
Regioselective Synthesis of 5- and 3-Hydroxy-N-Aryl-1H-Pyrazole-4-Carboxylates and Their Evaluation as Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase


 , , ,
, , ,  , and
, and
Abstract
:
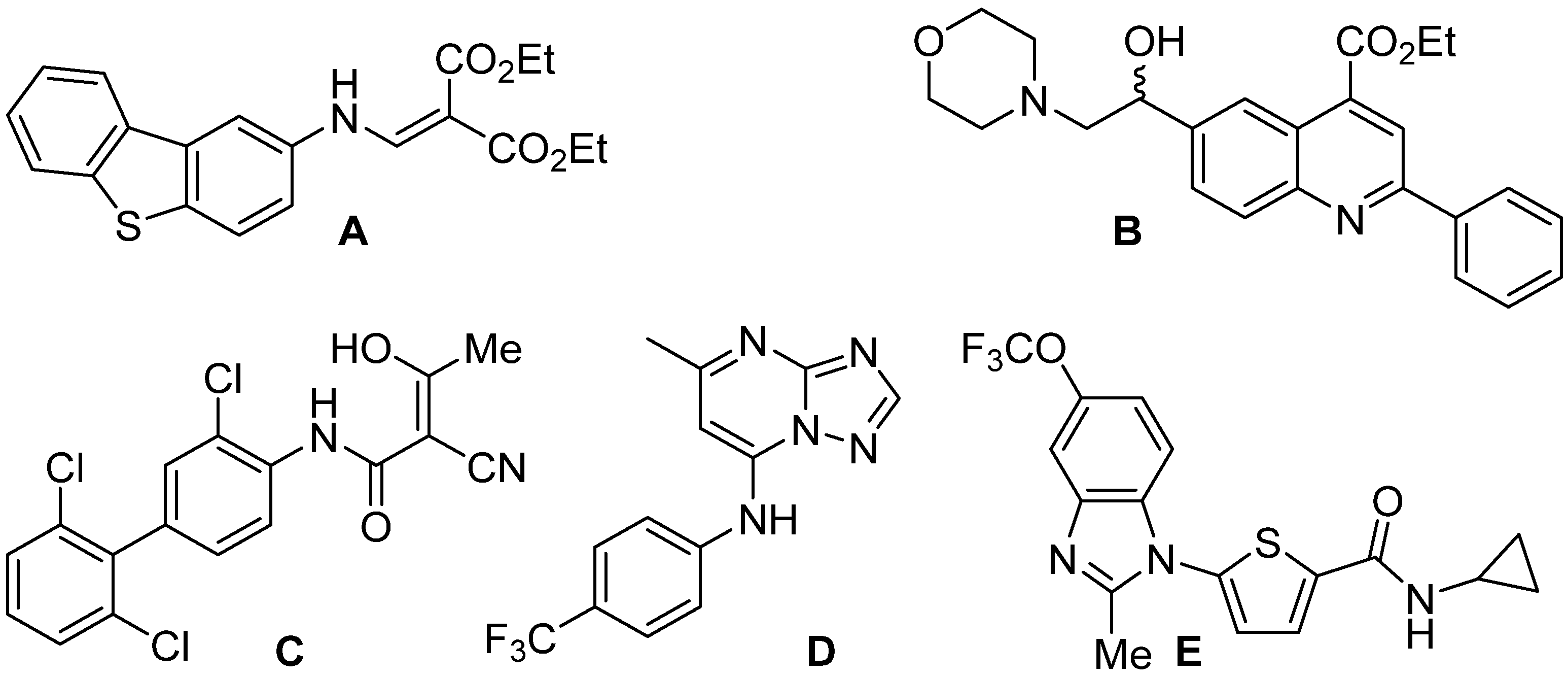
1. Introduction
2. Results and Discussion
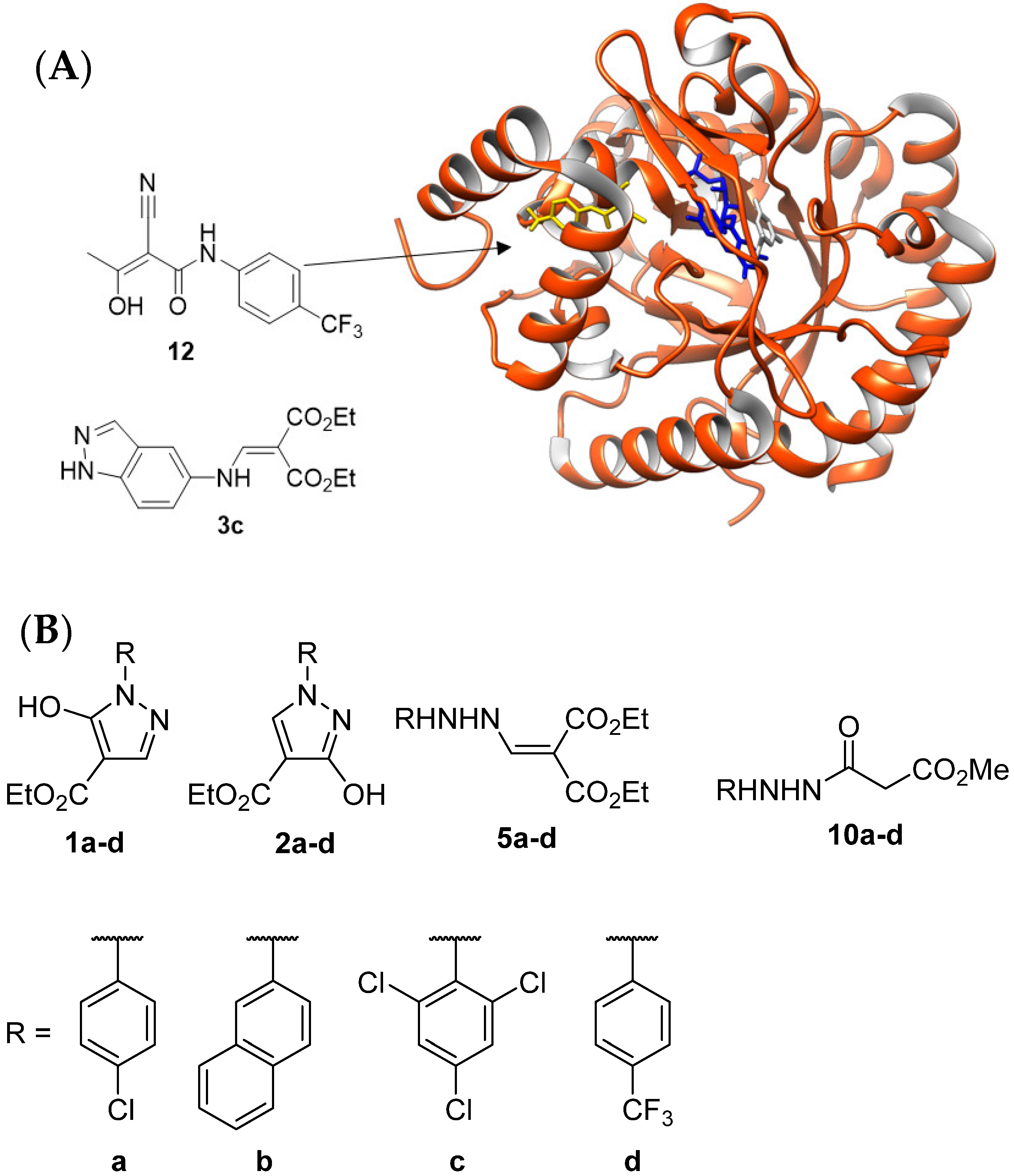
2.1. In Silico Evaluation of Compounds 3a, 1a–d, 2a–d, 5a–d, and 10a–d
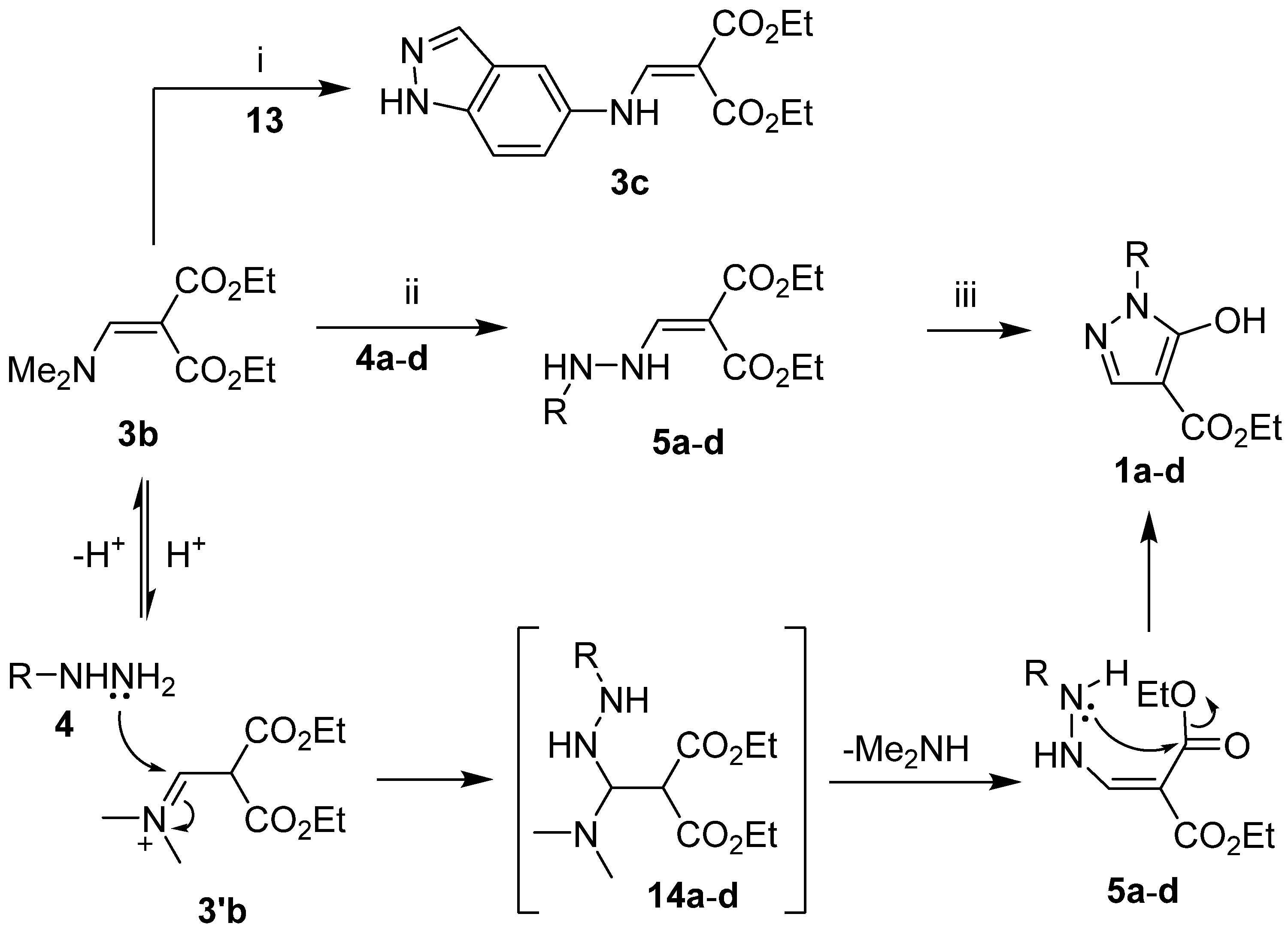
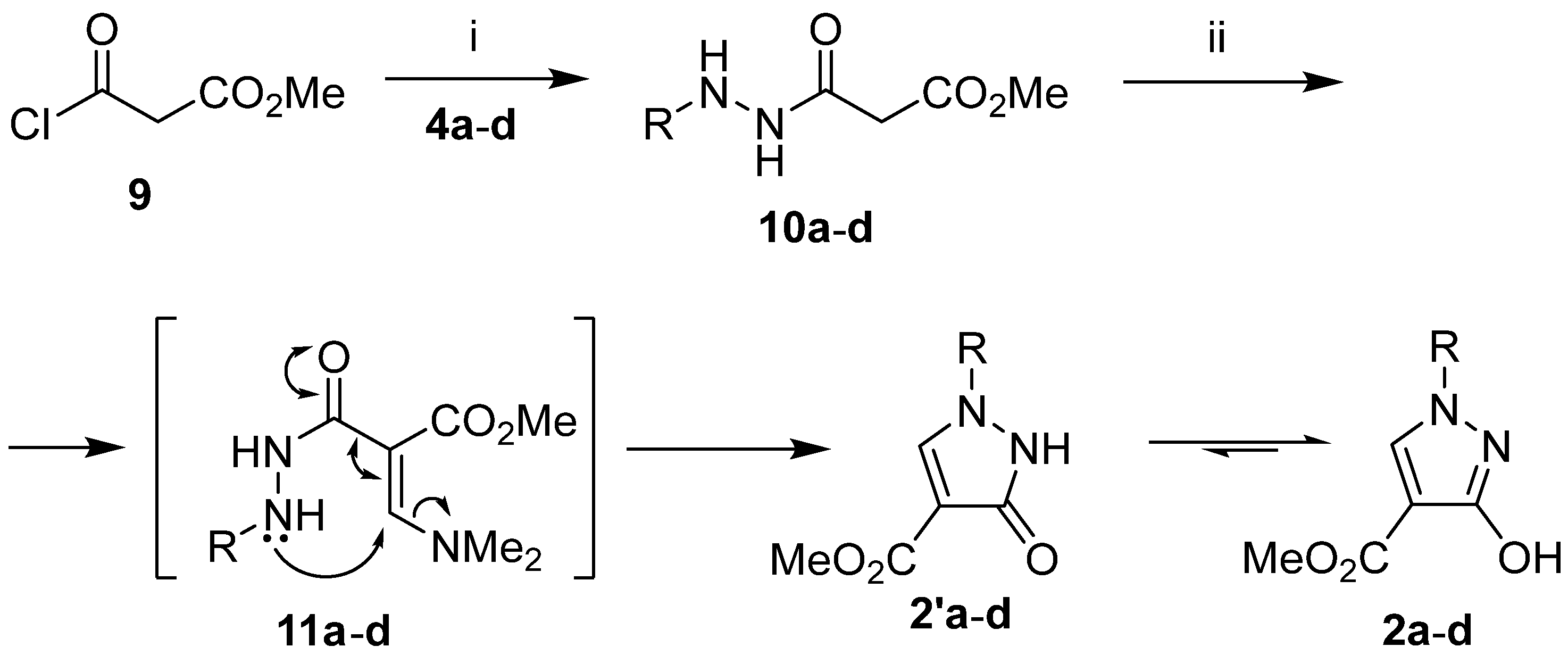
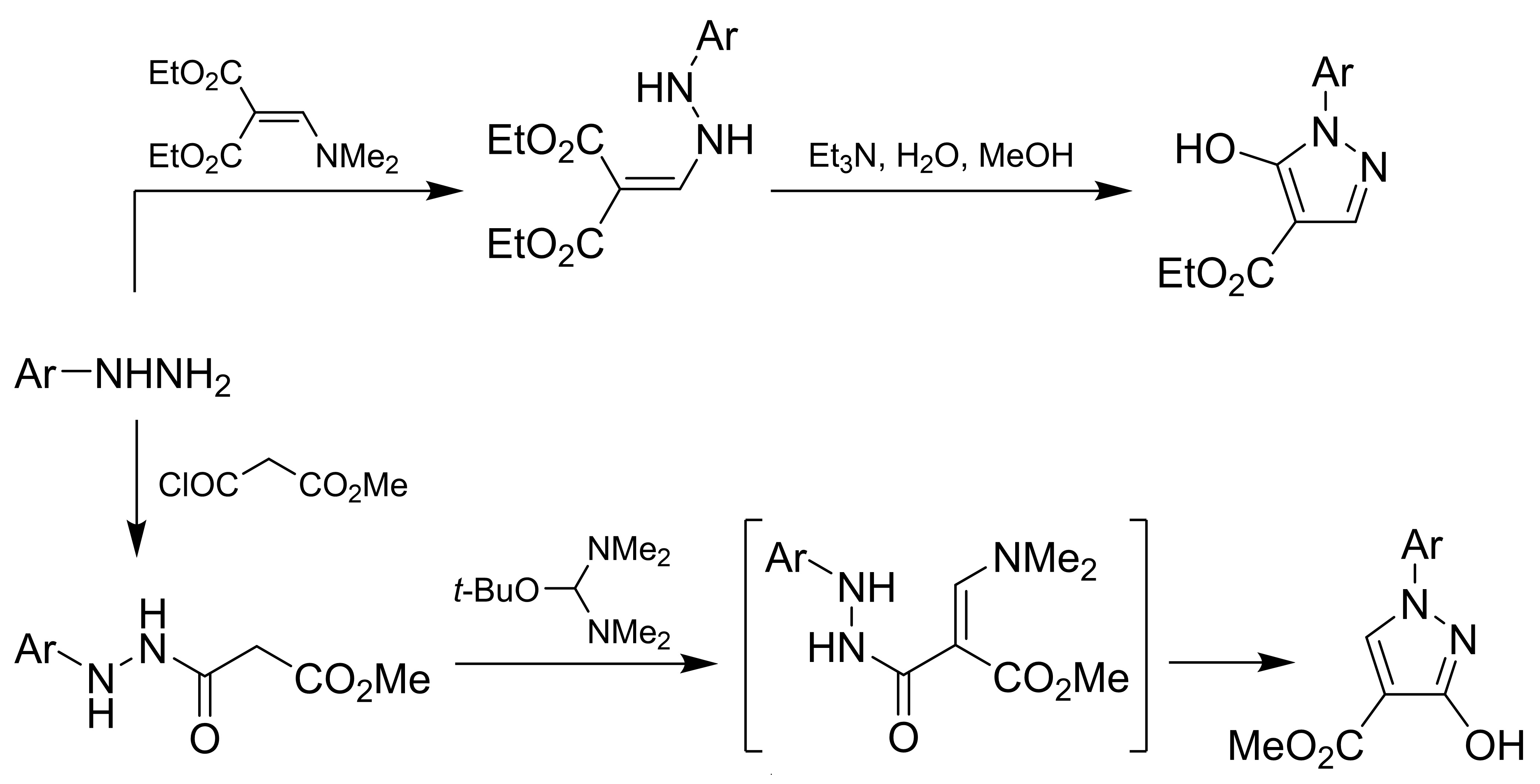
2.2. Synthesis of Compounds 1a–d, 2a–d, 3c, 5a–d, and 10a–d
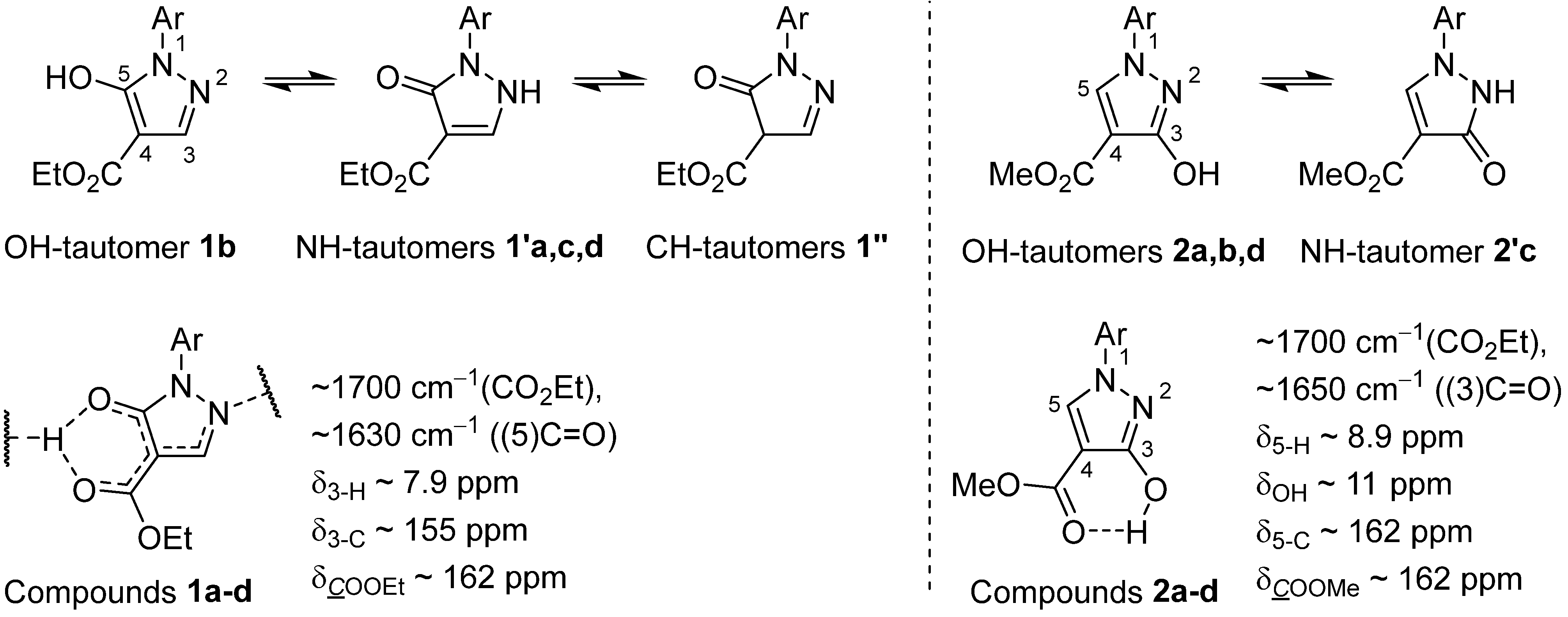
2.3. Structure Determination
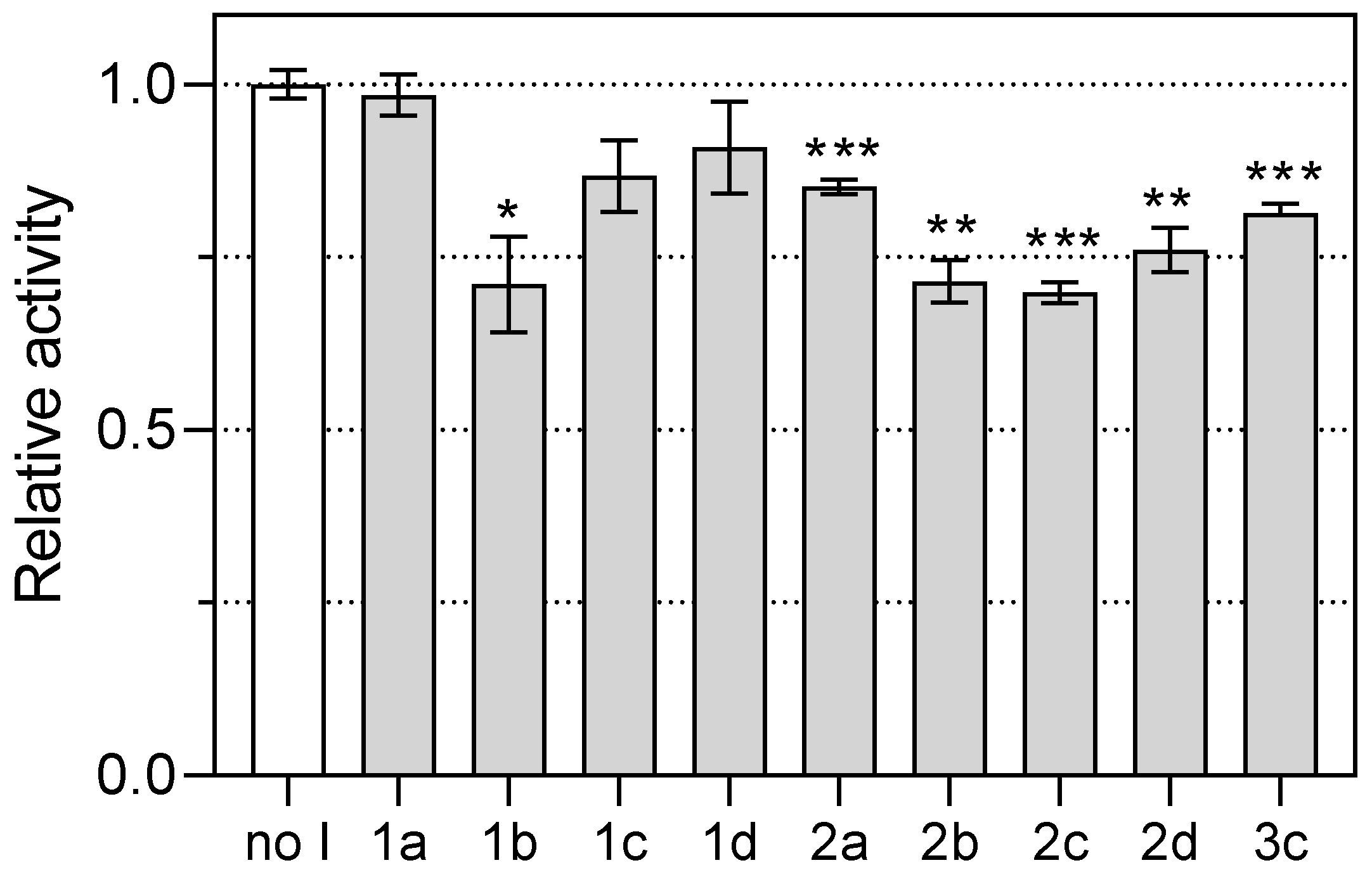
3. Inhibition of PfDHODH
4. Experimental Section
4.1. General Methods
4.2. Biochemistry
4.2.1. Expression and Purification of Recombinant DHODH
4.2.2. Enzyme Assays
4.3. General Procedure for the Synthesis of Enamine 3c and Enhydrazines 5a–d
4.3.1. Diethyl 2-{[(1H-Indazol-5-yl)amino]methylene}malonate (3c)
4.3.2. Diethyl 2-{[2-(4-Chlorophenyl)hydrazinyl]methylene}malonate (5a)
4.3.3. Diethyl 2-{[2-(Naphthalene-2-yl)hydrazinyl]methylene}malonate (5b)
4.3.4. Diethyl 2-{[2-(2,4,6-Trichlorophenyl)hydrazinyl]methylene}malonate (5c)
4.3.5. Diethyl 2-{[2-(4-Trifluoromethylphenyl)hydrazinyl]methylene}malonate (5d)
4.4. General Procedure for the Synthesis of 1-Aryl-5-hydroxy-1H-pyrazole-4-carboxylates 1a–d
4.4.1. Ethyl 1-(4-Chlorophenyl)-5-hydroxy-1H-pyrazole-4-carboxylate (1a)
4.4.2. Ethyl 1-(Naphthalen-2-yl)-5-hydroxy-1H-pyrazole-4-carboxylate (1b)
4.4.3. Ethyl 1-(2,4,6-Trichlorophenyl)-5-hydroxy-1H-pyrazole-4-carboxylate (1c)
4.4.4. Ethyl 1-(4-Trifluoromethylphenyl)-5-hydroxy-1H-pyrazole-4-carboxylate (1d)
4.5. General Procedure for the Synthesis of Hydrazides 10a–d
4.5.1. Methyl 3-[2-(4-Chlorophenyl)hydrazinyl]-3-oxopropanoate (10a)
4.5.2. Methyl 3-[2-(Naphthalene-2-yl)hydrazinyl]-3-oxopropanoate (10b)
4.5.3. Methyl 3-[2-(2,4,6-Trichlorophenyl)hydrazinyl]-3-oxopropanoate (10c)
