
Identification of 3-Oxindole Derivatives as Small Molecule HIV-1 Inhibitors Targeting Tat-Mediated Viral Transcription

 ,
,
Abstract
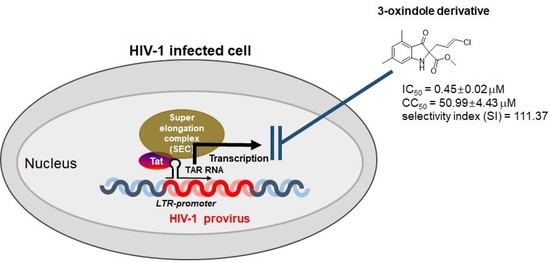
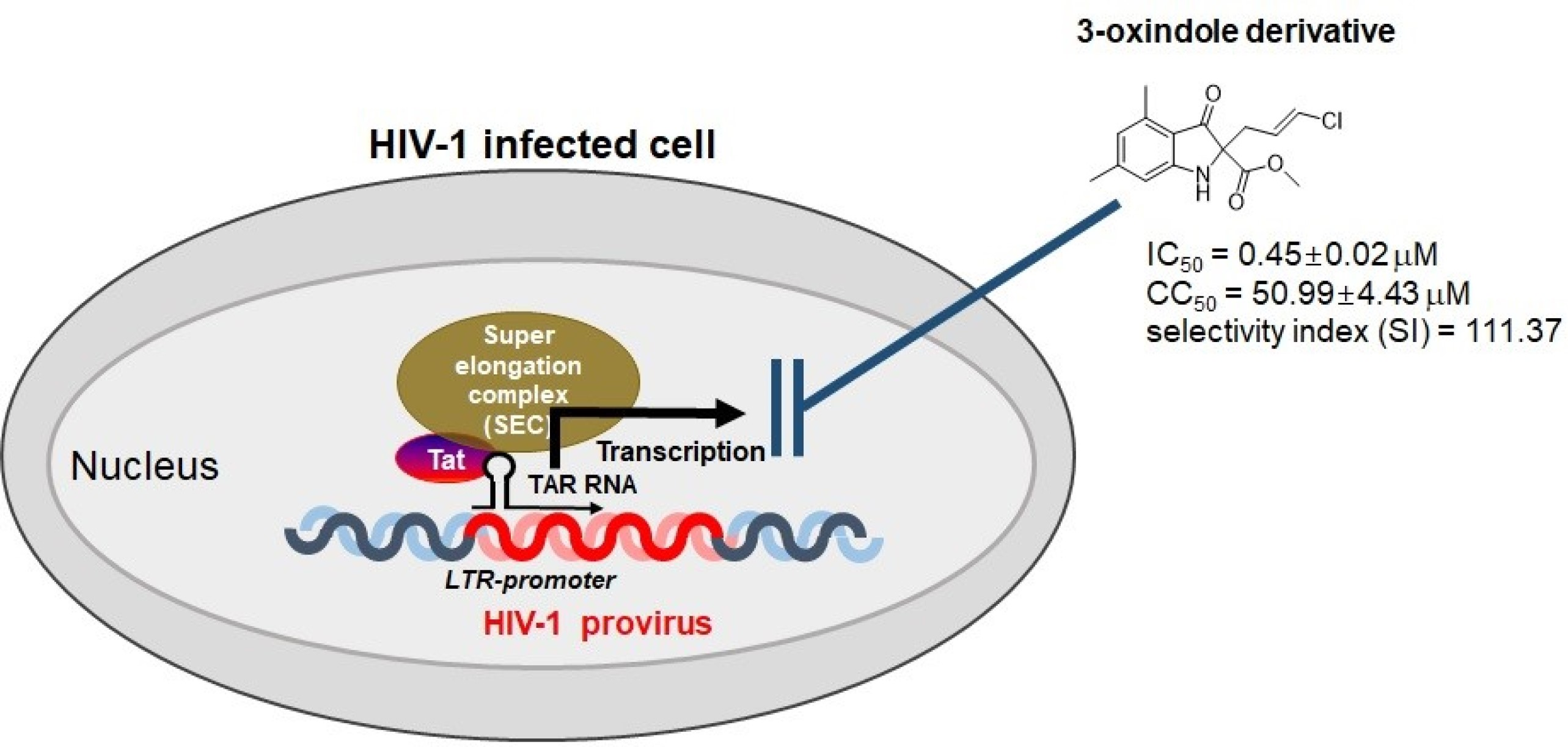
1. Introduction
2. Results and Discussion
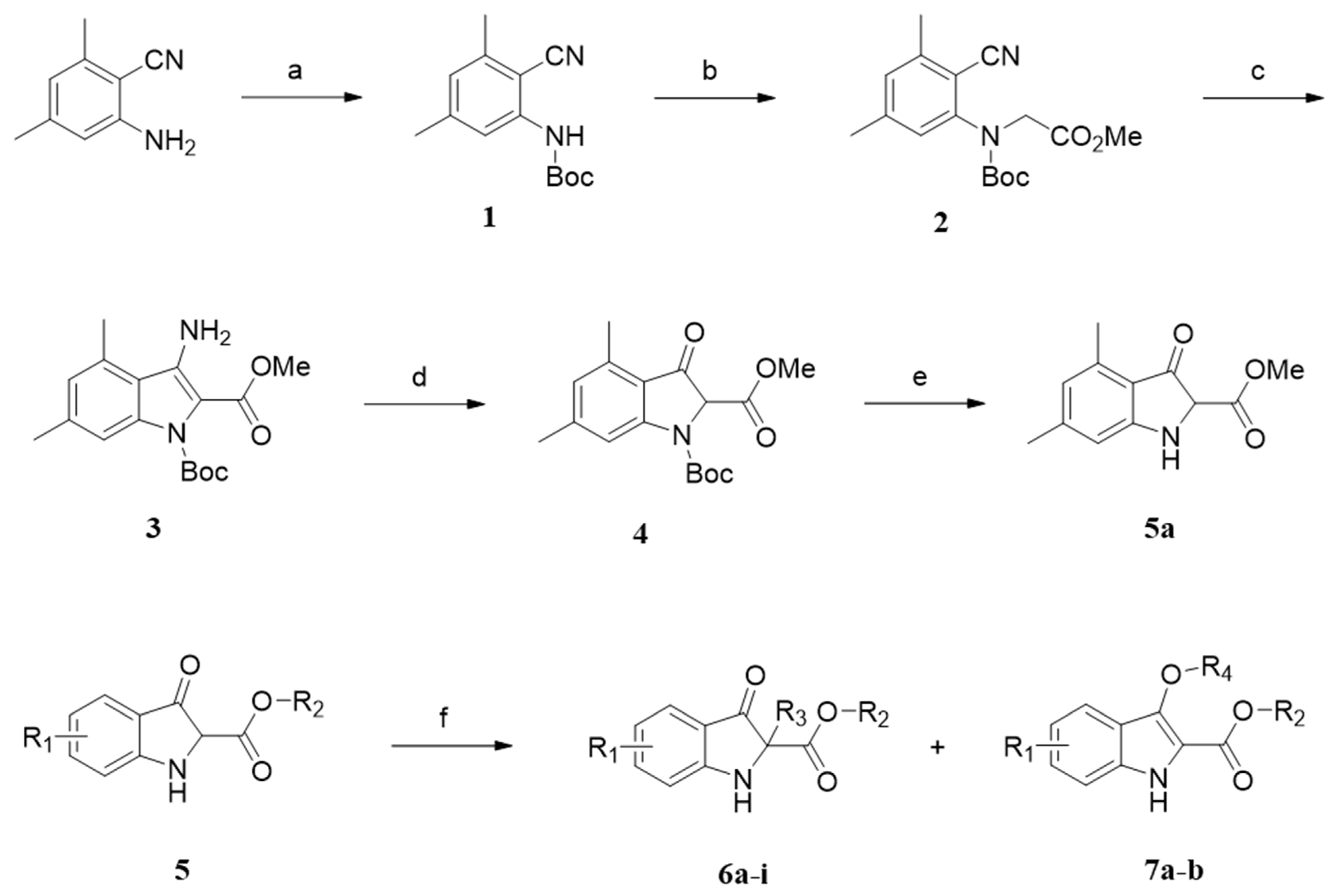
2.1. Chemistry
2.2. Evaluation of Anti-HIV-1 Activity
3. Materials and Methods
3.1. General Chemical Information
3.1.1. tert-Butyl (2-cyano-3,5-dimethylphenyl) carbamate (1)
3.1.2. Methyl-N-(tert-butoxycarbonyl)-N-(2-cyano-3,5-dimethylphenyl)-glycinate (2)
3.1.3. 1-(tert-Butyl) 2-methyl 4,6-dimethyl-3-oxindole-1,2-dicarboxylate (4)
3.1.4. Methyl 4,6-dimethyl-3-oxindole-2-carboxylate (5a)
3.1.5. Ethyl (E)-2-(3-chloroallyl)-4-methyl-3-oxindole-2-carboxylate (6a) and ethyl (e)-3-((3-chloroallyl)oxy)-4-methyl-1h-indole-2-carboxylate (7a)
3.1.6. Methyl (E)-2-(3-chloroallyl)-5-methoxy-3-oxindole-2-carboxylate (6b) and methyl (e)-3-((3-chloroallyl)oxy)-5-methoxy-1h-indole-2-carboxylate (7b)
3.1.7. Methyl 5-methoxy-3-oxo-2-propylindoline-2-carboxylate (6c)
3.1.8. Methyl (E)-2-(3-chloroallyl)-5-methyl-3-oxindole-2-carboxylate (6d)
3.1.9. Methyl (E)-2-(3-chloroallyl)-3-oxindole-2-carboxylate (6e)
3.1.10. Methyl (E)-2-(3-chloroallyl)-4,6-dimethyl-3-oxindole-2-carboxylate (6f)
3.1.11. Methyl 2-(3,3-dichloroallyl)-4,6-dimethyl-3-oxindole-2-carboxylate (6g)
3.1.12. Methyl 2-allyl-4,6-dimethyl-3-oxindole-2-carboxylate (6h)
3.1.13. Methyl 4,6-dimethyl-3-oxo-2-propylindoline-2-carboxylate (6i)
3.2. Biological Assays
3.2.1. Cells, Virus, and Reagents
3.2.2. Assay for HIV-1 Infectivity
3.2.3. Assay for Tat-Mediated HIV-1 Transcription
3.2.4. Assay for HIV-1 Reverse Transcription (RT) Activity
3.2.5. Assay for HIV-1 Integrase Activity
