
An Intramolecular Hydroaminomethylation-Based Approach to Pyrrolizidine Alkaloids under Microwave-Assisted Heating

 , ,
, ,  ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
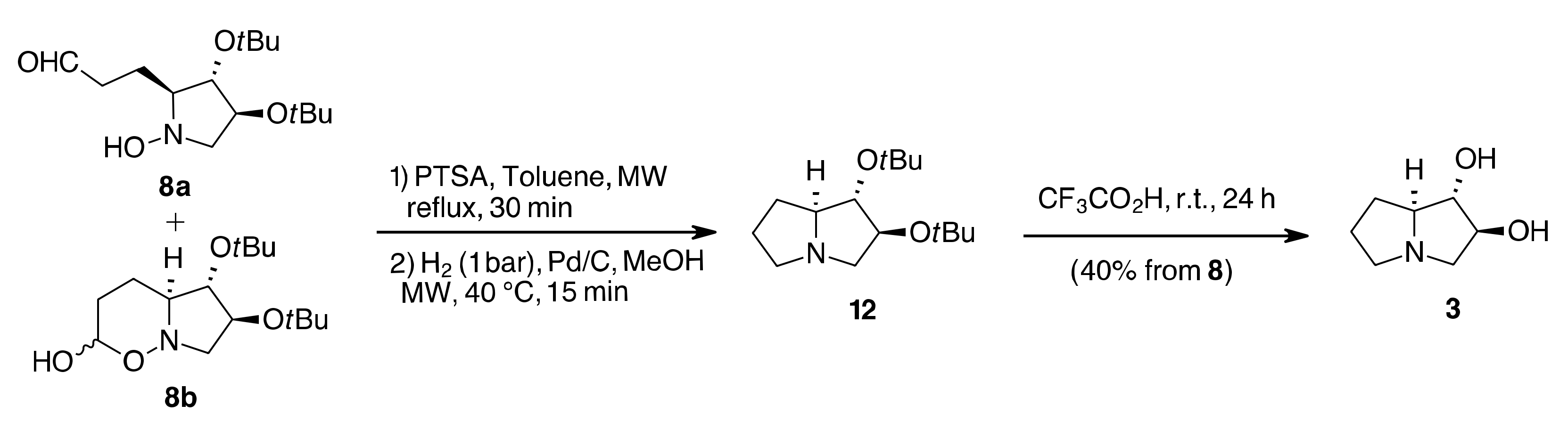
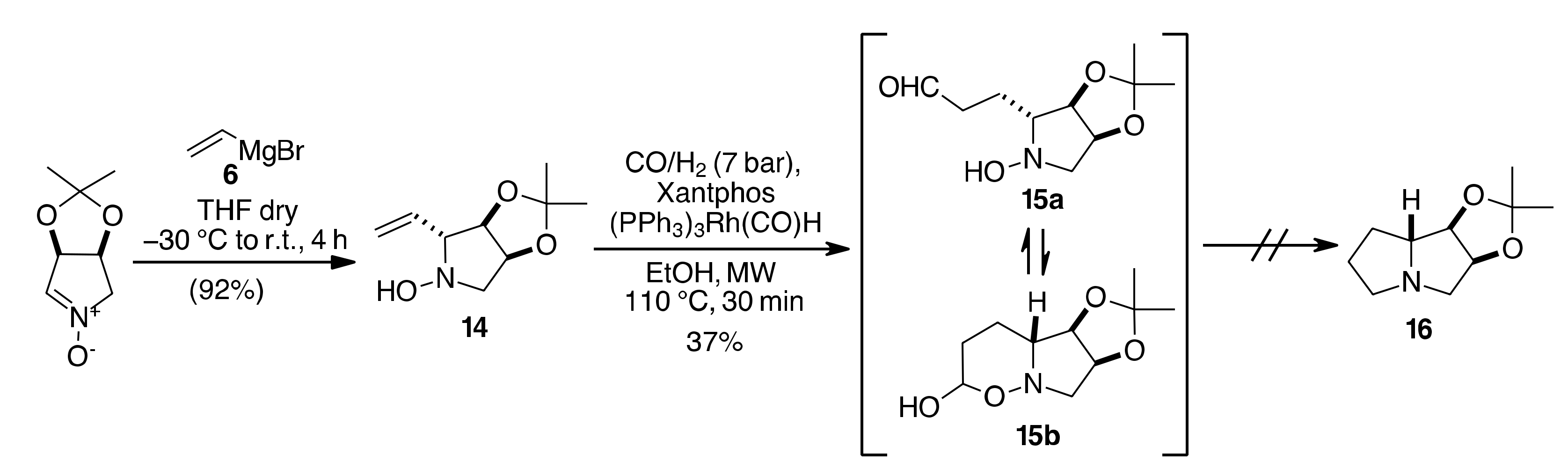
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Smith, L.W.; Culvenor, C.C.J. Plant Sources of Hepatotoxic Pyrrolizidine Alkaloids. J. Nat. Prod. 1981, 44, 129–152. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi Ziarani, G.; Jamasbi, N.; Mohajer, F. Recent Advances on the Synthesis of Natural Pyrrolizidine Alkaloids: Alexine, and Its Stereoisomers. Nat. Prod. Bioprospect. 2022, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Ruan, W.; Vrieling, K. Current Knowledge and Perspectives of Pyrrolizidine Alkaloids in Pharmacological Applications: A Mini-Review. Molecules 2021, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tropea, J.E.; Molyneux, R.J.; Kaushal, G.P.; Pan, Y.T.; Mitchell, M.; Elbein, A.D. Australine, a Pyrrolizidine Alkaloid That Inhibits Amyloglucosidase and Glycoprotein Processing. Biochemistry 1989, 28, 2027–2034. [Google Scholar] [CrossRef]
- Asano, N.; Ikeda, K.; Kasahara, M.; Arai, Y.; Kizu, H. Glycosidase-Inhibiting Pyrrolidines and Pyrrolizidines with a Long Side Chain in Scilla Peruviana. J. Nat. Prod. 2004, 67, 846–850. [Google Scholar] [CrossRef]
- Rousseaux, C.G.; Schachter, H. Regulatory Issues Concerning the Safety, Efficacy and Quality of Herbal Remedies. Birth Defects Res. Part B-Dev. Reprod. Toxicol. 2003, 68, 505–510. [Google Scholar] [CrossRef]
- EMA European Medicines Agency. Public Statement on the Use of Herbal Medicinal Products Containing Toxic, Unsaturated Pyrrolizidine Alkaloids (PAs); EMA: Amsterdam, The Netherlands, 2014; Volume 44. [Google Scholar]
- Stocker, B.L.; Dangerfield, E.M.; Win-Mason, A.L.; Haslett, G.W.; Timmer, M.S.M. Recent Developments in the Synthesis of Pyrrolidine-Containing Iminosugars. Eur. J. Org. Chem. 2010, 2010, 1615–1637. [Google Scholar] [CrossRef]