4.5.4. Methyl 3-[2-(4-Trifluoromethylphenyl)hydrazinyl]-3-oxopropanoate (10d)
4.6. General Procedure for the Synthesis of Methyl 1-Aryl-3-hydroxy-1H-pyrazole-4-carboxylates 2a–d
4.6.1. Methyl 1-(4-Chlorophenyl)-3-hydroxy-1H-pyrazole-4-carboxylate (2a)
4.6.2. Methyl 1-(Naphthalen-2-yl)-3-hydroxy-1H-pyrazole-4-carboxylate (2b)
4.6.3. Methyl 1-(2,4,6-Trichlorophenyl)-3-hydroxy-1H-pyrazole-4-carboxylate (2c)
4.6.4. Methyl 1-(4-Trifluoromethylphenyl)-3-hydroxy-1H-pyrazole-4-carboxylate (2d)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Morag, D. History of malaria and its treatment. In Antimalarial Agents Design and Mechanism of Action, 1st ed.; Patrick, G.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–48. ISBN 9780081012109. [Google Scholar]
- Patrick, G.L. (Ed.) Antimalarial Agents Design and Mechanism of Action, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–604. ISBN 9780081012109. [Google Scholar]
- Fidock, D.A.; Rosenthal, P.J.; Croft, S.L.; Brun, R.; Nwaka, S. Antimalarial drug discovery: Efficacy models for compound screening. Nat. Rev. Drug Disc. 2004, 3, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Su, X.Z. Discovery, mechanisms of action and combination therapy of artemisinin. Expert Rev. Anti-Infect. Ther. 2009, 7, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Pohlit, A.M.; Lima, R.B.; Frausin, G.; Silva, L.F.; Lopes, S.C.; Moraes, C.B.; Cravo, P.; Lacerda, M.V.; Siqueira, A.M.; Freitas-Junior, L.H.; et al. Amazonian Plant Natural Products: Perspectives for Discovery of New Antimalarial Drug Leads. Molecules 2013, 18, 9219–9240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, L.; Brown, G.V.; Genton, B.; Moorthy, V.S. A review of malaria vaccine clinical projects based on the WHO rainbow table. Malar. J. 2012, 11, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, T.; Dao, A.; Yaro, A.S.; Huestis, D.L.; Diallo, M.; Timbiné, S.; Kassogué, Y.; Adamou, A.; Traoré, A.I.; Samaké, D. Phenotypic divergence