
Intramolecular Phosphine-Promoted Knoevenagel Based Redox-Reaction



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
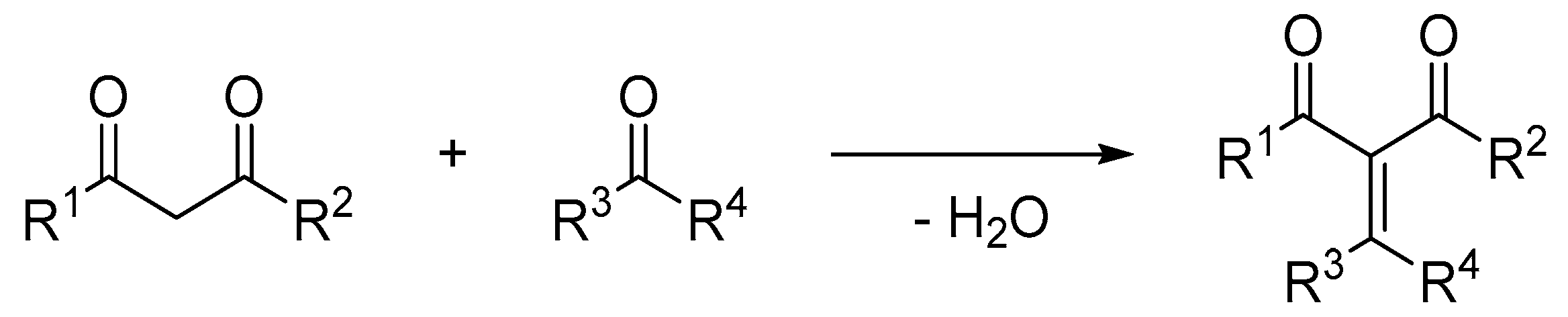
1. Introduction
2. Results and Discussion
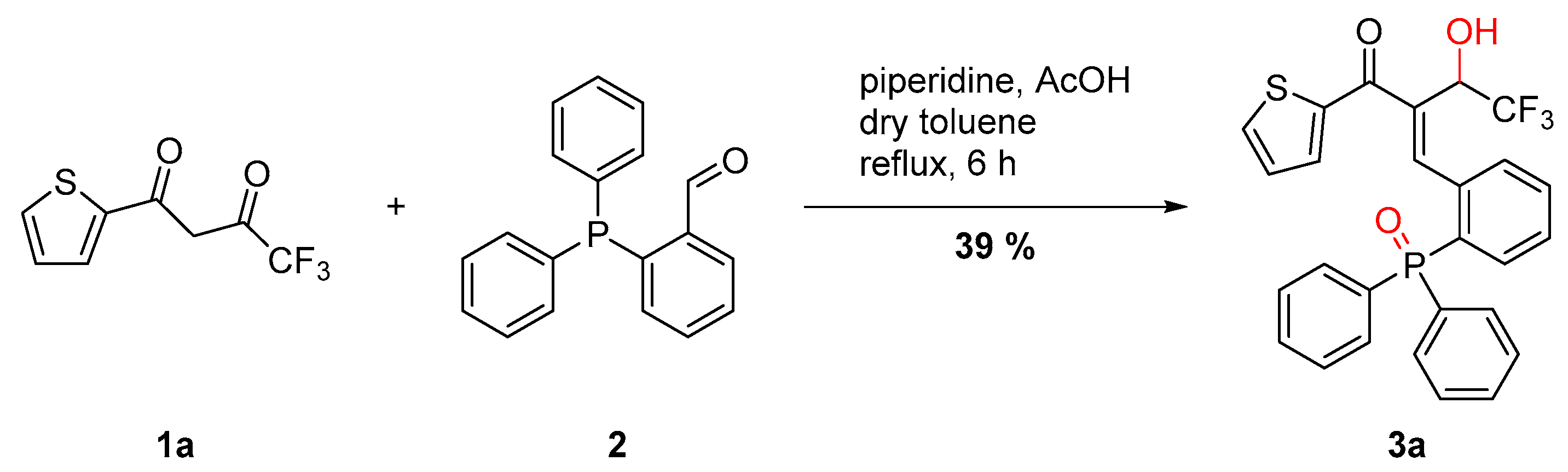
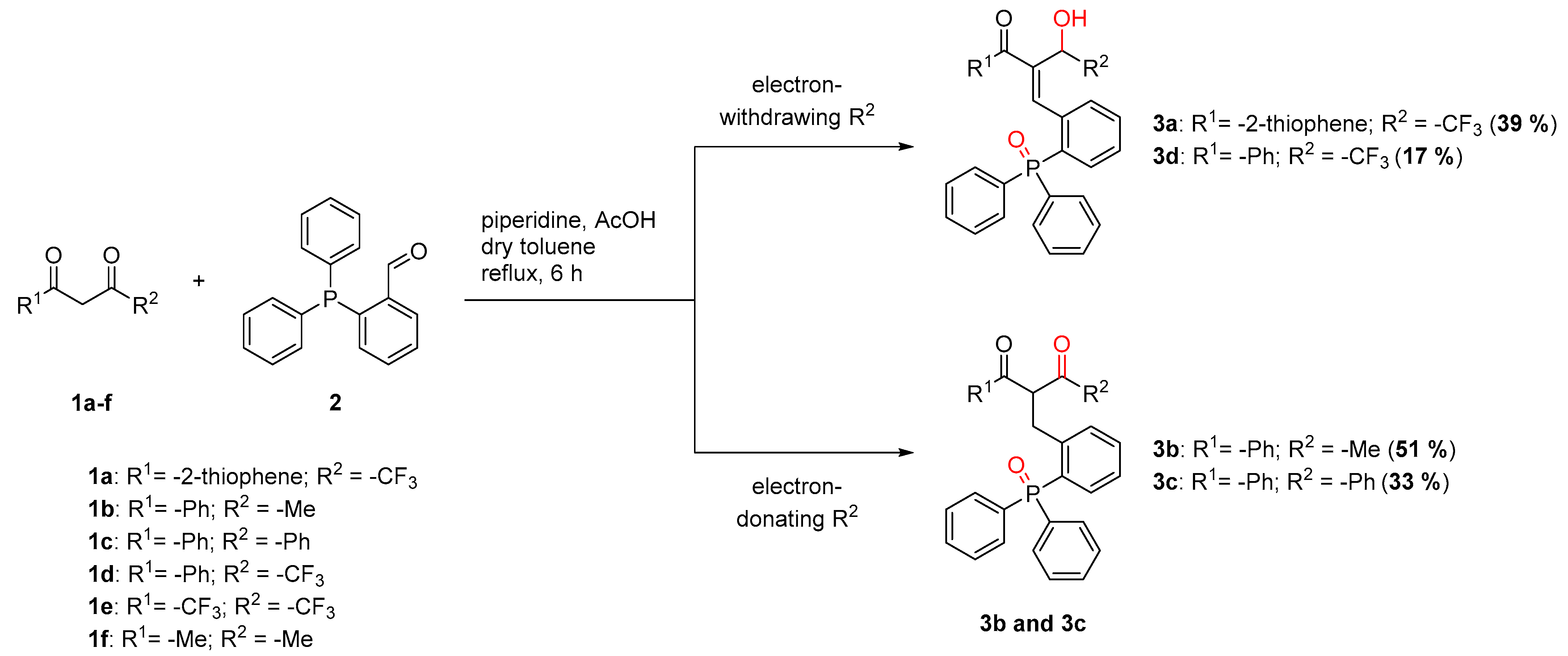
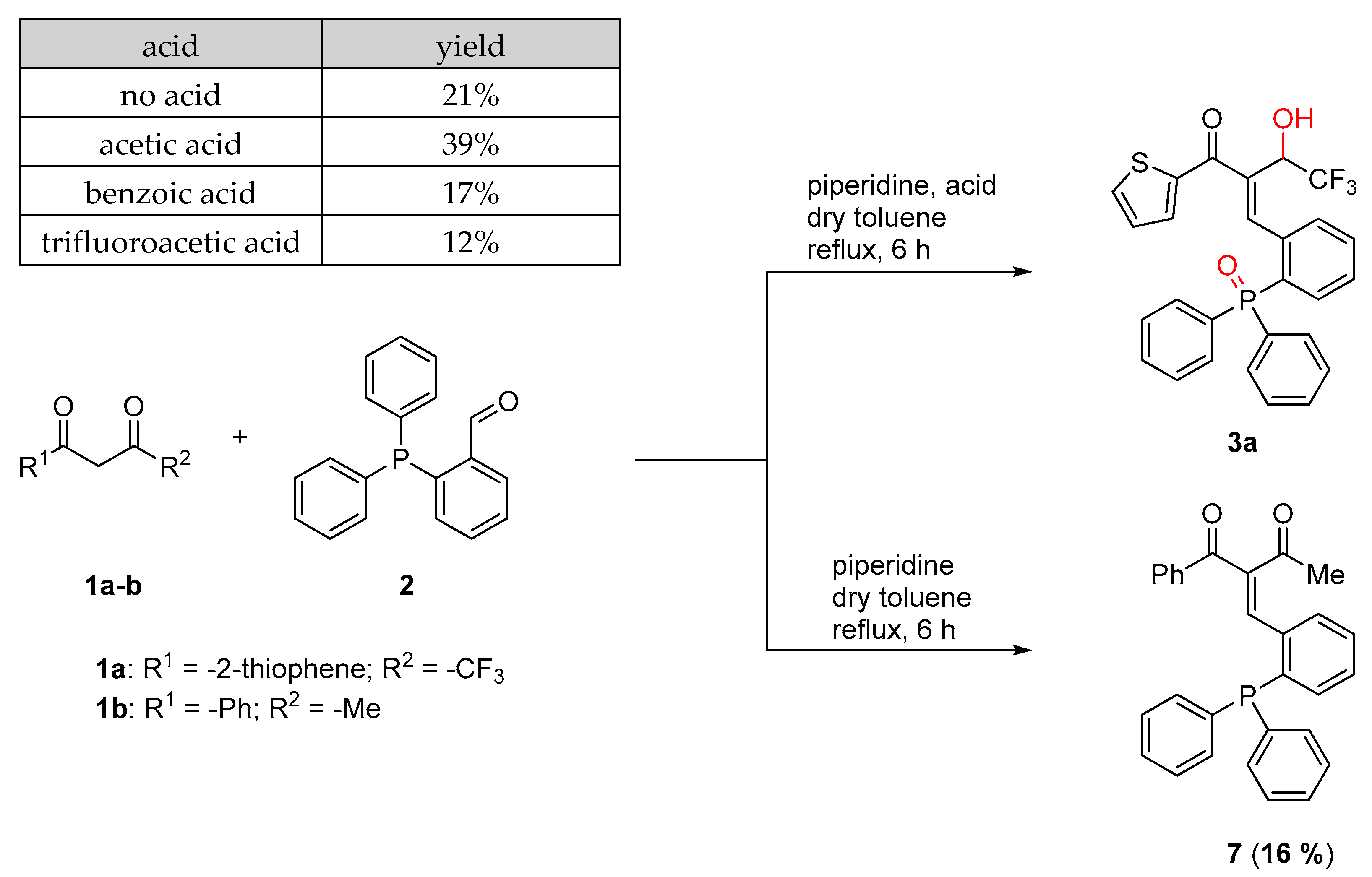
2.1. Scope of Diketones
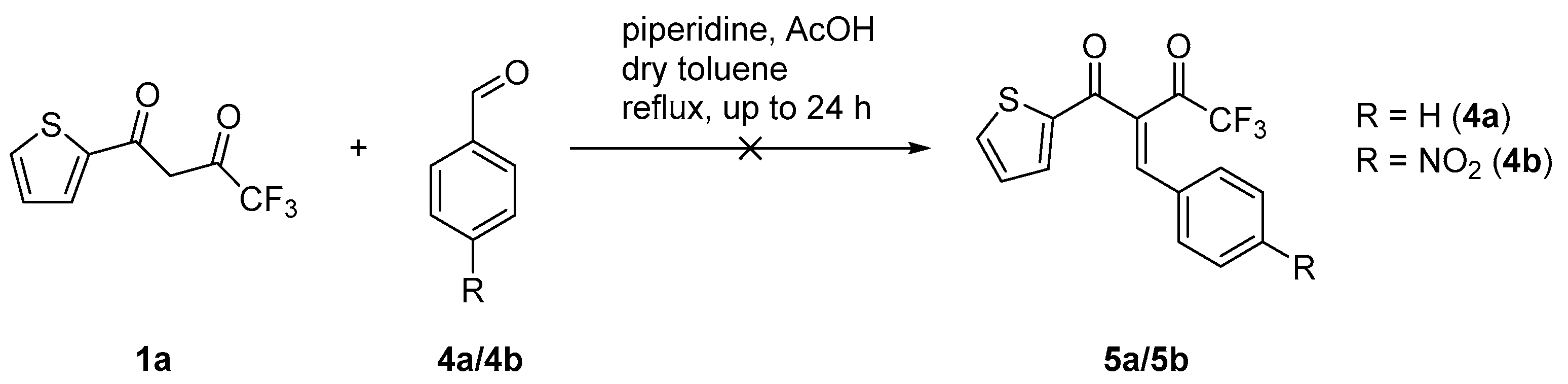
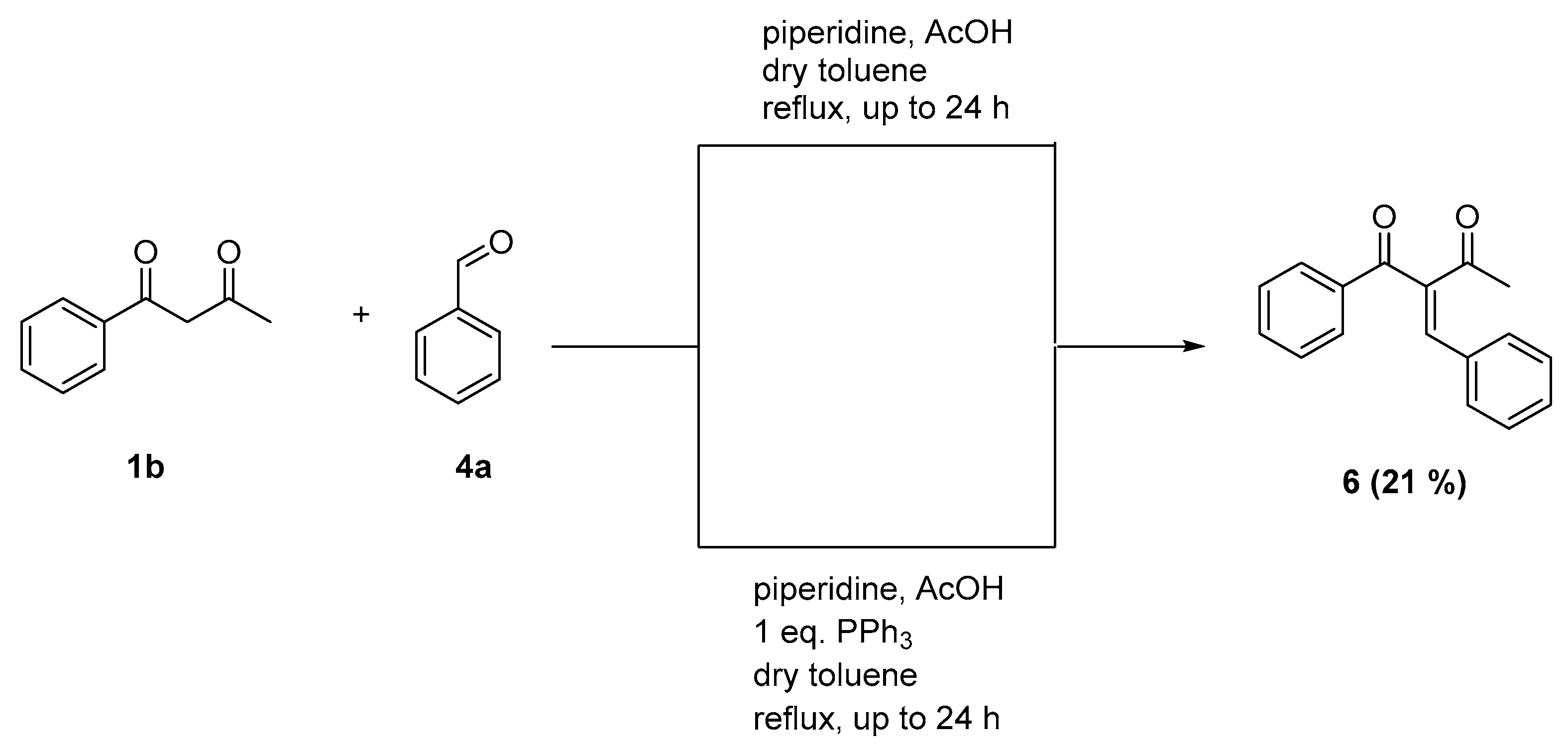
2.2. Reaction with Benzaldehyde
2.3. Scope of Acids
2.4. Scope of Bases
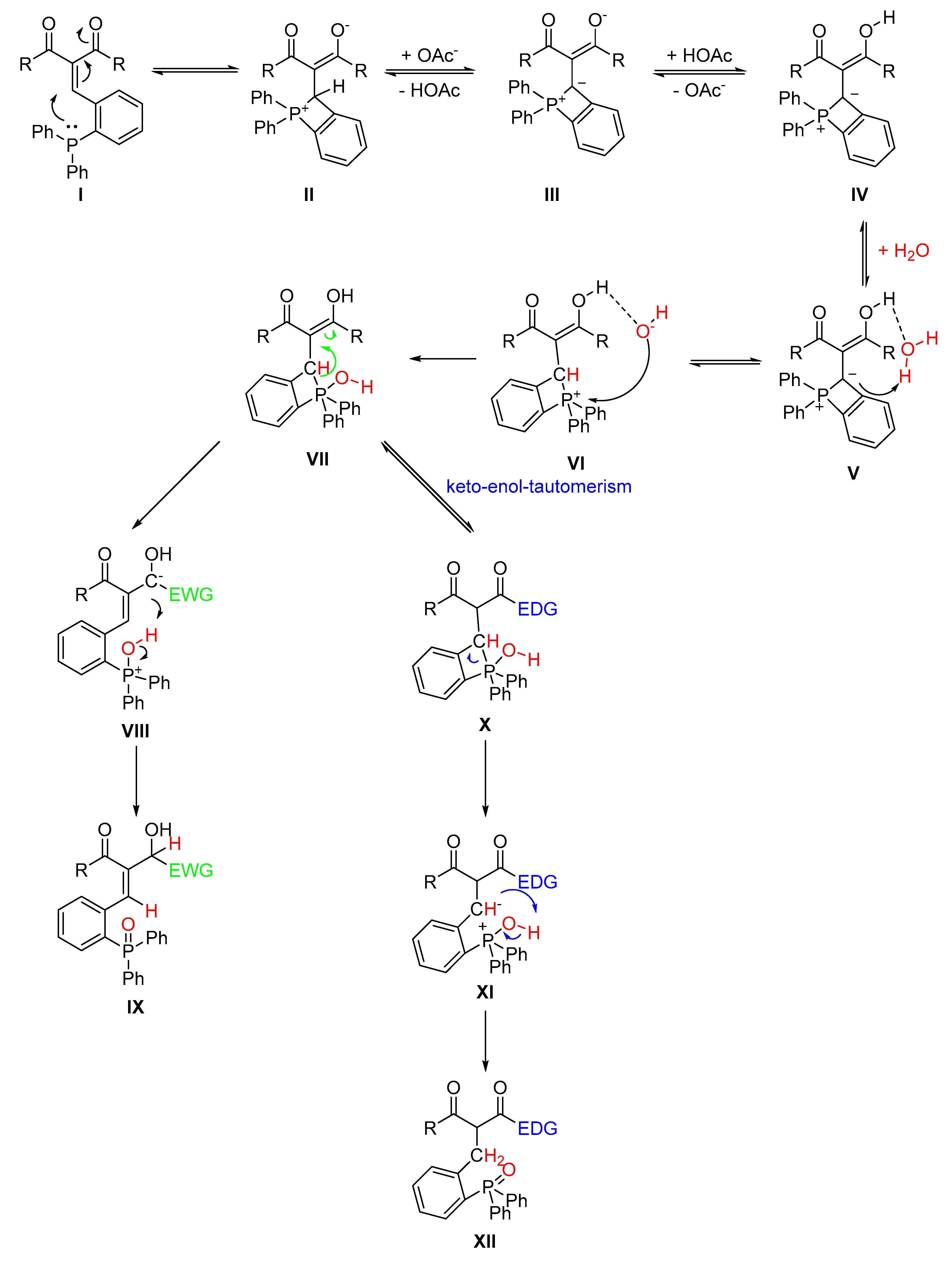
2.5. Proposed Mechanism
3. Materials and Methods
3.1. General Procedure for the Knoevenagel Reaction
3.2. 2-(2-(Diphenylphosphoryl)benzylidene)-4,4,4-trifluoro-3-hydroxy-1-(thiophen-2-yl)butan-1-one (3a)
3.3. 2-(2-(Diphenylphosphoryl)benzyl)-1-phenylbutane-1,3-dione (3b)
3.4. 2-(2-(Diphenylphosphoryl)benzyl)-1,3-diphenylpropane-1,3-dione (3c)
3.5. 2-(2-(Diphenylphosphoryl)benzylidene)-4,4,4-trifluoro-3-hydroxy-1-phenylbutan-1-one (3d)
3.6. 2-Benzylidene-1-phenylbutane-1,3-dione (6)
