
Polyphenols from the Peels of Punica granatum L. and Their Bioactivity of Suppressing Lipopolysaccharide-Stimulated Inflammatory Cytokines and Mediators in RAW 264.7 Cells via Activating p38 MAPK and NF-κB Signaling Pathways


Abstract
:1. Introduction
2. Results and Discussion
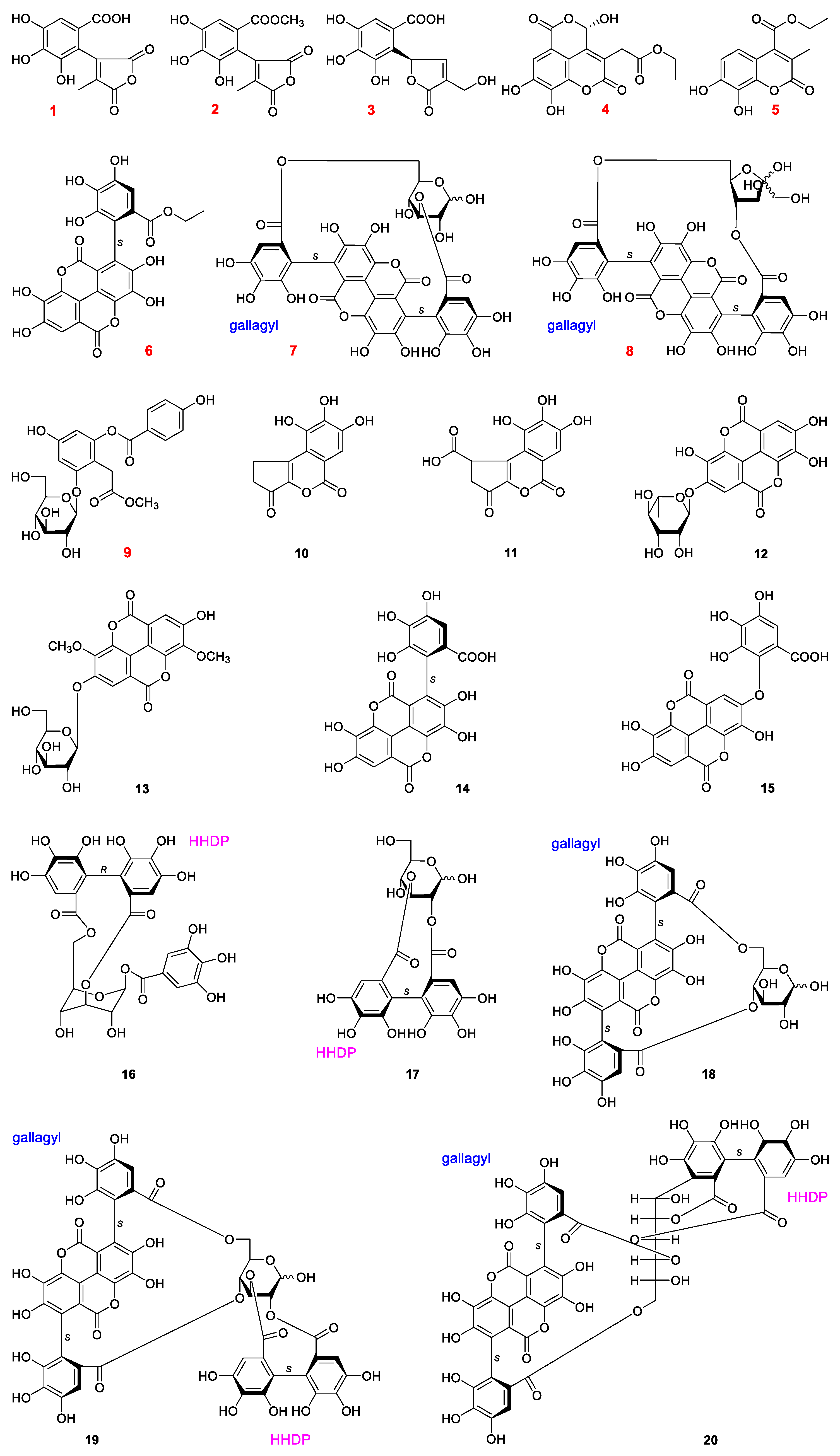
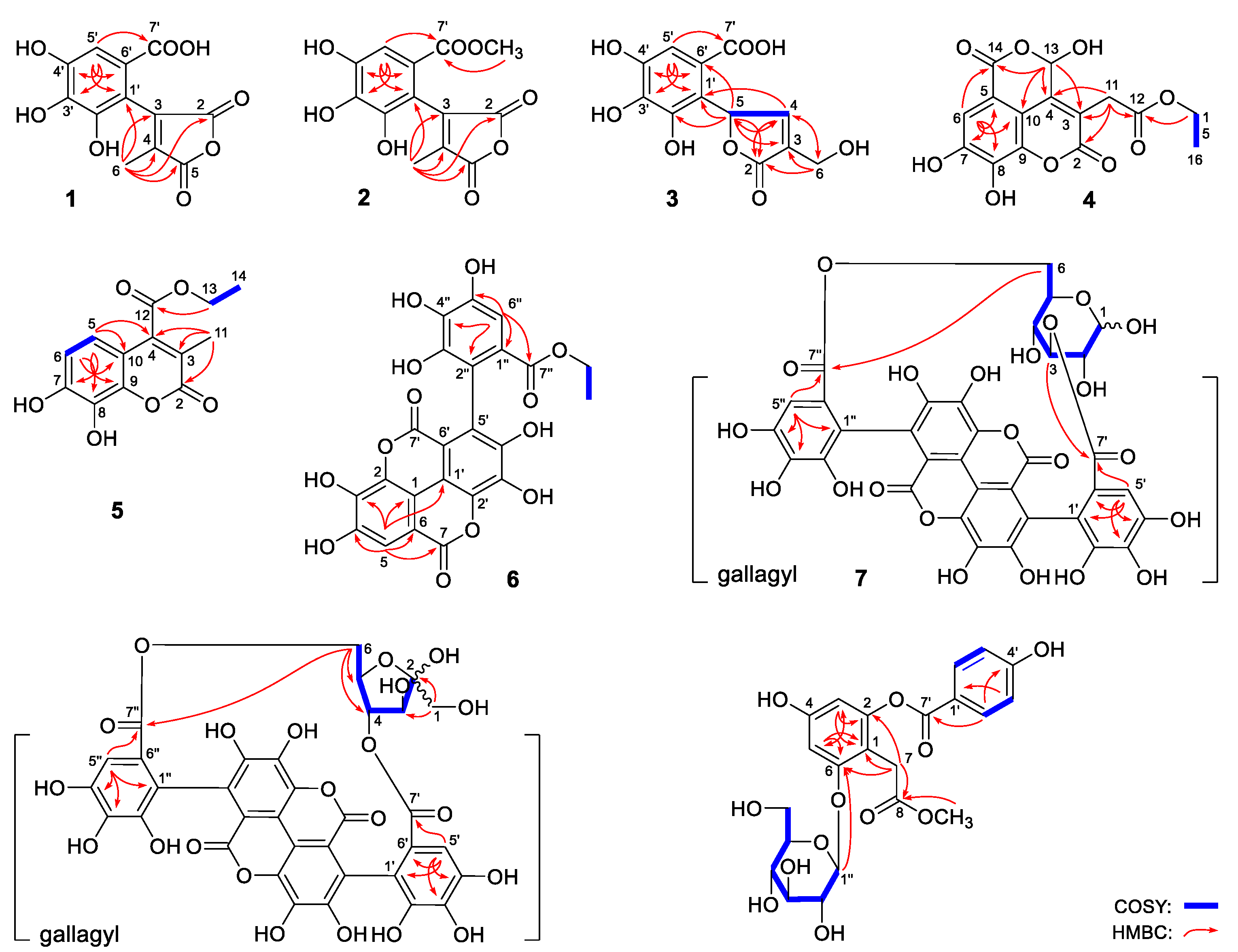
2.1. Structural Characterization