among the members of the African malaria mosquitoes and strategies of persistence throughout the dry season. In Challenges in Malaria Research: Core Science and Innovation. Malar. J. 2014, 13 (Suppl. S1), O2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnell, E.S. Drugs targeting mitochondrial functions. In Antimalarial Agents Design and Mechanism of Action, 1st ed.; Patrick, G.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 375–402. ISBN 9780081012109. [Google Scholar]
- Vyas, V.K.; Ghate, M. Recent Developments in the Medicinal Chemistry and Therapeutic Potential of Dihydroorotate Dehydrogenase (DHODH) Inhibitors. Mini-Rev. Med. Chem. 2011, 11, 1039–1055. [Google Scholar] [CrossRef]
- Nixon, G.L.; Pidathala, C.; Shone, A.E.; Antoine, T.; Fisher, N.; O’Neill, P.M.; Ward, S.A.; Biagini, G.A. Targeting the mitochondrial electron transport chain of Plasmodium falciparum: New strategies towards the development of improved antimalarials for the elimination era. Future Med. Chem. 2013, 5, 1573–1591. [Google Scholar] [CrossRef]
- Hoelz, L.V.B.; Calil, F.A.; Nonato, M.C.; Pinheiro, L.C.S.; Boechat, N. Plasmodium falciparum dihydroorotate dehydrogenase: A drug target against malaria. Future Med. Chem. 2018, 10, 1853–1874. [Google Scholar] [CrossRef]
- Heikkilä, T.; Ramsey, C.; Davies, M.; Galtier, C.; Stead, A.M.W.; Johnson, A.P.; Fishwick, C.W.G.; Boa, A.N.; McConke, G.A. Design and Synthesis of Potent Inhibitors of the Malaria Parasite Dihydroorotate Dehydrogenase. J. Med. Chem. 2007, 50, 186–191. [Google Scholar] [CrossRef]
- Boa, A.N.; Canavan, S.P.; Hirst, P.R.; Ramsey, C.; Stead, A.M.W.; McConkey, G.A. Synthesis of brequinar analogue inhibitors of malaria parasite dihydroorotate dehydrogenase. Bioorg. Med. Chem. 2005, 13, 1945–1967. [Google Scholar] [CrossRef]
- Davies, M.; Heikka, T.; McConkey, G.A.; Fishwick, C.W.G.; Parsons, M.R.; Johnson, A.P. Structure-based design, synthesis and characterization of inhibitors of human and Plasmodium falciparum dihydroorotate dehydrogenase. J. Med. Chem. 2009, 52, 2683–2693. [Google Scholar] [CrossRef]