- Ratmanova, N.K.; Andreev, I.A.; Leontiev, A.V.; Momotova, D.; Novoselov, A.M.; Ivanova, O.A.; Trushkov, I.V. Strategic Approaches to the Synthesis of Pyrrolizidine and Indolizidine Alkaloids. Tetrahedron 2020, 76, 131031. [Google Scholar] [CrossRef]
- Calveras, J.; Casas, J.; Parella, T.; Joglar, J.; Clapés, P. Chemoenzymatic Synthesis and Inhibitory Activities of Hyacinthacines A1 and A2 Stereoisomers. Adv. Synth. Catal. 2007, 349, 1661–1666. [Google Scholar] [CrossRef]
- Brandi, A.; Cardona, F.; Cicchi, S.; Cordero, F.M.; Goti, A. Stereocontrolled Cyclic Nitrone Cycloaddition Strategy for the Synthesis of Pyrrolizidine and Indolizidine Alkaloids. Chem.A Eur. J. 2009, 15, 7808–7821. [Google Scholar] [CrossRef]
- Brandi, A.; Cardona, F.; Cicchi, S.; Cordero, F.M.; Goti, A. [3 + 2] Dipolar Cycloadditions of Cyclic Nitrones with Alkenes. In Organic Reactions; Wiley & Sons: Hoboken, NJ, USA, 2017; Volume 94. [Google Scholar]
- Delso, I.; Marca, E.; Mannucci, V.; Tejero, T.; Goti, A.; Merino, P. Tunable Diastereoselection of Biased Rigid Systems by Lewis Acid Induced Conformational Effects: A Rationalization of the Vinylation of Cyclic Nitrones En Route to Polyhydroxylated Pyrrolidines. Chem.-A Eur. J. 2010, 16, 9901–9919. [Google Scholar] [CrossRef] [Green Version]
- Merino, P.; Revuelta, J.; Tejero, T.; Cicchi, S.; Goti, A. Fully Stereoselective Nucleophilic Addition to a Novel Chiral Pyrroline N-Oxide: Total Syntheses of (2S,3R)-3-Hydroxy-3-Methylproline and Its (2R)-Epimer. Eur. J. Org. Chem. 2004, 2004, 776–782. [Google Scholar] [CrossRef]
- Delso, I.; Tejero, T.; Goti, A.; Merino, P. Synthesis of D-Arabinose-Derived Polyhydroxylated Pyrrolidine, Indolizidine and Pyrrolizidine Alkaloids. Total Synthesis of Hyacinthacine A2. Tetrahedron 2010, 66, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- McCaig, A.E.; Meldrum, K.P.; Wightman, R.H. Synthesis of Trihydroxylated Pyrrolizidines and Indolizidines Using Cycloaddition Reactions of Functionalized Cyclic Nitrones, and the Synthesis of (+)- and (−)-Lentiginosine. Tetrahedron 1998, 54, 9429–9446. [Google Scholar] [CrossRef]