3.2.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pepin, J.; et al. The early spread and epidemic ignition of HIV-1 in human populations. Science 2014, 346, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Laskey, S.B.; Siliciano, R.F. A mechanistic theory to explain the efficacy of antiretroviral therapy. Nat. Rev. Microbiol. 2014, 12, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, J.; McWilliams, M.J.; Mott, B.T.; Thomas, C.J.; Smith, S.J.; Hughes, S.H. Drug resistant integrase mutants cause aberrant HIV integrations. Retrovirology 2016, 13, 71. [Google Scholar] [CrossRef]
- Ross, J.; Jiamsakul, A.; Kumarasamy, N.; Azwa, I.; Merati, T.P.; Do, C.D.; Lee, M.P.; Ly, P.S.; Yunihastuti, E.; Nguyen, K.V.; et al. Virological failure and HIV drug resistance among adults living with HIV on second-line antiretroviral therapy in the Asia-Pacific. HIV Med. 2021, 22, 201–211. [Google Scholar] [CrossRef] [PubMed]
- WHO. HIV Drug Resistance Report 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Dhokne, P.; Sakla, A.P.; Shankaraiah, N. Structural insights of oxindole-based kinase inhibitors as anticancer agents: Recent advances. Eur. J. Med. Chem. 2021, 216, 113334. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Jiang, Z.; Feng, X.Z. New oxindole alkaloid glycosides from Uncaria sinensis. Yao Xue Xue Bao = Acta Pharm. Sin. 1993, 28, 849–853. [Google Scholar]
- Mendes, V.I.S.; Bartholomeusz, G.A.; Ayres, M.; Gandhi, V.; Salvador, J.A.R. Synthesis and cytotoxic activity of novel A-ring cleaved ursolic acid derivatives in human non-small cell lung cancer cells. Eur. J. Med. Chem. 2016, 123, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Khetmalis, Y.M.; Shivani, M.; Murugesan, S.; Chandra Sekhar, K.V.G. Oxindole and its derivatives: A review on recent progress in biological activities. Biomed. Pharm. 2021, 141, 111842. [Google Scholar] [CrossRef] [PubMed]
- Chander, S.; Tang, C.R.; Penta, A.; Wang, P.; Bhagwat, D.P.; Vanthuyne, N.; Albalat, M.; Patel, P.; Sankpal, S.; Zheng, Y.T.; et al. Hit optimization studies of 3-hydroxy-indolin-2-one analogs as potential anti-HIV-1 agents. Bioorg. Chem. 2018, 79, 212–222. [Google Scholar] [CrossRef]