3.7. 2-(2-(Diphenylphosphaneyl)benzylidene)-1-phenylbutane-1,3-dione (7)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Heravi, M.M.; Janati, F.; Zadsirjan, V. Applications of Knoevenagel condensation reaction in the total synthesis of natural products. Monatsh. Chem. 2020, 151, 439–482. [Google Scholar] [CrossRef]
- Isobe, K.; Hoshi, T.; Suzuki, T.; Hagiwara, H. Knoevenagel reaction in water catalyzed by amine supported on silica gel. Mol. Divers. 2005, 9, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Kraus, G.A.; Krolski, M.E. Synthesis of a precursor to quassimarin. J. Org. Chem. 1986, 51, 3347–3350. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Pu, Y.-J.; Kurata, T.; Kido, J.; Nishide, H. Synthesis and electroluminescent property of poly(p-phenylenevinylene)s bearing triarylamine pendants. Polymer 2005, 46, 3767–3775. [Google Scholar] [CrossRef]
- Van Beurden, K.; de Koning, S.; Molendijk, D.; van Schijndel, J. The Knoevenagel reaction: A review of the unfinished treasure map to forming carbon–carbon bonds. Green Chem. Lett. Rev. 2020, 13, 349–364. [Google Scholar] [CrossRef]
- Van Schijndel, J.; Canalle, L.A.; Molendijk, D.; Meuldijk, J. The green Knoevenagel condensation: Solvent-free condensation of benzaldehydes. Green Chem. Lett. Rev. 2017, 10, 404–411. [Google Scholar] [CrossRef] [Green Version]