- Cardona, F.; Moreno, G.; Guarna, F.; Vogel, P.; Schuetz, C.; Merino, P.; Goti, A. New Concise Total Synthesis of (+)-Lentiginosine and Some Structural Analogues. J. Org. Chem. 2005, 70, 6552–6555. [Google Scholar] [CrossRef]
- Cardona, F.; Goti, A.; Picasso, S.; Vogel, P.; Brandi, A. Polyhydroxypyrrolidine Glycosidase Inhibitors Related to (+)-Lentiginosine1. J. Carbohydr. Chem. 2000, 19, 585–601. [Google Scholar] [CrossRef]
- Martella, D.; Cardona, F.; Parmeggiani, C.; Franco, F.; Tamayo, J.A.; Robina, I.; Moreno-Clavijo, E.; Moreno-Vargas, A.J.; Goti, A. Synthesis and Glycosidase Inhibition Studies of 5-Methyl-Substituted Tetrahydroxyindolizidines and -Pyrrolizidines Related to Natural Hyacinthacines B. Eur. J. Org. Chem. 2013, 2013, 4047–4056. [Google Scholar] [CrossRef]
- D’Adamio, G.; Sgambato, A.; Forcella, M.; Caccia, S.; Parmeggiani, C.; Casartelli, M.; Parenti, P.; Bini, D.; Cipolla, L.; Fusi, P.; et al. New Synthesis and Biological Evaluation of Uniflorine A Derivatives: Towards Specific Insect Trehalase Inhibitors. Org. Biomol. Chem. 2015, 13, 886–892. [Google Scholar] [CrossRef]
- Bonaccini, C.; Chioccioli, M.; Parmeggiani, C.; Cardona, F.; lo Re, D.; Soldaini, G.; Vogel, P.; Bello, C.; Goti, A.; Gratteri, P. Synthesis, Biological Evaluation and Docking Studies of Casuarine Analogues: Effects of Structural Modifications at Ring B on Inhibitory Activity towards Glucoamylase. Eur. J. Org. Chem. 2010, 2010, 5547–5585. [Google Scholar] [CrossRef]
- D’Adamio, G.; Parmeggiani, C.; Goti, A.; Moreno-Vargas, A.J.; Moreno-Clavijo, E.; Robina, I.; Cardona, F. 6-Azido Hyacinthacine A2 Gives a Straightforward Access to the First Multivalent Pyrrolizidine Architectures. Org. Biomol. Chem. 2014, 12, 6250–6266. [Google Scholar] [CrossRef]
- D’Adamio, G.; Goti, A.; Parmeggiani, C.; Moreno-Clavijo, E.; Robina, I.; Cardona, F. Total Synthesis of (+)-Hyacinthacine A1, (+)-7a-Epi- Hyacinthacine A1, (6R)-6-Hydroxyhyacinthacine A1 and (6S)-6-Hydroxy-7a-Epi-Hyacinthacine A1. Eur. J. Org. Chem. 2011, 2011, 7155–7162. [Google Scholar] [CrossRef]
- Pecchioli, T.; Cardona, F.; Reissig, H.U.; Zimmer, R.; Goti, A. Alkoxyallene-Based Stereodivergent Syntheses of (−)-Hyacinthacine B4 and of Putative Hyacinthacine C5 Epimers: Proposal of Hyacinthacine C5 Structure. J. Org. Chem. 2017, 82, 5835–5844. [Google Scholar] [CrossRef]
