
Synthesis, Pharmacokinetic Characterization and Antioxidant Capacity of Carotenoid Succinates and Their Melatonin Conjugates

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
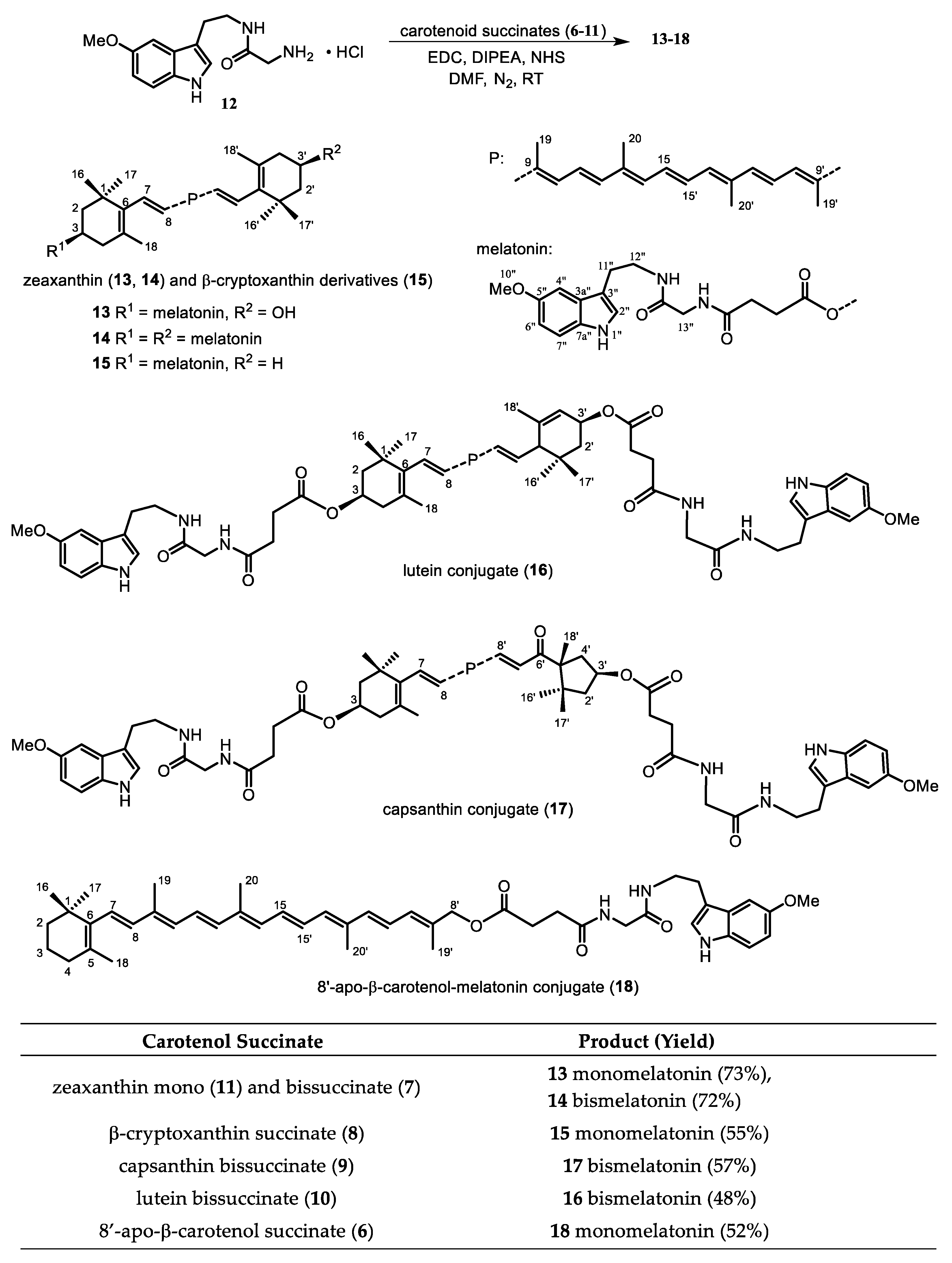
2.1. Synthesis
2.2. Physicochemical Characterization of Investigated Carotenol Derivatives
2.2.1. In Silico and in Vitro Physicochemical and Pharmacokinetic Characterization of Carotenol Derivatives
2.2.2. Aggregation
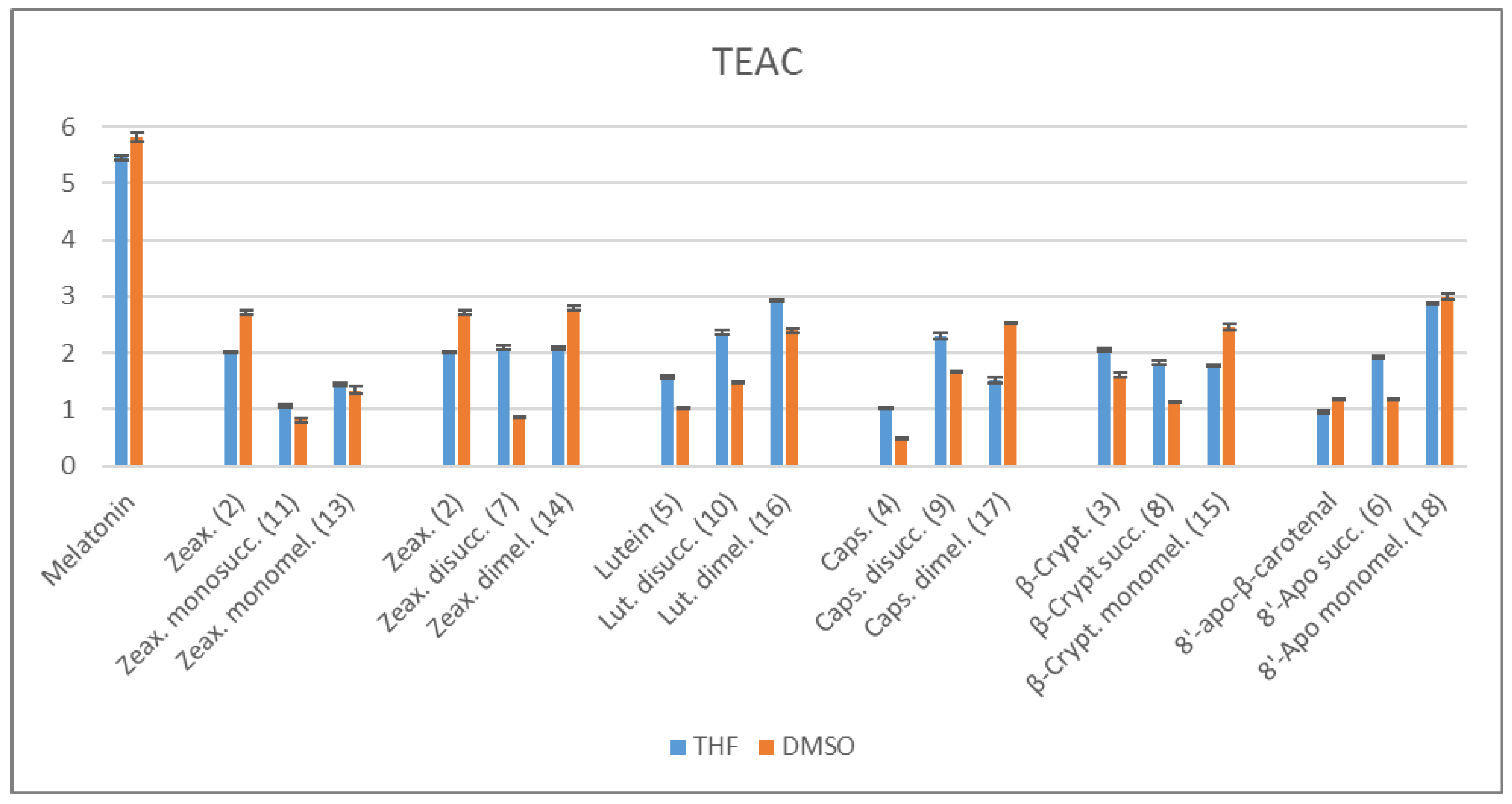
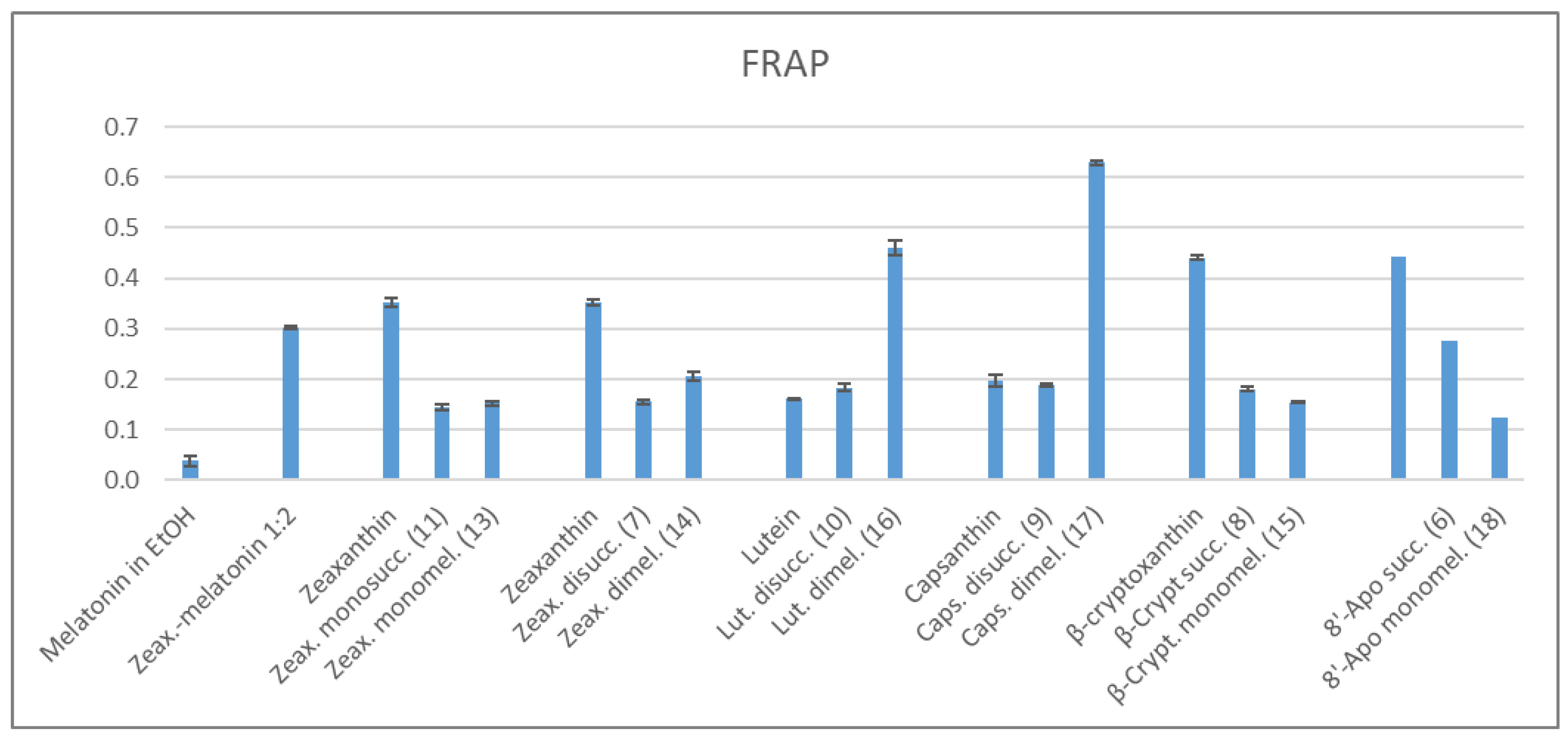
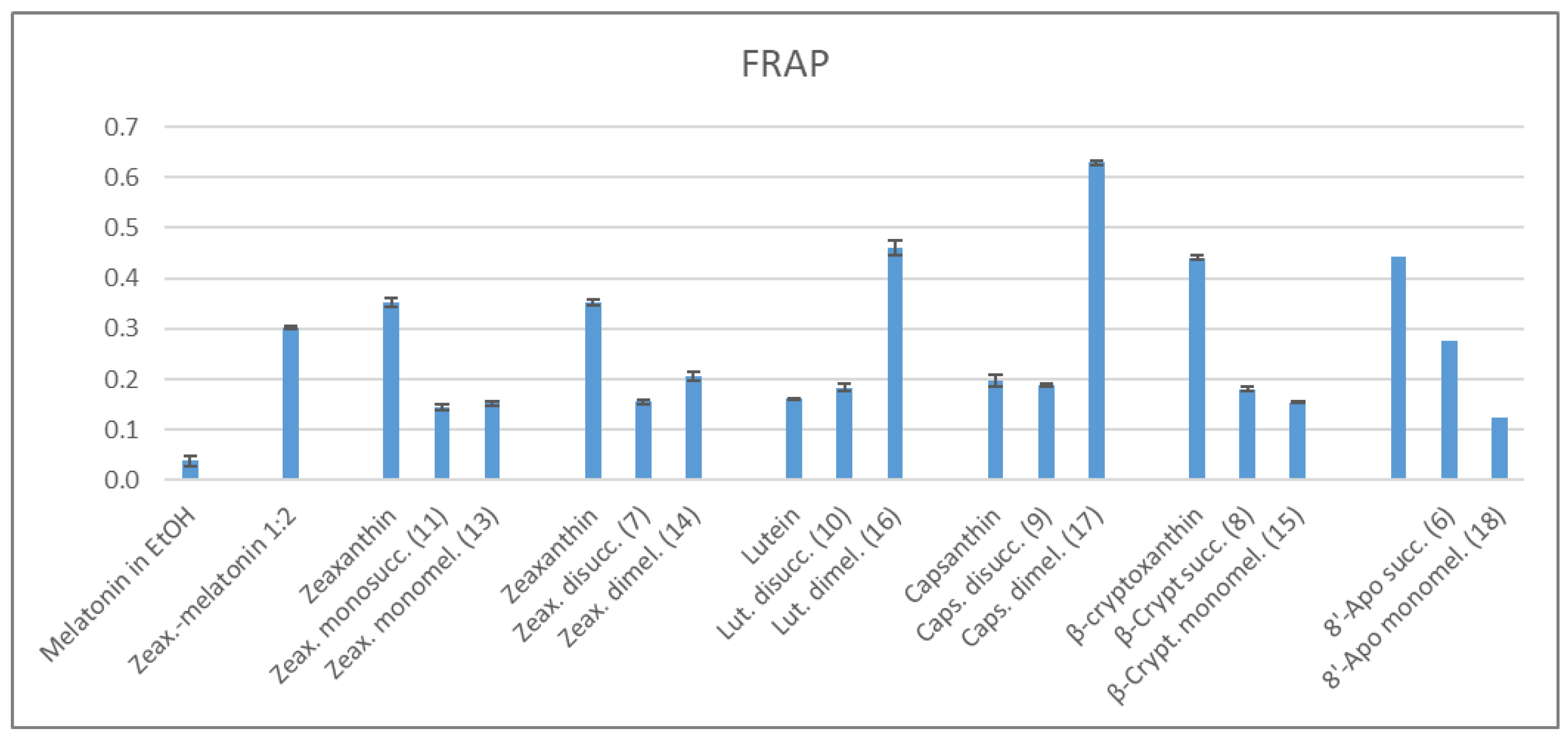
2.3. Antioxidant Properties
2.3.1. ABTS/TEAC Assays
2.3.2. FRAP Method
3. Materials and Methods
3.1. Determination of Kinetic Solubility
Determination of LOD, LOQ
3.2. BBB-Specific Permeability Assay
3.3. Determination of Water Dispersibility
3.4. Determination of Hydrodynamic Diameter and Colloidal Stability
3.5. Determination of Antioxidant Capacity
3.5.1. TEAC Assay Using ABTS Radical
3.5.2. FRAP Assay for the Antioxidant Capacity
3.5.3. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A. Experimental Section
References
- Rodriguez-Concepcion, M.; Avalos, J.; Bonet, M.L.; Boronat, A.; Gomez-Gomez, L.; Hornero-Mendez, D.; Limon, M.C.; Melendez-Martinez, A.J.; Olmedilla-Alonso, B.; Palou, A.; et al. A global perspective on carotenoids: Metabolism, biotechnology, and benefits for nutrition and health. Prog. Lipid Res. 2018, 70, 62–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.L.; Green, J.; Lewis, B. Lutein and zeaxanthin in eye and skin health. Clin. Dermatol. 