- Knoevenagel, E. Ueber eine Darstellungsweise der Glutarsäure. Ber. Dtsch. Chem. Ges. 1894, 27, 2345–2346. [Google Scholar] [CrossRef] [Green Version]
- Knochel, P.; Molander, G.A. (Eds.) Comprehensive Organic Synthesis II; Elsevier: Amsterdam, The Netherlands, 2014; ISBN 9780080977430. [Google Scholar]
- Bigi, F.; Quarantelli, C. The Knoevenagel Condensation in Water. Curr. Org. Synth. 2012, 9, 31–39. [Google Scholar] [CrossRef]
- Liu, Y.; Mao, Y.; Hu, Y.; Gui, J.; Wang, L.; Wang, W.; Zhang, S. The Employment of Sodium Hydride as a Michael Donor in Palladium-catalyzed Reductions of α, β-Unsaturated Carbonyl Compounds. Adv. Synth. Catal. 2019, 361, 1554–1558. [Google Scholar] [CrossRef]
- Saravanakumar, R.; Markopoulos, G.; Bahrin, L.; Jones, P.; Hopf, H. The Regiospecific Preparation of 2-Substituted Tribenzotriquinacenes. Synlett 2013, 24, 453–456. [Google Scholar] [CrossRef]
- Yuan, X.; Lin, L.; Chen, W.; Wu, W.; Liu, X.; Feng, X. Synthesis of Chiral Tetrahydrofurans via Catalytic Asymmetric 3 + 2 Cycloaddition of Heterosubstituted Alkenes with Oxiranes. J. Org. Chem. 2016, 81, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Sang, J.