- Cornils, B.; Herrmann, W.A.; Rasch, M. Otto Roelen, Pioneer in Industrial Homogeneous Catalysis. Angew. Chem. Int. Ed. 1994, 33, 2144–2163. [Google Scholar] [CrossRef]
- Raoufmoghaddam, S. Recent Advances in Catalytic C-N Bond Formation: A Comparison of Cascade Hydroaminomethylation and Reductive Amination Reactions with the Corresponding Hydroamidomethylation and Reductive Amidation Reactions. Org. Biomol. Chem. 2014, 12, 7179–7193. [Google Scholar] [CrossRef]
- Yang, J.; Delolo, F.G.; Spannenberg, A.; Jackstell, R.; Beller, M. A Selective and General Cobalt-Catalyzed Hydroaminomethylation of Olefins to Amines. Angew. Chem. Int. Ed. 2022, 61, e202112597. [Google Scholar] [CrossRef]
- Chen, C.; Dong, X.Q.; Zhang, X. Recent Progress in Rhodium-Catalyzed Hydroaminomethylation. Org. Chem. Front. 2016, 3, 1359–1370. [Google Scholar] [CrossRef]
- Kalck, P.; Urrutigoïty, M. Tandem Hydroaminomethylation Reaction to Synthesize Amines from Alkenes. Chem. Rev. 2018, 118, 3833–3861. [Google Scholar] [CrossRef]
- An, J.; Gao, Z.; Wang, Y.; Zhang, Z.; Zhang, J.; Li, L.; Tang, B.; Wang, F. Heterogeneous Ru/TiO2 for Hydroaminomethylation of Olefins: Multicomponent Synthesis of Amines. Green Chem. 2021, 23, 2722–2728. [Google Scholar] [CrossRef]
- Pizzetti, M.; Russo, A.; Petricci, E. Microwave-Assisted Aminocarbonylation of Ynamides by Using Catalytic [Fe3(CO)12] at Low Pressures of Carbon Monoxide. Chem.-A Eur. J. 2011, 17, 4523–4528. [Google Scholar] [CrossRef]
- Cardullo, F.; Donati, D.; Merlo, G.; Paio, A.; Petricci, E.; Taddei, M. Microwave-Assisted Aminocarbonylation of Aryl Bromides at Low Carbon Monoxide Pressure. Synlett 2009, 2009, 47–50. [Google Scholar] [CrossRef]
- Risi, C.; Cini, E.; Petricci, E.; Saponaro, S.; Taddei, M. In Water Markovnikov Hydration and One-Pot Reductive Hydroamination of Terminal Alkynes under Ruthenium Nanoparticle Catalysis. Eur. J. Inorg. Chem. 2020, 2020, 1000–1003. [Google Scholar] [CrossRef]