- Gujjar, R.; Marwaha, A.; Mazouni, F.E.; White, J.; White, K.; Creason, S.; Shackleford, D.; Baldwin, J.; Charman, W.N.; Buckner, F.S.; et al. Identification of a Metabolically Stable Triazolopyrimidine-Based Dihydroorotate Dehydrogenase Inhibitor with Antimalarial Activity in Mice. J. Med. Chem. 2009, 52, 1864–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skerlj, R.T.; Bastos, C.M.; Booker, M.L.; Kramer, M.L.; Barker, R.H.; Celatka, C.A.; O’Shea, T.J.; Munoz, B.; Sidhu, A.B.; Coretes, J.F.; et al. Optimization of Potent Inhibitors of P. falciparum Dihydroorotate Dehydrogenase for the Treatment of Malaria. ACS Med. Chem. Lett. 2011, 2, 708–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry, 5th ed.; Wiley-Blackwell: New York, NY, USA, 2010; pp. 1–718. ISBN 978-1-405-13300-5. [Google Scholar]
- Patrick, G.L. An Introduction to Medicinal Chemistry, 5th ed.; Oxford University Press: Oxford, UK, 2013; pp. 1–816. ISBN 9780199697397. [Google Scholar]
- Pernerstorfer, J. Molecular Design and Combinatorial Compound Libraries. In Handbook of Combinatorial Chemistry. Drugs, Catalysts, Materials; Nicolaou, K.C., Hanko, R., Hartwig, W., Eds.; Wiley-VCH: Weinheim, Germany, 2002; Volume 2, pp. 725–742. ISBN 0-471-49726-6. [Google Scholar]
- Petek, N.; Štefane, B.; Novinec, M.; Svete, J. Synthesis and biological evaluation of 7-(aminoalkyl)pyrazolo [1,5-a]pyrimidine derivatives as cathepsin K inhibitors. Bioorg. Chem. 2019, 84, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Prasher, P. An epigrammatic status of the ‘azole’-based antimalarial drugs. RSC Med. Chem. 2020, 11, 184–211. [Google Scholar] [CrossRef]
- Shamsuddin, M.A.; Ali, A.H.; Zakaria, N.H.; Mohammat, M.F.; Hamzah, A.S.; Shaameri, Z.; Lam, K.W.; Mark-Lee, W.F.; Agustar, H.K.; Razak, M.R.H.A.; et al. Synthesis, Molecular Docking, and Antimalarial Activity of Hybrid 4-Aminoquinoline-pyrano[2,3-c]pyrazole Derivatives. Pharmaceuticals 2021, 14, 1174. [Google Scholar] [CrossRef]