-W.; Xie, M.-S.; Wang, M.-M.; Qu, G.-R.; Guo, H.-M. Chemo- and regioselective ring-opening of donor-acceptor oxiranes with N-heteroaromatics. Chem. Comm. 2021, 57, 4552–4555. [Google Scholar] [CrossRef] [PubMed]
- Antonioletti, R.; Bovicelli, P.; Malancona, S. A new route to 2-alkenyl-1,3-dicarbonyl compounds, intermediates in the synthesis of dihydrofurans. Tetrahedron 2002, 58, 589–596. [Google Scholar] [CrossRef]
- Rioux, B.; Peyrot, C.; Mention, M.M.; Brunissen, F.; Allais, F. Sustainable Synthesis of p-Hydroxycinnamic Diacids through Proline-Mediated Knoevenagel Condensation in Ethanol: An Access to Potent Phenolic UV Filters and Radical Scavengers. Antioxidants 2020, 9, 331. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Xu, C.; Gao, J.; Gao, F.; Zhu, D.; Wang, M. Me3SiCF2Br-Self-Assisted Domino Reaction: Catalytic Synthesis of α,α-Difluorocyclopentanones from Methylvinylketones. Org. Lett. 2017, 19, 1850–1853. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, M.; Shu, W.-M.; Wu, L.-M.; Zhang, D.-X.; Wu, A.-X. Synthesis of α-iodoketals from methyl ketones via sustainable and orthogonal tandem catalysis. Org. Biomol. Chem. 2013, 11, 1226–1233. [Google Scholar] [CrossRef]