- Petricci, E.; Risi, C.; Ferlin, F.; Lanari, D.; Vaccaro, L. Avoiding Hot-Spots in Microwave-Assisted Pd/C Catalysed Reactions by Using the Biomass Derived Solvent γ-Valerolactone. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumde, V.R.; Petricci, E.; Petrucci, C.; Santillo, N.; Taddei, M.; Vaccaro, L. Domino Hydrogenation-Reductive Amination of Phenols, a Simple Process to Access Substituted Cyclohexylamines. Org. Lett. 2015, 17, 3990–3993. [Google Scholar] [CrossRef] [PubMed]
- Valentini, F.; Santillo, N.; Petrucci, C.; Lanari, D.; Petricci, E.; Taddei, M.; Vaccaro, L. Continuous-Flow Palladium-Catalyzed Synthesis of Cyclohexanones from Phenols Using Sodium Formate as a Safe Hydrogen Source. ChemCatChem 2018, 10, 1277–1281. [Google Scholar] [CrossRef]
- Verheyen, T.; Santillo, N.; Marinelli, D.; Petricci, E.; de Borggraeve, W.M.; Vaccaro, L.; Smet, M. An Effective and Reusable Hyperbranched Polymer Immobilized Rhodium Catalyst for the Hydroformylation of Olefins. ACS Appl. Polym. Mater. 2019, 1, 1496–1504. [Google Scholar] [CrossRef]
- Airiau, E.; Chemin, C.; Girard, N.; Lonzi, G.; Mann, A.; Petricci, E.; Salvadori, J.; Taddei, M. Microwave-Assisted Domino Hydroformylation/Cyclization Reactions: Scope and Limitations. Synthesis 2010, 2010, 2901–2914. [Google Scholar] [CrossRef]
- Pizzetti, M.; de Luca, E.; Petricci, E.; Porcheddu, A.; Taddei, M. A General Approach to Substituted Benzimidazoles and Benzoxazoles via Heterogeneous Palladium-Catalyzed Hydrogen-Transfer with Primary Amines. Adv. Synth. Catal. 2012, 354, 2453–2464. [Google Scholar] [CrossRef]
- Arena, G.; Cini, E.; Petricci, E.; Randino, R.; Taddei, M. A Highly Stereo-Controlled Protocol to Prepare Pipecolic Acids Based on Heck and Cyclohydrocarbonylation Reactions. Org. Chem. Front. 2015, 2, 526–530. [Google Scholar] [CrossRef]
- Balducci, E.; Bellucci, L.; Petricci, E.; Taddei, M.; Tafi, A. Microwave-Assisted Intramolecular Huisgen Cycloaddition of Azido Alkynes Derived from α-Amino Acids. J. Org. Chem. 2009, 74, 1314–1321. [Google Scholar] [CrossRef]