- Boger, D.L.; Nishi, T. Diastereoselective Dieckmann condensation suitable for introduction of the duocarmycin A C6 center: Development of a divergent strategy for the total synthesis of duocarmycins A and SA. Bioorg. Med. Chem. 1995, 3, 67–77. [Google Scholar] [CrossRef]
- Park, C.S.; Choi, E.B.Y.G.H.; Lee, H.G.; Yang, H.C.; Lee, Y.S.; Cho, S.H.; Shin, D.W.; Choi, K.J. Fungicidal composition for agriculture and horticulture having a 3-alkoxyindole-2-carboxylate derivative. South Korea patent 100434814, 27 May 2004. [Google Scholar]
- Lee, J.H.; So, J.H.; Jeon, J.H.; Choi, E.B.; Lee, Y.R.; Chang, Y.T.; Kim, C.H.; Bae, M.A.; Ahn, J.H. Synthesis of a new fluorescent small molecule probe and its use for in vivo lipid imaging. Chem. Commun. 2011, 47, 7500–7502. [Google Scholar] [CrossRef] [PubMed]
- Le Van, K.; Cauvin, C.; de Walque, S.; Georges, B.; Boland, S.; Martinelli, V.; Demonte, D.; Durant, F.; Hevesi, L.; Van Lint, C. New pyridinone derivatives as potent HIV-1 nonnucleoside reverse transcriptase inhibitors. J. Med. Chem. 2009, 52, 3636–3643. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mitsuya, H.; Weinhold, K.J.; Furman, P.A.; St Clair, M.H.; Lehrman, S.N.; Gallo, R.C.; Bolognesi, D.; Barry, D.W.; Broder, S. 3’-Azido-3′-deoxythymidine (BW A509U): An antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/ lymphadenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. USA 1985, 82, 7096–7100. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Kim, H.G.; Park, C.M.; Choi, M.S.; Kim, D.E.; Choi, B.S.; Kim, K.; Yoon, C.H. Identification of novel compounds against Tat-mediated human immunodeficiency virus-1 transcription by high-throughput functional screening assay. Biochem. Biophys. Res. Commun. 