- Kumar, G.; Tanwar, O.; Kumar, J.; Akhter, M.; Sharma, S.; Pillai, C.R.; Alam, M.M.; Zama, M.S. Pyrazole-pyrazoline as promising novel antimalarial agents: A mechanistic study. Eur. J. Med. Chem. 2018, 149, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Bekhit, A.A.; Nasralla, S.N.; Bekhit, S.A.; Bekhit, A.E.-D.A. Novel Dual Acting Antimalarial Antileishmanial Agents Derived from Pyrazole Moiety. Biointerface Res. Appl. Chem. 2022, 12, 6225–6233. [Google Scholar] [CrossRef]
- Aggarwal, S.; Paliwal, D.; Kaushik, D.; Kumar Gupta, G.; Kumar, A. Pyrazole Schiff Base Hybrids as Anti-Malarial Agents: Synthesis, In Vitro Screening and Computational Study. Comb. Chem. High Throughput Screen. 2018, 21, 194–203. [Google Scholar] [CrossRef]
- Stanovnik, B.; Svete, J. Pyrazoles. In Science of Synthesis, Houben-Weyl Methods of Molecular Transformations; Neier, R., Bellus, D., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 2002; Volume 12, pp. 15–225. ISBN 3-13-112271-4. [Google Scholar]
- Taylor, A.W.; Cook, R.T. A direct preparation of 2-aryl-4-ethoxycarbonyl-3-pyrazolin-5-ones from aryl hydrazines. Tetrahedron 1987, 43, 607–616. [Google Scholar] [CrossRef]
- Kralj, D.; Mecinović, J.; Bevk, D.; Grošelj, U.; Stanovnik, B.; Svete, J. 3-(Dimethylamino)propenoate-based regioselective synthesis of 1,4-disubstituted 5-hydroxy-1H-pyrazoles. Heterocycles 2006, 68, 897–914. [Google Scholar] [CrossRef]
- Michaelis, A.; Remy, E. Über die Darstellung des 1-Phenyl-3-pyrazolons. Chem. Ber. 1907, 40, 1020–1021. [Google Scholar] [CrossRef]
- Downloaded from RSCB PDB Protein Data Bank. Available online: https://www.rcsb.org/structure/1TV5 (accessed on 27 April 2021).