2009, 27, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Hammond, B.R.; Miller, L.S.; Bello, M.O.; Lindbergh, C.A.; Mewborn, C.; Renzi-Hammond, L.M. Effects of Lutein/Zeaxanthin Supplementation on the Cognitive Function of Community Dwelling Older Adults: A Randomized, Double-Masked, Placebo-Controlled Trial. Front. Aging Neurosci. 2017, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Renzi-Hammond, L.M.; Bovier, E.R.; Fletcher, L.M.; Miller, L.S.; Mewborn, C.M.; Lindbergh, C.A.; Baxter, J.H.; Hammond, B.R. Effects of a Lutein and Zeaxanthin Intervention on Cognitive Function: A Randomized, Double-Masked, Placebo-Controlled Trial of Younger Healthy Adults. Nutrients 2017, 9, 1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, P.S.; Li, B.; Vachali, P.P.; Gorusupudi, A.; Shyam, R.; Henriksen, B.S.; Nolan, J.M. Lutein, zeaxanthin, and meso-zeaxanthin: The basic and clinical science underlying carotenoid-based nutritional interventions against ocular disease. Prog. Retin. Eye Res. 2016, 35, 34–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo, A.G.; Hu, S.Q.; Fernandez, M.L. Zeaxanthin: Metabolism, Properties, and Antioxidant Protection of Eyes, Heart, Liver, and Skin. Antioxidants 2019, 8, 390. [Google Scholar] [CrossRef] [Green Version]
- Murillo, E.; Nagy, V.; Agócs, A.; Deli, J. Carotenoids with κ-end group. In Carotenoids: Food Sources, Production and Health Benefits; Yamaguchi, M., Ed.; Nova Science Publishers Inc.: Hauppauge, NY, USA, 2013; Chapter 3; pp. 49–78. ISBN 978-1-62808-622-5. [Google Scholar]