2020, 523, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Park, C.M.; Kim, H.G.; Kim, D.E.; Choi, M.S.; Kim, J.A.; Choi, B.S.; Yoon, C.H. Identification of Aristolactam Derivatives that Act as Inhibitors of Human Immunodeficiency Virus Type 1 Infection and Replication by Targeting Tat-Mediated Viral Transcription. Virol. Sin. 2021, 36, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Choi, B.S.; Kim, K.C.; Kang, C.; Kim, K.; Yoon, C.H. Development of a dual reporter screening assay for distinguishing the inhibition of HIV Tat-mediated transcription from off-target effects. J. Virol. Methods 2017, 249, 1–9. [Google Scholar] [CrossRef]
- Chun, T.W.; Moir, S.; Fauci, A.S. HIV reservoirs as obstacles and opportunities for an HIV cure. Nat. Immunol. 2015, 16, 584–589. [Google Scholar] [CrossRef]
- Garg, V.; Maurya, R.K.; Thanikachalam, P.V.; Bansal, G.; Monga, V. An insight into the medicinal perspective of synthetic analogs of indole: A review. Eur. J. Med. Chem. 2019, 180, 562–612. [Google Scholar]
- Hassam, M.; Basson, A.E.; Liotta, D.C.; Morris, L.; van Otterlo, W.A.; Pelly, S.C. Novel Cyclopropyl-Indole Derivatives as HIV Non-Nucleoside Reverse Transcriptase Inhibitors. ACS Med. Chem. Lett. 2012, 3, 470–475. [Google Scholar] [CrossRef]
- Wang, T.; Ueda, Y.; Zhang, Z.; Yin, Z.; Matiskella, J.; Pearce, B.C.; Yang, Z.; Zheng, M.; Parker, D.D.; Yamanaka, G.A.; et al. Discovery of the Human Immunodeficiency Virus Type 1 (HIV-1) Attachment Inhibitor Temsavir and Its Phosphonooxymethyl Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 6308–6327. [Google Scholar] [CrossRef]
- Ferro, S.; De Luca, L.; Barreca, M.L.; De Grazia, S.; Christ, F.; Debyser, Z.; Chimirri, A. New chloro,fluorobenzylindole derivatives as integrase strand-transfer inhibitors (INSTIs) and their mode of action. Bioorg. Med. Chem. 2010, 18, 5510–5518. [Google Scholar] [CrossRef] [PubMed]