- Hurt, D.E.; Widom, J.; Clardy, J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Cryst. 2006, D62, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswandhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Nadin, A.; Hattotuwagama, C.; Churcher, I. Lead-oriented synthesis: A new opportunity for synthetic chemistry. Angew. Chem. Int. Ed. 2012, 51, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Hardegger, L.A.; Kuhn, B.; Spinnler, B.; Anselm, L.; Ecabert, R.; Stihle, M.; Gsell, B.; Thoma, R.; Diez, J.; Benz, J.; et al. Systematic Investigation of Halogen Bonding in Protein–Ligand Interactions. Angew. Chem. Int. Ed. 2011, 50, 314–318. [Google Scholar] [CrossRef]
- Paulini, R.; Müller, K.; Diedrich, F. Orthogonal Multipolar Interactions in Structural Chemistry and Biology. Angew. Chem. Int. Ed. 2011, 50, 314–318. [Google Scholar] [CrossRef]
- Roehrig, S.; Straub, A.; Pohlmann, J.; Lampe, T.; Pernerstorfer, J.; Schlemmer, K.-H.; Reinemer, P.; Perzborn, E.J. Discovery of the Novel Antithrombotic Agent 5-Chloro-N-({(5S)-2-oxo-3- [4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene- 2-carboxamide (BAY 59-7939): An Oral, Direct Factor Xa Inhibitor. Med. Chem. 2005, 48, 5900–5908. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.D. A New Reagent for the Synthesis of Diethyl Arylaminomethylenemalonates. Synthesis 1971, 1971, 220. [Google Scholar] [CrossRef]
- Desimoni, G.; Righetti, P.P.; Selva, E.; Tacconi, G.; Riganti, V.; Specchiarello, M. Heterodiene syntheses–XIX1: Correlation of the kinetic data with lumo energies in the reaction between 1-aryl-4-benzylidene-5-pyrazolones and isopropyl vinyl ether. Tetrahedron 1977, 33, 2829–2836. [Google Scholar] [CrossRef]
- Gehring, R.; Schallner, O.; Stetter, J.; Santel, H.J.; Schmidt, R.R. 1-Aryl-4-nitropyrazole. DE 3501323 A1 19860717 (1986). Chem. Abstr. 1986, 105, 191074. [Google Scholar]
- Kundu, M.; Nadkarni, S.M.; Gullapalli, S.; Joshi, N.K.; Karnik, P.V. Preparation of pyrazole amides as cannabinoid receptor ligands. WO 2007026215A1 20070308 (2007). Chem. Abstr. 2007, 146, 316911. [Google Scholar]