- Burri, B.J.; La Frano, M.R.; Zhu, C. Absorption, metabolism, and functions of β-cryptoxanthin. Nutr. Rev. 2016, 74, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Stahl, W.; Ale-Agha, N.; Polidori, M.C. Non-Antioxidant Properties of Carotenoids. Biol. Chem. 2002, 383, 553–558. [Google Scholar] [CrossRef]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin and Related Compounds: Chemical Insights into their Protective Effects against Oxidative Stress. Curr. Org. Chem. 2017, 21, 2077–2095. [Google Scholar] [CrossRef]
- Favero, G.; Franceschetti, L.; Buffoli, B.; Moghadasian, M.H.; Reiter, R.J.; Rodella, L.F.; Rezzani, R. Melatonin: Protection against age-related cardiac pathology. Ageing Res. Rev. 2017, 35, 336–349. [Google Scholar] [CrossRef]
- Aranda, M.L.; Fleitas, M.F.G.; Dieguez, H.; Iaquinandi, A.; Sande, P.H.; Dorfman, D.; Rosenstein, R.E. Melatonin as a Therapeutic Resource for Inflammatory Visual Diseases. Curr. Neuropharmacol. 2017, 15, 951–962. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.F.; Xu, K. Melatonin, a full service anti-cancer agent: Inhibition of initiation, progression and metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef] [Green Version]
- Shneider, A.; Kudriavtsev, A.; Vakhrusheva, A. Can melatonin reduce the severity of COVID-19 pandemic? Int. Rev. Immunol. 2020, 39, 153–162. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, T.; Lee, T.H. Cellular Mechanisms of Melatonin: Insight from Neurodegenerative Diseases. Biomolecules 2020, 10, 1158. [Google Scholar] [CrossRef]