- Guendel, I.; Iordanskiy, S.; Van Duyne, R.; Kehn-Hall, K.; Saifuddin, M.; Das, R.; Jaworski, E.; Sampey, G.C.; Senina, S.; Shultz, L.; et al. Novel neuroprotective GSK-3beta inhibitor restricts Tat-mediated HIV-1 replication. J. Virol. 2014, 88, 1189–1208. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Kuhen, K.L.; Wolff, K.; Yin, H.; Bieza, K.; Caldwell, J.; Bursulaya, B.; Tuntland, T.; Zhang, K.; Karanewsky, D.; et al. Design, synthesis, and biological evaluations of novel oxindoles as HIV-1 non-nucleoside reverse transcriptase inhibitors. Part 2. Bioorg. Med. Chem. Lett. 2006, 16, 2109–2112. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Shin, Y.; Yoon, C.H. Molecular Mechanism Underlying HIV-1 Tat Inhibition by 3-Oxindole-Derivatives; Korea National Institute of Health: Cheongwon-gun, South Korea, 2022; manuscript in preparation. [Google Scholar]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Sarzotti-Kelsoe, M.; Bailer, R.T.; Turk, E.; Lin, C.L.; Bilska, M.; Greene, K.M.; Gao, H.; Todd, C.A.; Ozaki, D.A.; Seaman, M.S.; et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J. Immunol. Methods 2014, 409, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Vicenzi, E.; Poli, G. Infection of CD4+ Primary T Cells and Cell Lines, Generation of Chronically Infected Cell Lines, and Induction of HIV Expression. Current Protocols in Immunology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkyl 3-Oxindole-2-Carboxylates (5) | Product | Yield (%) | Inhibition of HIV-1 Infectivity (%) | Cell Viability (%) | |
|---|---|---|---|---|---|
 5a |  | 6a | 46% | 20.42 ± 1.69 | 106.26 ± 9.33 |
 | 7a | 26% | 29.58 ± 8.08 | 116.76 ± 2.70 | |
 5b |  | 6b | 50% | 0.55 ± 4.34 | 99.64 ± 14.19 |
 | 7b | 30% | 28.97 ± 3.78 | 110.47 ± 4.96 | |
 5c |  | 6c | 45% | 16.20 ± 5.52 | 92.70 ± 5.77 |
 5d |  | 6d | 39% | 0.00 ± 10.66 | 96.96 ± 9.15 |
 5e |  | 6e | 50% | 11.49 ± 5.66 | 95.66 ± 7.32 |