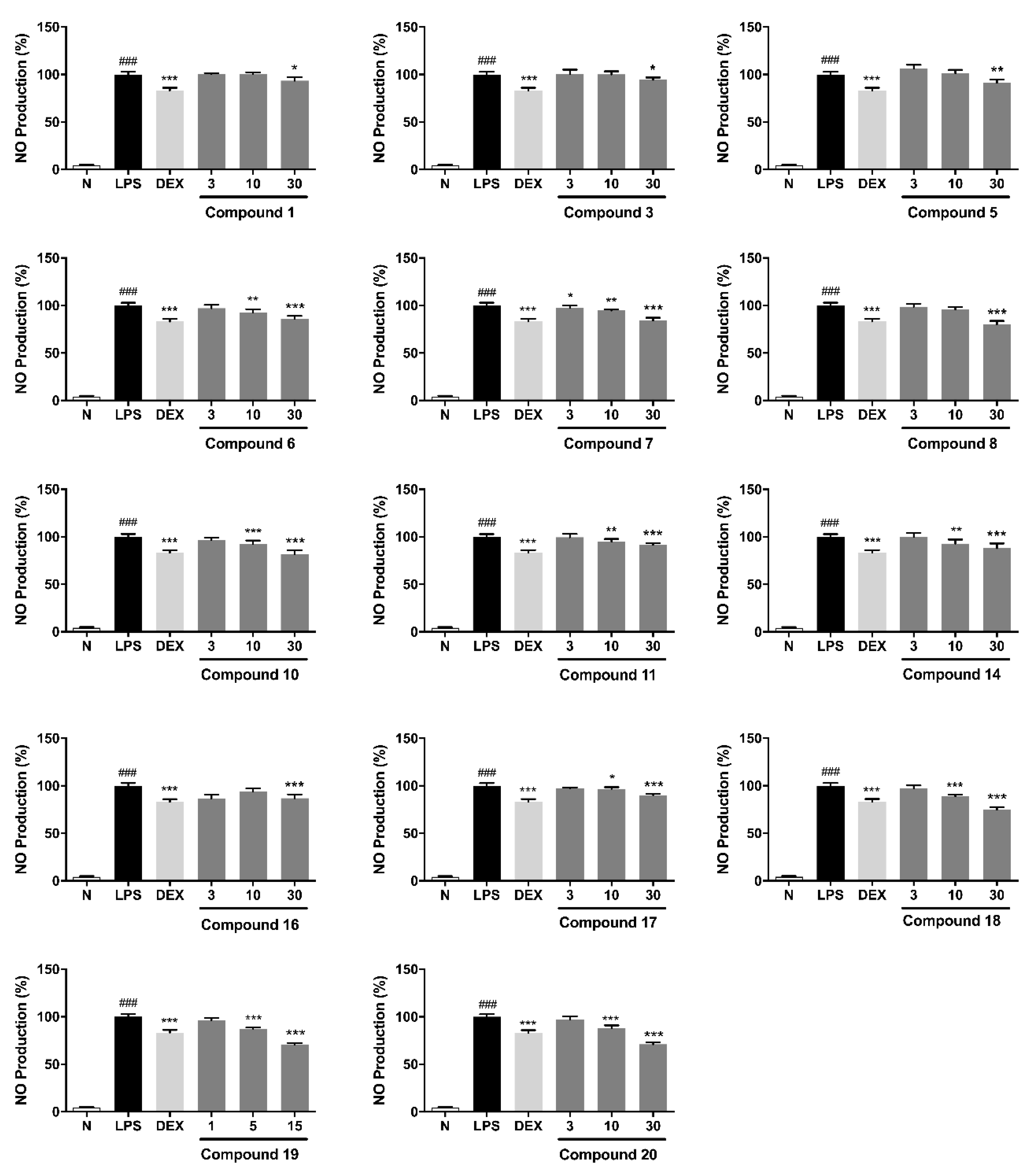
2.2. In Vitro Anti-Inflammatory Evaluation of Compounds 1–20
- (1)
- Tannins containing gallagyl (6–8 and 18–20) had good inhibitory activity on NO release production. Compound 19 displayed stronger activity than 17. Both of the results suggested gallagyl group may be a positive factor for NO inhibitory activity.
- (2)
- The HHDP group in tannins also played an important role in inhibiting NO release (18 vs. 19).
- (3)
- The activities of flavogallonic acid derivatives formed by the condensation of ellagic acid and gallic acid through carbon-carbon bonds were significantly stronger than those of valoneic acid derivatives produced by the condensation of ellagic acid and gallic acid through carbon-oxygen-carbon bond (6 and 14 vs. 15).