- Bedini, A.; Fraternale, A.; Crinelli, R.; Mari, M.; Bartolucci, S.; Chiarantini, L.; Spadoni, G. Design, Synthesis, and Biological Activity of Hydrogen Peroxide Responsive Arylboronate Melatonin Hybrids. Chem. Res. Toxicol. 2019, 32, 100–112. [Google Scholar] [CrossRef]
- Pachón-Angona, I.; Refouvelet, B.; Andrýs, R.; Martin, H.; Luzet, V.; Iriepa, I.; Moraleda, I.; Diez-Iriepa, D.; Oset-Gasque, M.-J.; Marco-Contelles, J.; et al. Donepezil + chromone + melatoninhybrids as promising agents for Alzheimer’s disease therapy. J. Enzym. Inhib. Med. Chem. 2019, 34, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Łozińska, I.; Świerczyńska, A.; Molęda, Z.; Hartman, A.M.; Hirsch, A.K.H.; Czarnocki, Z. Donepezil–melatonin hybrids as butyrylcholinesterase inhibitors: Improving binding affinity through varying mode of linking fragments. Arch. Pharm. 2018, 351, e1800194. [Google Scholar] [CrossRef]
- Gerenu, G.; Liu, K.; Chojnacki, J.E.; Saathoff, J.M.; Martínez-Martín, P.; Perry, G.; Zhu, X.; Lee, H.; Zhang, S. Curcumin/Melatonin Hybrid 5-(4-Hydroxy-phenyl)-3-oxo-pentanoic Acid [2-(5-Methoxy-1H-indol-3-yl)-ethyl]-amide Ameliorates AD-Like Pathology in the APP/PS1 Mouse Model. ACS Chem. Neurosci. 2015, 6, 1393–1399. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J. Pineal Melatonin—Cell Biology of Its Synthesis and of Its Physiological Interactions. Endocr. Rev. 1991, 12, 151–180. [Google Scholar] [CrossRef] [Green Version]