- Abiola, T.T.; Rioux, B.; Toldo, J.M.; Alarcan, J.; Woolley, J.M.; Turner, M.A.P.; Coxon, D.J.L.; Telles do Casal, M.; Peyrot, C.; Mention, M.M.; et al. Towards developing novel and sustainable molecular light-to-heat converters. Chem. Sci. 2021, 12, 15239–15252. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.L.; Melnikov, M.Y.; Budynina, E.M. Reductive Knoevenagel Condensation with the Zn–AcOH System. Synthesis 2021, 53, 1285–1291. [Google Scholar] [CrossRef]
- Arai, H.; Ohno, A.; Tani, Y.; Imachi, S.; Mukaiyama, T. A Synthesis of AB-Ring Model System of Taxane Framework by Way of Intramolecular Knoevenagel Cyclization. Chem. Lett. 2002, 31, 92–93. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Wang, Z.; Zhou, J.; Fan, X.; Fu, Y. Biosynthesis of α-Substituted β-Ketoesters via the Tandem Knoevenagel Condensation–Reduction Reaction Using a Single Enzyme. ACS Sustain. Chem. Eng. 2020, 8, 8206–8213. [Google Scholar] [CrossRef]
- Li, X.; Zhang, B.; Fang, Y.; Sun, W.; Qi, Z.; Pei, Y.; Qi, S.; Yuan, P.; Luan, X.; Goh, T.W.; et al. Metal-Organic-Framework-Derived Carbons: Applications as Solid-Base Catalyst and Support for Pd Nanoparticles in Tandem Catalysis. Chemistry 2017, 23, 4266–4270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyon, C.; Duclos, M.-C.; Sutter, M.; Métay, E.; Lemaire, M. Reductive alkylation of active methylene compounds with carbonyl derivatives, calcium hydride and a heterogeneous catalyst. Org. Biomol. Chem. 2015, 13, 7067–7075. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Kazmaier, U. Internal redox catalyzed by triphenylphosphine. J. Am. Chem. Soc. 1992, 114, 7933–7935. [Google Scholar] [CrossRef]
- Dutta, L.; Mondal, A.; Ramasastry, S.S.V. Metal-Free Reductive Aldol Reactions. Asian J. Org. Chem. 2021, 10, 680–691. [Google Scholar] [CrossRef]
- Gu, J.; Xiao, B.-X.; Chen, Y.-R.; Li, Q.-Z.; Ouyang, Q.; Du, W.; Chen, Y.-C. Interrupted Morita-Baylis-Hillman-Type Reaction of α-Substituted Activated Olefins. Org. Lett. 2018, 20, 2088–2091. [Google Scholar] [CrossRef] [PubMed]
- Satpathi, B.; Dutta, L.; Ramasastry, S.S.V. Metal- and Hydride-Free Pentannulative Reductive Aldol Reaction. Org. Lett. 2019, 21, 170–174. [Google Scholar] [CrossRef]