- (4)
- Moreover, it was found that ethyl or methyl esterification of carboxyl group could affect the activity slightly only (2 vs. 1 and 6 vs. 14).
- (5)
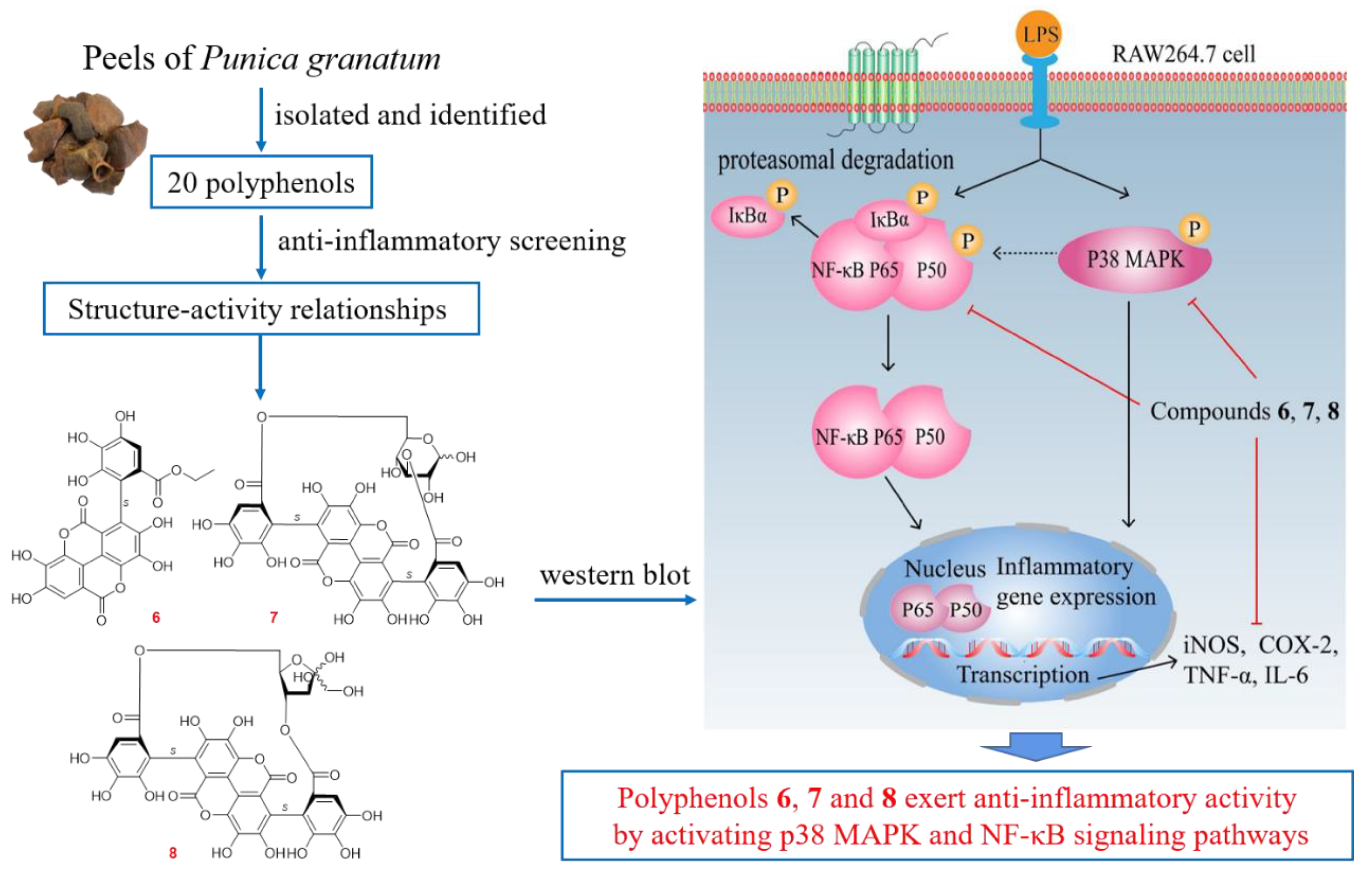
2.3. Polyphenols from P. granatum Peels Exerted Anti-Inflammatory Effects through p38 MAPK and NF-κB Signaling Pathway
3. Conclusions
4. Experimental
4.1. Materials and Methods for Phytochemistry Research
4.1.1. General Experimental Procedures
4.1.2. Plant Material
4.1.3. Extraction and Isolation
4.1.4. Acid Hydrolysis of 7–9
4.2. Materials and Methods for Anti-Inflammatory Assay
4.2.1. Cell Culture
4.2.2. Cytotoxicity Assay
4.2.3. Determination of NO Production
4.2.4. Western Blot Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Olejnik, A.; Kowalska, K.; Olkowicz, M.; Rychlik, J.; Juzwa, W.; Myszka, K.; Dembczyński, R.; Wojciech Białas, W. Antiinflammatory effects of gastrointestinal digested Sambucus nigra L. fruit extract analysed in co-cultured intestinal epithelial cells and lipopolysaccharide-stimulated macrophages. J. Funct. Foods 2015, 19, 649–660. [Google Scholar] [CrossRef]
- Hollebeeck, S.; Winand, J.; Hérent, M.F.; During, A.; Leclercq, J.; Larondelle, Y.; Schneider, Y.J. Anti-inflammatory effects of pomegranate (Punica granatum L.) husk ellagitannins in Caco-2 cells, an in vitro model of human intestine. Food Funct. 2012, 3, 875–885. [Google Scholar] [CrossRef]
- Guo, Y.; Li, Y.; Liu, F.; Zhang, G. Review of active components of traditional Chinese medicine to reduce the lipopolysaccharide-induced inflammation. Huaxi Yaoxue Zazhi 2021, 36, 331–335. [Google Scholar]
- Hu, X.; Yu, Q.; Hou, K.; Ding, X.; Chen, Y.; Xie, J.; Nie, S.; Xie, M. Regulatory effects of Ganoderma atrum polysaccharides on LPS-induced inflammatory macrophages model and intestinal-like Caco-2/macrophages co-culture inflammation model. Food Chem. Toxicol. 2020, 140, 111321. [Google Scholar] [CrossRef] [PubMed]