- Shida, C.S.; Castrucci, A.M.L.; Lamy-Freund, M.T. High Melatonin Solubility in Aqueous-Medium. J. Pineal Res. 1994, 16, 198–201. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Cabrera, J.; D’Arpa, D.; Sainz, R.M.; Mayo, J.C.; Ramos, S. The oxidant/antioxidant network: Role of melatonin. Neurosignals 1999, 8, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Lebars, D.; Thivolle, P.; Vitte, P.A.; Bojkowski, C.; Chazot, G.; Arendt, J.; Frackowiak, R.S.J.; Claustrat, B. Pet and Plasma Pharmacokinetic Studies after Bolus Intravenous Administration of [C-11] Melatonin in Humans. Int. J. Radiat. Appl. Instrum. Part B Nucl. Med. Biol. 1991, 18, 357–362. [Google Scholar]
- Hada, M.; Nagy, V.; Takatsy, A.; Deli, J.; Agocs, A. Dicarotenoid esters of bivalent acids. Tetrahedron Lett. 2008, 49, 3524–3526. [Google Scholar] [CrossRef]
- Hada, M.; Nagy, V.; Gulyas-Fekete, G.; Deli, J.; Agocs, A. Towards Carotenoid Dendrimers: Carotenoid Diesters and Triesters with Aromatic Cores. Helv. Chim. Acta 2010, 93, 1149–1155. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, L.; Guo, Y.; Zhang, T.; Qiao, X.; Wang, J.; Xu, J.; Xue, C. Hydrophilic Astaxanthin: PEGylated Astaxanthin Fights Diabetes by Enhancing the Solubility and Oral Absorbability. J. Agric. Food Chem. 2020, 68, 3649–3655. [Google Scholar] [CrossRef]
- Frey, D.A.; Kataisto, E.W.; Ekmanis, J.L.; O’Malley, S.; Lockwood, S.F. The efficient synthesis of disodium disuccinate astaxanthin (Cardax). Org. Process Res. Dev. 2004, 8, 796–801. [Google Scholar] [CrossRef]
- Gloor, A.; Simon, W. Salts of Astaxanthin Esters. U.S. Patent 2008/0008798 Al, 10 January 2008. [Google Scholar]
- Ester von Karotinoiden zum Gebrauch bei der Verhütung und Behandlung von Augenkrankheiten. DE 19950327 A1, 20 April 2000.