- Kralj, D.; Grošelj, U.; Meden, A.; Dahmann, G.; Stanovnik, B.; Svete, J. A simple synthesis of 4-(2-aminoethyl)-5-hydroxy-1H-pyrazoles. Tetrahedron 2007, 63, 11213–11222. [Google Scholar] [CrossRef]
- Dorn, H. Tautomerie und Nomenklatur der “Pyrazolone” und Aminopyrazole. J. Prakt. Chem. 1973, 315, 382–418. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Karelson, M.; Harris, P.A. Prototropic tautomerism of heteroaromatic compounds. Heterocyles 1991, 32, 329–369. [Google Scholar] [CrossRef]
- Yet, L. Pyrazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Joule, J., Eds.; Elsevier: Oxford, UK, 2008; Volume 4, pp. 1–141. ISBN 9780080449913. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, J.; Michnoff, C.H.; Malmquist, N.A.; White, J.; Roth, M.G.; Rathod, P.K.; Phillips, M.A. High-throughput Screening for Potent and Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. J. Biol. Chem. 2005, 280, 21847–21853. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | Dscore 1,2 | Glide 2 | Penalty 2 |
|---|---|---|---|---|
| 1 | 1a | −7.80/−6.20 | −7.89/−6.29 | 0.09/0.09 |
| 2 | 1b | −6.87/−6.01 | −7.07/−6.75 | 0.20/0.74 |
| 3 | 1c | −6.13/−5.52 | −7.52/−6.90 | 1.39/1.39 |
| 4 | 1d | −7.68/−6.99 | −7.93/−7.24 | 0.24/0.24 |
| 5 | 2a | −8.39/−6.65 | −8.82/−7.07 | 0.43/0.42 |
| 6 | 2b | −7.68/−6.36 | −8.17/−6.85 | 0.49/0.49 |
| 7 | 2c | −7.97/−7.88 | −8.40/−8.28 | 0.44/0.40 |
| 8 | 2d | −8.28/−6.90 | −8.90/−7.27 | 0.61/0.38 |
| 9 | 3c | −6.44/−6.40 | −6.44/−6.40 | 0.00/0.00 |
| 10 | 12 | −9.96/−11.38 | −9.97/−11.38 | 0.01/0.00 |
| Entry | Compound | R | Yield (%) 1 |
|---|---|---|---|
| 1 | 3c | 1H-Indazol-5-yl | 83 [12] |
| 2 | 5a | 4-Chlorophenyl | 70 [39] |
| 3 | 5b | Naphthalen-2-yl | 86 |
| 4 | 5c | 2,4,6-Trichlorophenyl | 79 [40] |
| 5 | 5d | 4-Trifluoromethylphenyl | 94 |
| 6 | 1a | 4-Chlorophenyl | 60 [39] |
| 7 | 1b | Naphthalen-2-yl | 78 |
| 8 | 1c | 2,4,6-Trichlorophenyl | 86 [40] |
| 9 | 1d | 4-Trifluoromethylphenyl | 80 [41] |
| Entry | Compound | R | Yield (%) 1 |
|---|---|---|---|