- Salvadori, J.; Balducci, E.; Zaza, S.; Petricci, E.; Taddei, M. Microwave-Assisted Carbonylation and Cyclocarbonylation of Aryl Iodides under Ligand Free Heterogeneous Catalysis. J. Org. Chem. 2010, 75, 1841–1847. [Google Scholar] [CrossRef]
- Migliorini, F.; Dei, F.; Calamante, M.; Maramai, S.; Petricci, E. Micellar Catalysis for Sustainable Hydroformylation. ChemCatChem 2021, 13, 2794–2806. [Google Scholar] [CrossRef]
- Petricci, E.; Santillo, N.; Castagnolo, D.; Cini, E.; Taddei, M. Iron-Catalyzed Reductive Amination of Aldehydes in Isopropyl Alcohol/Water Media as Hydrogen Sources. Adv. Synth. Catal. 2018, 360, 2560–2565. [Google Scholar] [CrossRef]
- Petricci, E.; Mann, A.; Schoenfelder, A.; Rota, A.; Taddei, M. Microwaves Make Hydroformylation a Rapid and Easy Process. Org. Lett. 2006, 8, 3725–3727. [Google Scholar] [CrossRef]
- Petricci, E.; Mann, A.; Salvadori, J.; Taddei, M. Microwave Assisted Hydroaminomethylation of Alkenes. Tetrahedron Lett. 2007, 48, 8501–8504. [Google Scholar] [CrossRef]
- Cicchi, S.; Höld, I.; Brandi, A. New Synthesis of Five-Membered Cyclic Nitrones from Tartaric Acid. J. Org. Chem. 1993, 58, 5274–5275. [Google Scholar] [CrossRef]
- Matassini, C.; Bonanni, M.; Marradi, M.; Cicchi, S.; Goti, A. On the Virtue of Indium in Reduction Reactions. A Comparison of Reductions Mediated by Indium and Zinc: Is Indium Metal an Effective Catalyst for Zinc Induced Reductions? Eur. J. Inorg. Chem. 2020, 2020, 1106–1113. [Google Scholar] [CrossRef] [Green Version]
- Bini, D.; Forcella, M.; Cipolla, L.; Fusi, P.; Matassini, C.; Cardona, F. Synthesis of Novel Iminosugar-Based Trehalase Inhibitors by Cross-Metathesis Reactions. Eur. J. Org. Chem. 2011, 2011, 3995–4000. [Google Scholar] [CrossRef]
- Cicchi, S.; Bonanni, M.; Cardona, F.; Revuelta, J.; Goti, A. Indium-Mediated Reduction of Hydroxylamines to Amines. Org. Lett. 2003, 5, 1773–1776. [Google Scholar] [CrossRef]
- Cardona, F.; Faggi, E.; Liguori, F.; Cacciarini, M.; Goti, A. Total Syntheses of Hyacinthacine A2 and 7-Deoxycasuarine by Cycloaddition to a Carbohydrate Derived Nitrone. Tetrahedron Lett. 2003, 44, 2315–2318. [Google Scholar] [CrossRef]