- Lockwood, S.F.; Penn, M.S.; Hazen, S.L.; Bikadi, Z.; Zsila, F. The effects of oral CardaxTM (disodium disuccinate astaxanthin) on multiple independent oxidative stress markers in a mouse peritoneal inflammation model: Influence on 5-lipoxygenase in vitro and in vivo. Life Sci. 2006, 79, 162–174. [Google Scholar] [CrossRef]
- Cardounel, A.J.; Dumitrescu, C.; Zweier, J.L.; Lockwood, S.F. Direct superoxide anion scavenging by a disodium disuccinate astaxanthin derivative: Relative efficacy of individual stereoisomers versus the statistical mixture of stereoisomers by electron paramagnetic resonance imaging. Biochem. Biophys. Res. Comm. 2003, 307, 704–712. [Google Scholar] [CrossRef]
- Hada, M.; Petrovics, D.; Nagy, V.; Boddi, K.; Deli, J.; Agocs, A. The first synthesis of PEG-carotenoid conjugates. Tetrahedron Lett. 2011, 52, 3195–3197. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- ACD/Labs Percepta, Version: v2020 Build 3382. Available online: https://www.acdlabs.com/products/percepta/ (accessed on 18 June 2020).
- Lanevskij, K.; Didziapetris, R. Physicochemical QSAR Analysis of Passive Permeability Across Caco-2 Monolayers. J. Pharm. Sci. 2019, 108, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Lanevskij, K.; Dapkunas, J.; Juska, L.; Japertas, P.; Didziapetris, R. QSAR analysis of blood-brain distribution: The influence of plasma and brain tissue binding. J. Pharm. Sci. 2011, 100, 2147–2160. [Google Scholar] [CrossRef]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Esső, K.; Dargó, G.; Könczöl, Á.; Balogh, G.T. Tuning the predictive capacity of the PAMPA-BBB model. Eur. J. Pharm. Sci. 2015, 79, 53–60. [Google Scholar] [CrossRef]
- Köhn, S.; Kolbe, H.; Korger, M.; Köpsel, C.; Mayer, B.; Auweter, H.; Lüddecke, E.; Bettermann, H.; Martin, H.-D. Chapter 5: Aggregation and interface behaviour of carotenoids. In Carotenoids, Vol. 4: Natural Functions; Britton, G., Liaaen-Jensen, S., Pfander, H., Eds.; Birkhäuser Verlag: Basel, Switzerland, 2008; Volume 4, pp. 53–98. [Google Scholar]
- Simonyi, M.; Bikadi, Z.; Zsila, F.; Deli, J. Supramolecular exciton chirality of carotenoid aggregates. Chirality 2003, 15, 680–698. [Google Scholar] [CrossRef]
- Ruban, A.V.; Horton, P.; Young, A.J. Aggregation of higher plant xanthophylls: Differences in absorption spectra and in the dependency on solvent polarity. J. Photochem. Photobiol. B Biol. 1993, 21, 229–234. [Google Scholar] [CrossRef]
- Tay-Agbozo, S.; Street, S.; Kispert, L.D. The carotenoid bixin: Optical studies of aggregation in polar/water solvents. J. Photochem. Photobiol. A Chem. 2018, 362, 31–39. [Google Scholar] [CrossRef]
- Línzembold, I.; Czett, D.; Böddi, K.; Kurtán, T.; Király, B.S.; Gulyás-Fekete, G.; Takátsy, A.; Lóránd, T.; Deli, J.; Agócs, A.; et al. Study on the Synthesis, Antioxidant Properties, and Self-Assembly of Carotenoid–Flavonoid Conjugates. Molecules 2020, 25, 636. [Google Scholar] [CrossRef] [Green Version]
