3C-Like Protease of SARS-CoV-2: An In Silico Analysis. Molecules. 2022; 27(15):4739. https://doi.org/10.3390/molecules27154739
Chicago/Turabian StyleMahmood, Raisul Awal, Anamul Hasan, Mohammed Rahmatullah, Alok K. Paul, Rownak Jahan, Khoshnur Jannat, Tohmina Afroze Bondhon, Tooba Mahboob, Veeranoot Nissapatorn, Maria de Lourdes Pereira, and et al. 2022. "Solanaceae Family Phytochemicals as Inhibitors of 3C-Like Protease of SARS-CoV-2: An In Silico Analysis" Molecules 27, no. 15: 4739. https://doi.org/10.3390/molecules27154739
APA StyleMahmood, R. A., Hasan, A., Rahmatullah, M., Paul, A. K., Jahan, R., Jannat, K., Bondhon, T. A., Mahboob, T., Nissapatorn, V., de Lourdes Pereira, M., Paul, T. K., Rumi, O. H., Wiart, C., & Wilairatana, P. (2022). Solanaceae Family Phytochemicals as Inhibitors of 3C-Like Protease of SARS-CoV-2: An In Silico Analysis. Molecules, 27(15), 4739. https://doi.org/10.3390/molecules27154739