- Desvergnes, S.; Py, S.; Vallee, Y. Total Synthesis of (+)-Hyacinthacine A2 (I) Based on SmI2-Induced Nitrone Umpolung. ChemInform 2005, 36. [Google Scholar] [CrossRef]
- Rambaud, L.; Compain, P.; Martin, O.R. First Total Synthesis of (+)-Hyacinthacine A2. Tetrahedron Asymmetry 2001, 12, 1807–1809. [Google Scholar] [CrossRef]
- Martella, D.; D’Adamio, G.; Parmeggiani, C.; Cardona, F.; Moreno-Clavijo, E.; Robina, I.; Goti, A. Cycloadditions of Sugar-Derived Nitrones Targeting Polyhydroxylated Indolizidines. Eur. J. Org. Chem. 2016, 2016, 1588–1598. [Google Scholar] [CrossRef]
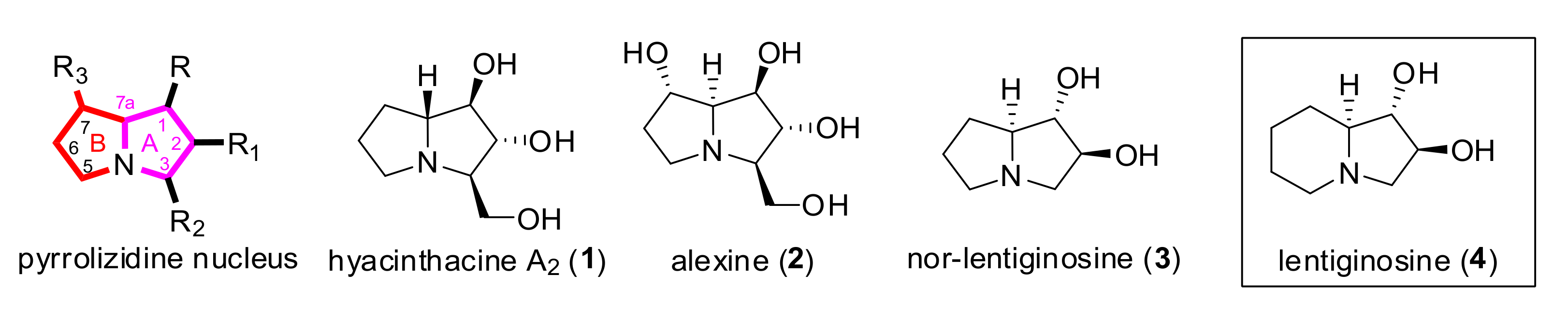


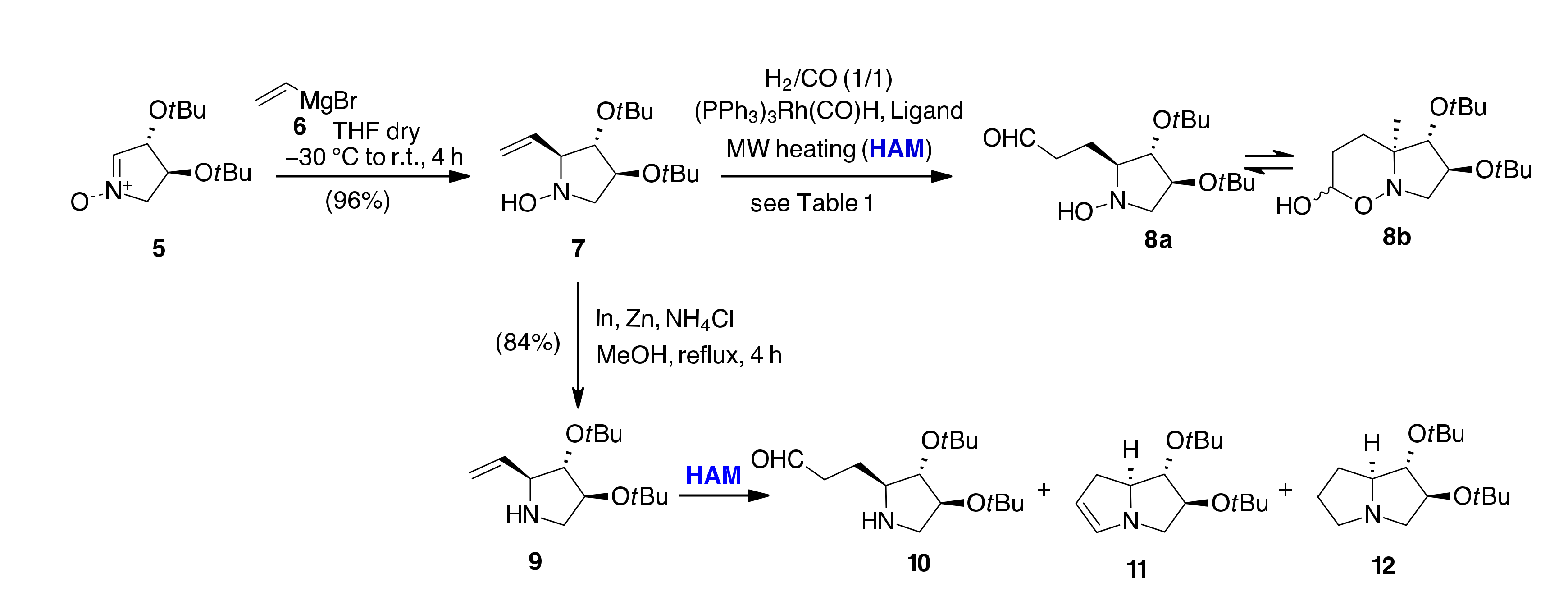
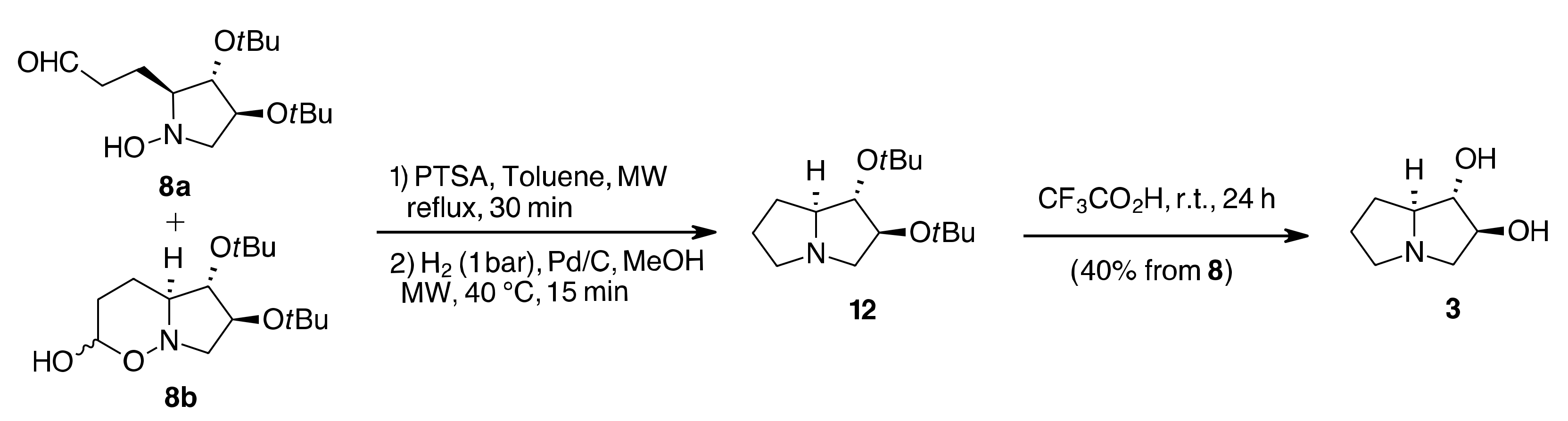
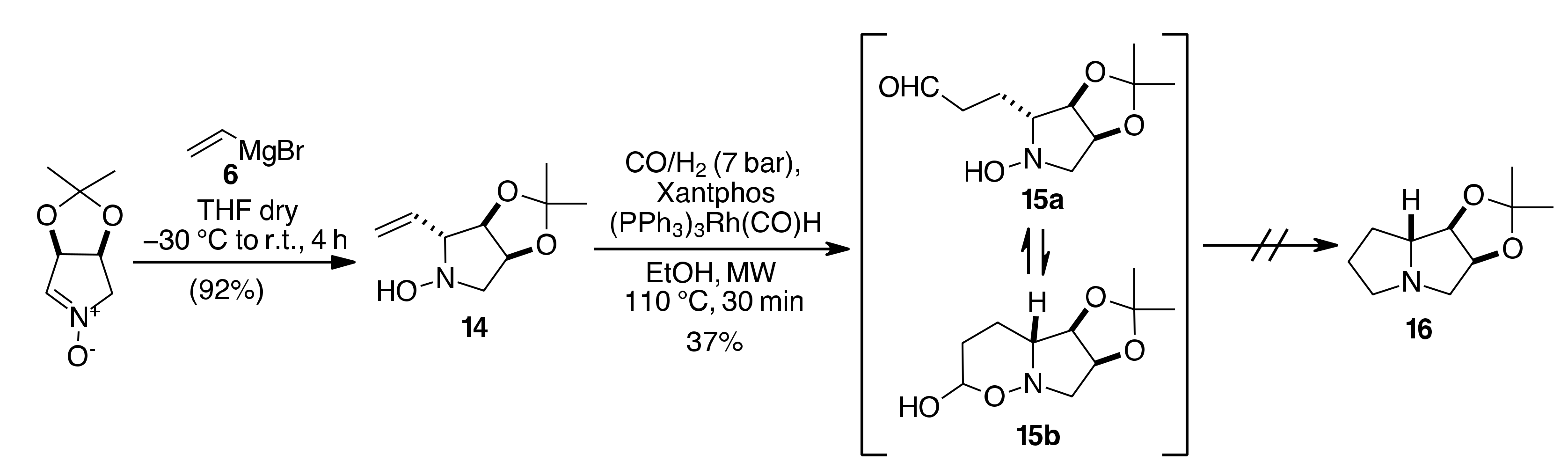
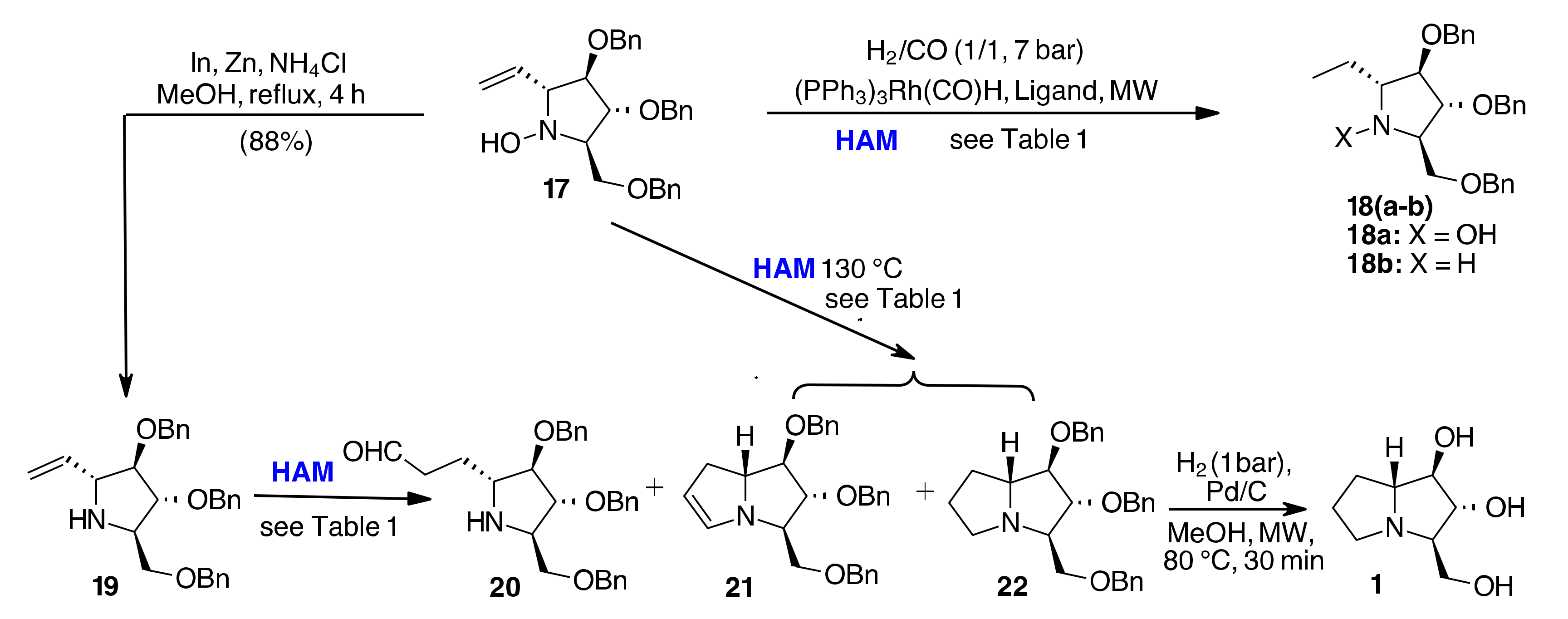

{kind=link}
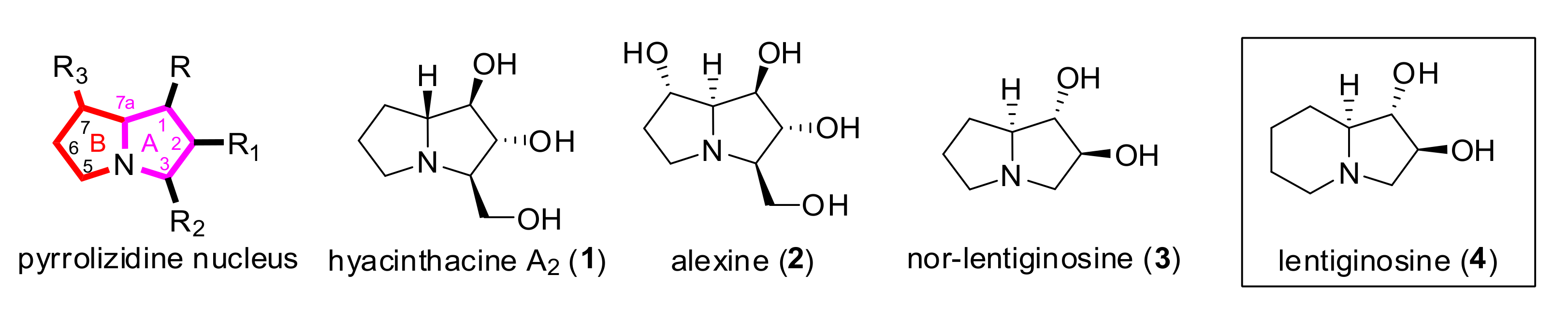
{kind=link}
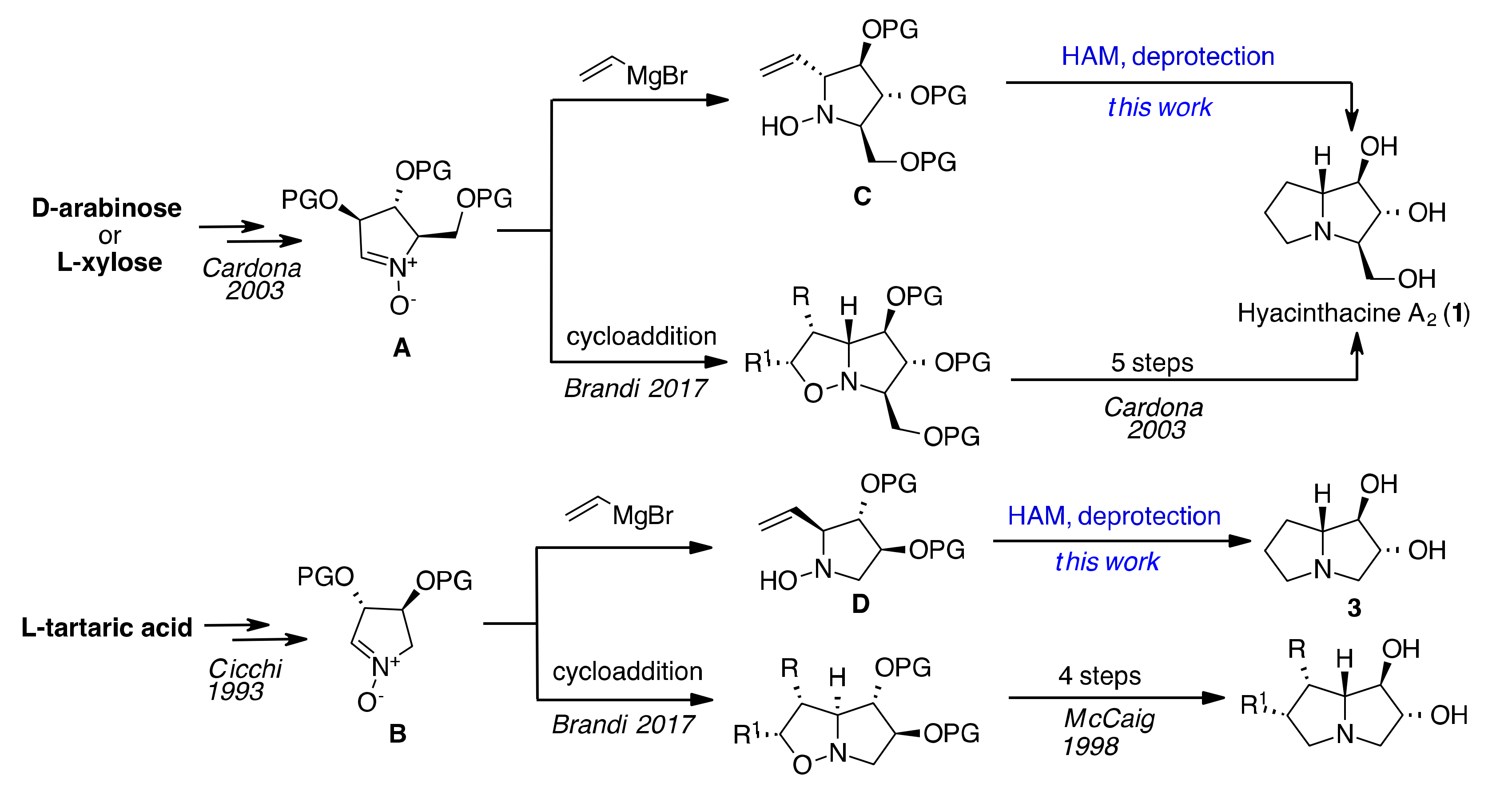
{kind=link}
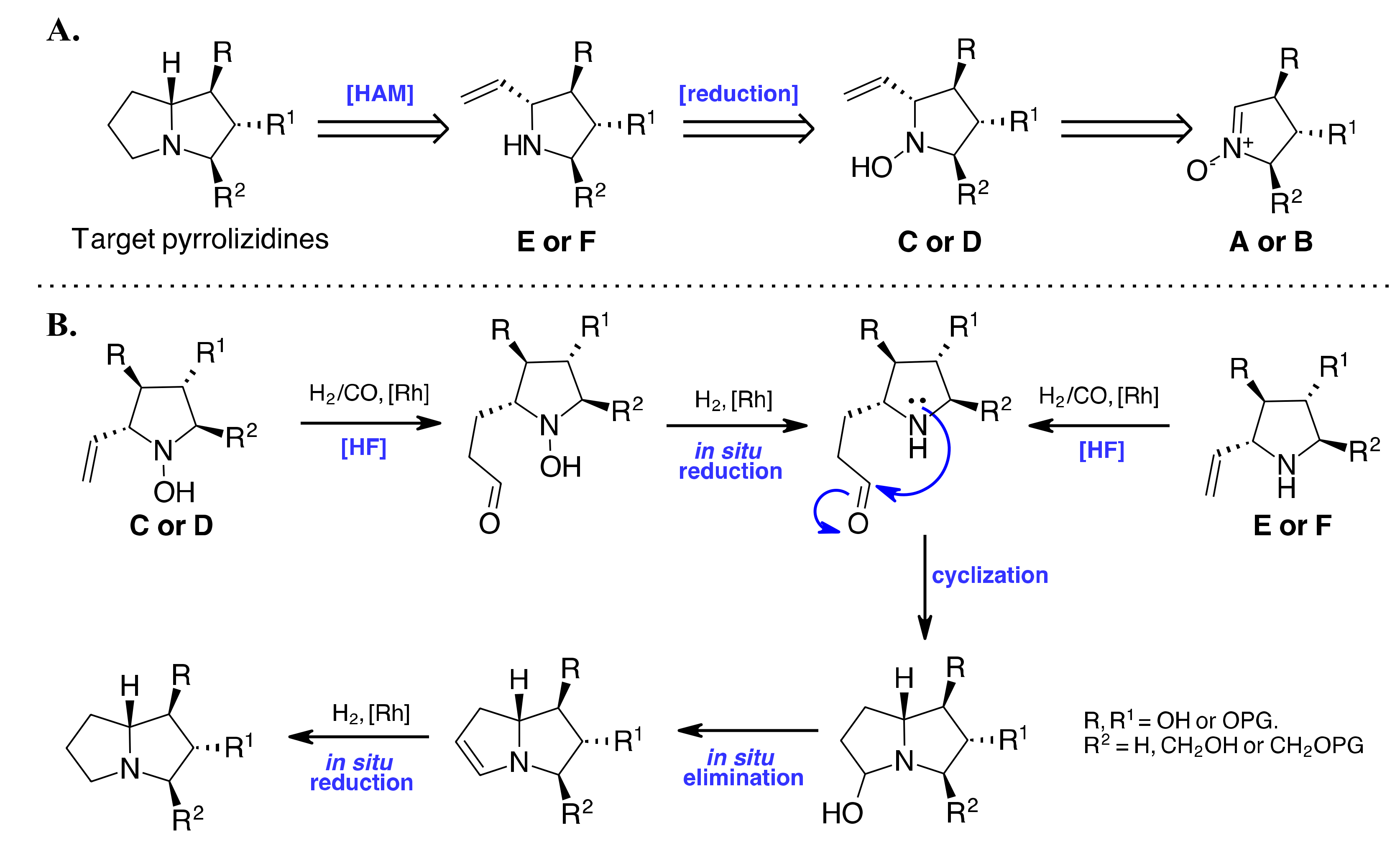
{kind=link}
{kind=link}
{kind=link}
{kind=link}
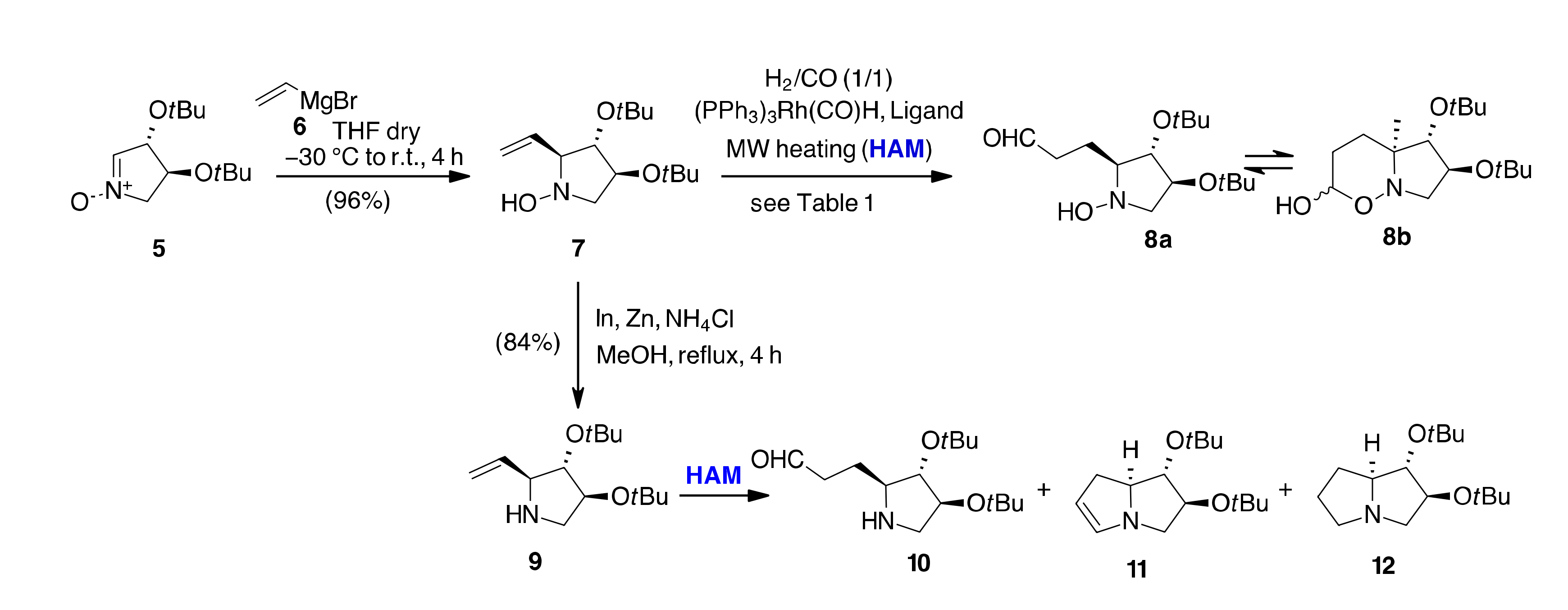
| En | SM | Conditions [a] | Rec SM (%) | 10 (%) | 11 (%) | 12 (%) | 13 (%) | 8 (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 7 | CO/H2 (7 bar), Xantphos/(PPh3)3Rh(CO)H, Toluene/[bmim][BF4], MW, 110 °C, 30 min | 19 | 44 | 5 | 10 | - | - |
| 2 | 9 | 70 | - | 27 | - | - | - | |
| 3 | 7 | CO/H2 (7 bar), Xantphos/(PPh3)3Rh(CO)H EtOH, MW, 110 °C, 30 min | 12 | - | - | 5 | - | 65 |
| 4 | 9 | 72 | 5 | 20 | - | - | - | |
| 5 | 7 | CO/H2 (7 bar), Xantphos/(PPh3)3Rh(CO)H Toluene, MW, 80 °C, 30 min | 88 | - | - | - | - | - |
| 6 | 9 | 22 | - | - | - | 74 | - | |
| 7 | 7 | CO/H2 (7 bar), Xantphos/(PPh3)3Rh(CO)H Toluene/[bmim][BF4], MW, 130 °C, 30 min | 54 | 10 | 20 | 10 | - | - |
| 8 | 9 | 63 | - | 27 | 2 | 5 | - | |
| 9 | 7 | CO/H2 (7 bar), Biphephos/(PPh3)3Rh(CO)H EtOH, MW, 110 °C, 30 min | - | - | - | - | 99 | - |
| 10 | 9 | 2 | - | - | - | 90 | - | |
| 11 | 7 | CO/H2 (7 bar), PPh3/(PPh3)3Rh(CO)H, EtOH, MW, 110 °C, 30 min | 67 | - | - | - | 31 | - |
| 12 | 9 | 5 | - | - | - | 92 | - | |
| 13 | 7 | CO/H2 (7 bar), PPh3/(PPh3)3Rh(CO)H Toluene, MW, 80 °C, 30 min | 55 | - | - | - | - | 38 |