- van den Berg, R.; Haenen, G.R.M.M.; van den Berg, H.; Bast, A. Applicability of an improved Trolox equivalent antioxidant capacity (TEAC) assay for evaluation of antioxidant capacity measurements of mixtures. Food Chem. 1999, 66, 511–517. [Google Scholar] [CrossRef]
- Nagane, M.; Yamashita, T.; Vörös, P.; Kálai, T.; Hideg, K.; and Bognár, B. Synthesis and evaluation of paramagnetic caffeic acid phenethyl ester (CAPE) analogs. Mon. Chem. Chem. Mon. 2019, 150, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Benzie, I.F.F.; Strain, J.J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of ‘‘Antioxidant Power’’: The FRAP Assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliwka, H.R.; Melø, T.-B.; Foss, B.J.; Abdel-Hafez, S.H.; Partali, V.; Nadolski, G.; Jackson, H.; Lockwood, S.F. Electron- and Energy-Transfer Properties of Hydrophilic Carotenoids. Chem. Eur. J. 2007, 13, 4458–4466. [Google Scholar] [CrossRef] [PubMed]
- Deli, J.; Molnár, P.; Matus, Z.; Tóth, G. Carotenoid Composition in the Fruits of Red Paprika (Capsicum annuum var. Lycopersiciforme rubrum) during Ripening; Biosynthesis of Carotenoids in Red Paprika J. Agric. Food Chem. 2001, 49, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Agócs, A.; Bokor, E.; Takátsy, A.; Lórand, T.; Deli, J.; Somsák, L.; Nagy, V. Synthesis of carotenoid-monosaccharide conjugates via azide-alkyne click-reaction. Tetrahedron 2017, 73, 519–526. [Google Scholar] [CrossRef]
- Avdeef, A. Permeability Equations. In Absorption and Drug Development: Solubility, Permeability, and Charge State; Avdeef, A., Ed.; Wiley Interscience: Hoboken, NJ, USA, 2012; pp. 465–481. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
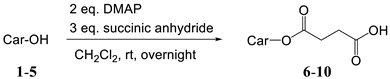 | |
|---|---|
| Carotenoid (Car-OH) | Product (Yield) |
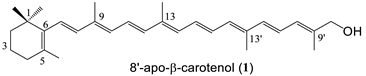 | 6 succinate (92%) 1 |
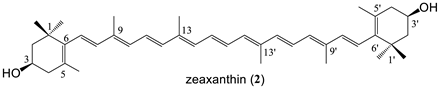 | 7 bissuccinate (82%) |
 | 8 succinate (78%) |
 | 9 bissuccinate (75%) |
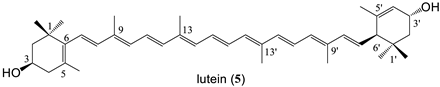 | 10 bissuccinate (79%) |
| Predicted Parameters | Experimental Data | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Mw | Strongest pKa,acid a | logP/logD7.4 a | HBD/ HBA | TPSA Å2 | Aq.Sol. b mg/mL | Caco-2 Permeability Pe 10−6 cm/s [35] | logBB [36] | Kin.Sol. c μM | BBB-PAMPA Pe 10−6 cm/s |
| 1 | 417 | - | 8.6 | 1/1 | 17.1 | 5 × 10−5 | 3.2 | 0.03 | ND | ND |
| 2 | 569 | - | 10.8 | 2/2 | 40.5 | 7 × 10−5 | 0.2 | −0.96 | ND | ND |
| 3 | 553 | - | 12.1 | 1/1 | 20.2 | 3 × 10−5 | 0.1 | −2 | ND | ND |
| 4 | 585 | - | 9.3 | 2/3 | 57.5 | 2 × 10−5 | 0.3 | −0.58 | ND | ND |
| 5 | 569 | - | 10.4 | 2/2 | 40.5 | 1 × 10−4 | 0.2 | −0.66 | ND | ND |
| 6 | 519 | 4.4 | 9.3/6.4 | 1/3 | 63.6 | 2 × 10−2 | 5.8 | −1.08 | 52.3 ± 2.4 | ND |
| 7 | 769 | 4.2 | 11.4/6.7 | 2/7 | 127.2 | 4 × 10−1 | 2.2 | −2 | 0.6 ± 0.1 | ND |
| 8 | 653 | 4.4 | 12.5/9.6 | 1/3 | 63.6 | 2 × 10−3 | 0.3 | −2 | 1.3 ± 0.1 | ND |
| 9 | 785 | 4.4 | 10.1/5.3 | 2/8 | 144.3 | 5 × 10−2 | 6.7 | −2 | ND | ND |
| 10 | 769 | 4.2 | 11.2/6.4 | 2/7 | 127.2 | 2 × 10−1 | 2.9 | −2 | ND | ND |
| 11 | 669 | 4.4 | 11.0/8.1 | 2/4 | 83.8 | 4 × 10−3 | 0.4 | −2 | ND | ND |
| 13 | 898 | - | 12.2 | 4/5 | 129.8 | 2 × 10−7 | 0.1 | −2 | ND | ND |