| 1 | 10a | 4-Chlorophenyl | 47 |
| 2 | 10b | Naphthalen-2-yl | 48 |
| 3 | 10c | 2,4,6-Trichlorophenyl | 53 |
| 4 | 10d | 4-Trifluoromethylphenyl | 47 |
| 5 | 2a | 4-Chlorophenyl | 74 |
| 6 | 2b | Naphthalen-2-yl | 57 |
| 7 | 2c | 2,4,6-Trichlorophenyl | 48 |
| 8 | 2d | 4-Trifluoromethylphenyl | 48 |
| Entry | Compound | Inhibition (%) 1 |
|---|---|---|
| 1 | 1a | n.i. 2 |
| 2 | 1b | 29 ± 7 |
| 3 | 1c | n.i.2 |
| 4 | 1d | n.i.2 |
| 5 | 2a | 15 ± 2 |
| 6 | 2b | 29 ± 3 |
| 7 | 2c | 30 ± 2 |
| 8 | 2d | 24 ± 3 |
| 9 | 3c | 19 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vah, L.; Medved, T.; Grošelj, U.; Klemenčič, M.; Podlipnik, Č.; Štefane, B.; Wagger, J.; Novinec, M.; Svete, J. Regioselective Synthesis of 5- and 3-Hydroxy-N-Aryl-1H-Pyrazole-4-Carboxylates and Their Evaluation as Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. Molecules 2022, 27, 4764. https://doi.org/10.3390/molecules27154764
Vah L, Medved T, Grošelj U, Klemenčič M, Podlipnik Č, Štefane B, Wagger J, Novinec M, Svete J. Regioselective Synthesis of 5- and 3-Hydroxy-N-Aryl-1H-Pyrazole-4-Carboxylates and Their Evaluation as Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. Molecules. 2022; 27(15):4764. https://doi.org/10.3390/molecules27154764
Chicago/Turabian StyleVah, Luka, Tadej Medved, Uroš Grošelj, Marina Klemenčič, Črtomir Podlipnik, Bogdan Štefane, Jernej Wagger, Marko Novinec, and Jurij Svete. 2022. "Regioselective Synthesis of 5- and 3-Hydroxy-N-Aryl-1H-Pyrazole-4-Carboxylates and Their Evaluation as Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase" Molecules 27, no. 15: 4764. https://doi.org/10.3390/molecules27154764
APA StyleVah, L., Medved, T., Grošelj, U., Klemenčič, M., Podlipnik, Č., Štefane, B., Wagger, J., Novinec, M., & Svete, J. (2022). Regioselective Synthesis of 5- and 3-Hydroxy-N-Aryl-1H-Pyrazole-4-Carboxylates and Their Evaluation as Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. Molecules, 27(15), 4764. https://doi.org/10.3390/molecules27154764