| 14 | 7 | CO/H2 (7 bar), Biphephos/[RhCl(COD)]2 EtOH, MW, 110 °C, 30 min | 32 | - | 15 | 5 | 32 | 10 |
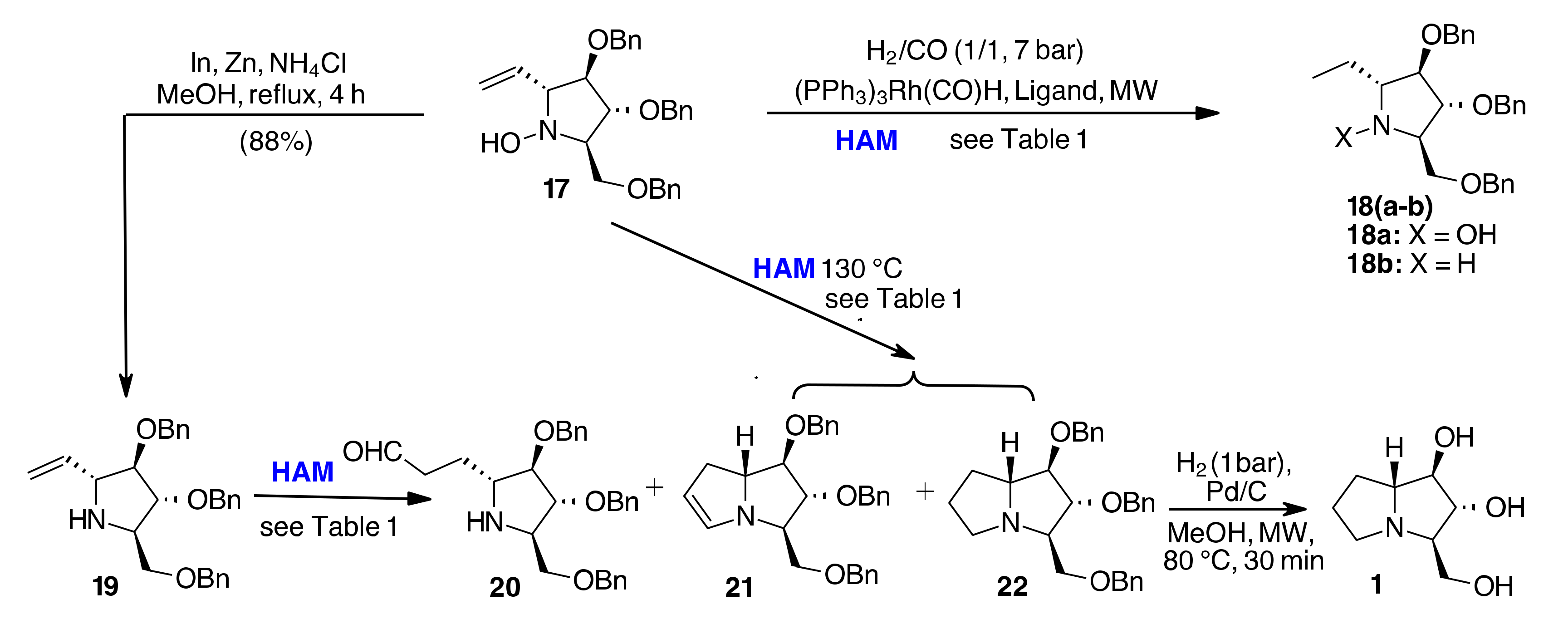
| En | SM | Conditions | Rec SM | 18 | 20 | 21 | 22 |
|---|---|---|---|---|---|---|---|
| 1 | 17 | Xantphos, Toluene, [bmim][BF4], 110 °C, 30 min | 74% | 28% [a] | - | - | |
| 2 | 17 | Xantphos, Toluene, [bmim][BF4], 130 °C, 30 min | 56% | 5% [a] | - | 2% | 27% |
| 3 | 17 | Xantphos, EtOH, MW, 110 °C, 30 min | 99% | - | - | - | - |
| 4 | 17 | Biphephos, EtOH, MW, 110 °C, 30 min | - | 96% [a] | - | - | - |
| 5 | 19 | Xantphos, Toluene, [bmim][BF4], 110 °C, 30 min | 42% | - | 43% | - | |
| 6 | 19 | Xantphos, Toluene, [bmim][BF4], 130 °C, 30 min | 42% | - | 10% | 10% | 20% |
| 7 | 19 | Xantphos, EtOH, 110 °C, 30 min | - | 5% [b] | - | - | 82% |
| 8 | 19 | Biphephos, EtOH, 110 °C, 30 min | - | 99% [b] | - | - | - |
| 9 | 19 | PPh3, EtOH, 110 °C, 30 min | 31% | 57% [b] | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petricci, E.; Zurzolo, S.; Matassini, C.; Maramai, S.; Cardona, F.; Goti, A.; Taddei, M. An Intramolecular Hydroaminomethylation-Based Approach to Pyrrolizidine Alkaloids under Microwave-Assisted Heating. Molecules 2022, 27, 4762. https://doi.org/10.3390/molecules27154762
Petricci E, Zurzolo S, Matassini C, Maramai S, Cardona F, Goti A, Taddei M. An Intramolecular Hydroaminomethylation-Based Approach to Pyrrolizidine Alkaloids under Microwave-Assisted Heating. Molecules. 2022; 27(15):4762. https://doi.org/10.3390/molecules27154762
Chicago/Turabian StylePetricci, Elena, Simone Zurzolo, Camilla Matassini, Samuele Maramai, Francesca Cardona, Andrea Goti, and Maurizio Taddei. 2022. "An Intramolecular Hydroaminomethylation-Based Approach to Pyrrolizidine Alkaloids under Microwave-Assisted Heating" Molecules 27, no. 15: 4762. https://doi.org/10.3390/molecules27154762
APA StylePetricci, E., Zurzolo, S., Matassini, C., Maramai, S., Cardona, F., Goti, A., & Taddei, M. (2022). An Intramolecular Hydroaminomethylation-Based Approach to Pyrrolizidine Alkaloids under Microwave-Assisted Heating. Molecules, 27(15), 4762. https://doi.org/10.3390/molecules27154762