| 14 | 1228 | - | 13.5 | 6/8 | 219.0 | 3 × 10−10 | 0.1 | −2 | ND | ND |
| 15 | 882 | - | 13.7 | 3/4 | 109.5 | 6 × 10−8 | 0.1 | −2 | ND | ND |
| 16 | 1228 | - | 13.1 | 6/8 | 219.0 | 4 × 10−10 | 0.1 | −2 | ND | ND |
| 17 | 1244 | - | 11.8 | 6/9 | 231.1 | 8 × 10−11 | 0.1 | −2 | ND | ND |
| 18 | 748 | - | 10.2 | 3/4 | 109.5 | 1 × 10−5 | 0.1 | −1 | ND | ND |
| Melatonin | 232 | - | 1.7 | 2/2 | 54.1 | 9 × 10−1 | 58.2 | −0.22 | 123.2 ± 9.4 | 2.97 ± 1.18 |
| Succinate | Water Dispersibility (mg/100 mL) |
|---|---|
| 8′-apo-β-carotenol succinate (6) | 1.5 |
| zeaxanthin bissuccinate (7) | 1.3 |
| β-cryptoxanthin succinate (8) | less than 0.5 |
| capsanthin bissuccinate (9) | less than 0.6 |
| lutein bissuccinate (10) | 1.5 |
| zeaxanthin monosuccinate (11) | less than 0.3 |
| underivatized carotenoids (1–5) | 0.05–0.1 |
| Compound | λmax (nm) | Slope of Fitted Calibration Curve (s) | R2 of Fitted Calibration Curve | LOD (μM) | LOQ (μM) |
|---|---|---|---|---|---|
| 1 | 467 | 0.0119 | 0.999 | 2.20 | 6.67 |
| 2 | 467 | 0.0009 | 0.995 | 29.08 | 88.14 |
| 3 | 457 | 0.0013 | 0.999 | 20.14 | 61.02 |
| 4 | 472 | 0.0256 | 0.991 | 1.02 | 3.10 |
| 5 | 452 | 0.0084 | 0.995 | 3.12 | 9.44 |
| 6 | 459 | 0.0252 | 0.998 | 1.04 | 3.15 |
| 7 | 456 | 0.0246 | 0.993 | 1.06 | 3.22 |
| 8 | 483 | 0.0190 | 0.985 | 1.38 | 4.17 |
| 9 | 459 | 0.0124 | 0.998 | 2.11 | 6.40 |
| 10 | 452 | 0.0347 | 0.999 | 0.75 | 2.29 |
| 11 | 434 | 0.0267 | 0.999 | 0.98 | 2.97 |
| 12 | 280 | 0.0021 | 0.999 | 12.46 | 37.77 |
| 13 | 458 | 0.0258 | 0.996 | 1.01 | 3.07 |
| 14 | 457 | 0.0380 | 0.999 | 0.69 | 2.09 |
| 15 | 452 | 0.0085 | 0.987 | 3.08 | 9.33 |
| 16 | 452 | 0.0282 | 0.999 | 0.93 | 2.81 |
| 17 | 478 | 0.0126 | 0.999 | 2.08 | 6.30 |
| 18 | 431 | 0.0015 | 0.998 | 17.45 | 52.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czett, D.; Böddi, K.; Nagy, V.; Takátsy, A.; Deli, J.; Tone, P.; Balogh, G.T.; Vincze, A.; Agócs, A. Synthesis, Pharmacokinetic Characterization and Antioxidant Capacity of Carotenoid Succinates and Their Melatonin Conjugates. Molecules 2022, 27, 4822. https://doi.org/10.3390/molecules27154822
Czett D, Böddi K, Nagy V, Takátsy A, Deli J, Tone P, Balogh GT, Vincze A, Agócs A. Synthesis, Pharmacokinetic Characterization and Antioxidant Capacity of Carotenoid Succinates and Their Melatonin Conjugates. Molecules. 2022; 27(15):4822. https://doi.org/10.3390/molecules27154822
Chicago/Turabian StyleCzett, Dalma, Katalin Böddi, Veronika Nagy, Anikó Takátsy, József Deli, Paul Tone, György T. Balogh, Anna Vincze, and Attila Agócs. 2022. "Synthesis, Pharmacokinetic Characterization and Antioxidant Capacity of Carotenoid Succinates and Their Melatonin Conjugates" Molecules 27, no. 15: 4822. https://doi.org/10.3390/molecules27154822
APA StyleCzett, D., Böddi, K., Nagy, V., Takátsy, A., Deli, J., Tone, P., Balogh, G. T., Vincze, A., & Agócs, A. (2022). Synthesis, Pharmacokinetic Characterization and Antioxidant Capacity of Carotenoid Succinates and Their Melatonin Conjugates. Molecules, 27(15), 4822. https://doi.org/10.3390/molecules27154822