- Wang, K.; McConnachie, J.M.; Stiefel, E.I. Syntheses of Metal Dithiolene Complexes from Thiometalates by Induced Internal Redox Reactions. Inorg. Chem. 1999, 38, 4334–4341. [Google Scholar] [CrossRef]
- Yamaki, Y.; Shigenaga, A.; Tomita, K.; Narumi, T.; Fujii, N.; Otaka, A. Synthesis of Fluoroalkene Dipeptide Isosteres by an Intramolecular Redox Reaction Utilizing N -Heteorocyclic Carbenes (NHCs). J. Org. Chem. 2009, 74, 3272–3277. [Google Scholar] [CrossRef]
- Ma, L.; Seidel, D. Intramolecular Redox-Mannich Reactions: Facile Access to the Tetrahydroprotoberberine Core. Chemistry 2015, 21, 12908–12913. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, N.T.; Read de Alaniz, J.; Rovis, T. Conversion of alpha-haloaldehydes into acylating agents by an internal redox reaction catalyzed by nucleophilic carbenes. J. Am. Chem. Soc. 2004, 126, 9518–9519. [Google Scholar] [CrossRef]
- Lee, S.W.; Trogler, W.C. Nucleophilic addition of phosphines to carbonyl groups. Isolation of 1-hydroxy phosphonium and 1-(trimethylsiloxy) phosphonium salts and the crystal structure of (1-hydroxy-1-methylethyl)triethylphosphonium bromide. J. Org. Chem. 1990, 55, 2644–2648. [Google Scholar] [CrossRef]