 5f |  | 6f | 24% | 91.87 ± 1.78 | 92.95 ± 5.39 |
 | 6g | 7% | 85.39 ± 1.44 | 87.90 ± 0.82 | |
 | 6h | 50% | 23.56 ± 4.46 | 107.13 ± 4.73 | |
 | 6i | 50% | 9.15 ± 0.96 | 99.37 ± 8.83 | |
| Azidothymidine (AZT) | 99.65 ± 0.048 | 94.75 ± 7.21 | |||
| Compound | IC50(μM) a | CC50(μM) b | SI c |
|---|---|---|---|
| 6a | 13.685 ± 2.5526 | 87.3833 ± 12.6685 | 6.38 |
| 6f | 0.4578 ± 0.0227 | 50.99 ± 4.4357 | 111.37 |
| 6g | 1.276 ± 0.9589 | 43.6357 ± 10.5195 | 34.19 |
| EFV | <0.05 | 34.1933 ± 0.59 | >683.86 |
| AZT | 0.079 ± 0.0237 | >100 | >1265.5 |
| Compound | IC50 (μM) a |
|---|---|
| 6a | 9.53 ± 0.48 |
| 6f | 1.20 ± 0.04 |
| 6g | 2.85 ± 0.63 |
| seliciclib | 2.41 ± 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-E.; Shin, Y.H.; Cho, J.-E.; Myung, S.; Kim, H.G.; Kim, K.-C.; Park, C.M.; Yoon, C.-H. Identification of 3-Oxindole Derivatives as Small Molecule HIV-1 Inhibitors Targeting Tat-Mediated Viral Transcription. Molecules 2022, 27, 4921. https://doi.org/10.3390/molecules27154921
Kim D-E, Shin YH, Cho J-E, Myung S, Kim HG, Kim K-C, Park CM, Yoon C-H. Identification of 3-Oxindole Derivatives as Small Molecule HIV-1 Inhibitors Targeting Tat-Mediated Viral Transcription. Molecules. 2022; 27(15):4921. https://doi.org/10.3390/molecules27154921
Chicago/Turabian StyleKim, Dong-Eun, Young Hyun Shin, Jung-Eun Cho, Subeen Myung, Hong Gi Kim, Kyung-Chang Kim, Chul Min Park, and Cheol-Hee Yoon. 2022. "Identification of 3-Oxindole Derivatives as Small Molecule HIV-1 Inhibitors Targeting Tat-Mediated Viral Transcription" Molecules 27, no. 15: 4921. https://doi.org/10.3390/molecules27154921
APA StyleKim, D.-E., Shin, Y. H., Cho, J.-E., Myung, S., Kim, H. G., Kim, K.-C., Park, C. M., & Yoon, C.-H. (2022). Identification of 3-Oxindole Derivatives as Small Molecule HIV-1 Inhibitors Targeting Tat-Mediated Viral Transcription. Molecules, 27(15), 4921. https://doi.org/10.3390/molecules27154921