- Heim, U.; Pritzkow, H.; Fleischer, U.; Grützmacher, H.; Sanchez, M.; Réau, R.; Bertrand, G. λ5-Phosphetes, Benzo-λ5-Phosphetes, Naphtho-λ5-Phosphetes: Four-π-, Eight-π-, and Twelve-π-Electron Systems. Chem. Eur. J. 1996, 2, 68–74. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feuge, N.; Namyslo, J.C.; Kaufmann, D.E.; Wilhelm, R. Intramolecular Phosphine-Promoted Knoevenagel Based Redox-Reaction. Molecules 2022, 27, 4875. https://doi.org/10.3390/molecules27154875
Feuge N, Namyslo JC, Kaufmann DE, Wilhelm R. Intramolecular Phosphine-Promoted Knoevenagel Based Redox-Reaction. Molecules. 2022; 27(15):4875. https://doi.org/10.3390/molecules27154875
Chicago/Turabian StyleFeuge, Niklas, Jan C. Namyslo, Dieter E. Kaufmann, and René Wilhelm. 2022. "Intramolecular Phosphine-Promoted Knoevenagel Based Redox-Reaction" Molecules 27, no. 15: 4875. https://doi.org/10.3390/molecules27154875
APA StyleFeuge, N., Namyslo, J. C., Kaufmann, D. E., & Wilhelm, R. (2022). Intramolecular Phosphine-Promoted Knoevenagel Based Redox-Reaction. Molecules, 27(15), 4875. https://doi.org/10.3390/molecules27154875