- Aly, E.; López-Nicolás, R.; Darwish, A.A.; Ros-Berruezo, G.; Frontela-Saseta, C. In vitro effectiveness of recombinant human lactoferrin and its hydrolysate in alleviating LPS-induced inflammatory response. Food Res. Int. 2019, 118, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Chen, J.; Ren, G.; Zhang, Y.; Tan, X.; Yang, L. Punicalagin prevents inflammation in LPS-induced RAW 264.7 macrophages by inhibiting FoxO3a/autophagy signaling pathway. Nutrients 2019, 11, 2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canuti, V.; Cecchi, L.; Khatib, M.; Guerrini, L.; Mulinacci, N.; Zanoni, B. A new extract from Pomegranate (Punica granatum L.) by-products as a potential oenological tannin: Preliminary characterization and comparison with existing commercial products. Molecules 2020, 25, 4460. [Google Scholar] [CrossRef]
- Nikfarjam, M.; Rashki Ghaleno, L.; Shahverdi, A.H.; Mirshahvalad, S.H.; Ghoreishi, S.M.; Alizadeh, A.R. Effects of dietary pomegranate peel on antioxidant gene expression and DJ-1 protein abundance in ram testes. Int. J. Fertil. Steril. 2021, 15, 258–262. [Google Scholar]
- Pan, Z.; Qu, W.; Ma, H.; Atungulu, G.G.; McHugh, T.H. Continuous and pulsed ultrasound-assisted extractions of antioxidants from pomegranate peel. Ultrason. Sonochem. 2012, 19, 365–372. [Google Scholar] [CrossRef]
- Laurindo, L.F.; Barbalho, S.M.; Marquess, A.R.; Grecco, A.I.S.; Goulart, R.A.; Tofano, R.J.; Bishayee, A. Pomegranate (Punica granatum L.) and metabolic syndrome risk factors and outcomes: A systematic review of clinical studies. Nutrients 2022, 14, 1665. [Google Scholar] [CrossRef]
- Eghbali, S.; Askari, S.F.; Avan, R.; Sahebkar, A. Therapeutic effects of Punica granatum (Pomegranate): An updated review of clinical trials. J. Nutr. Metab. 2021, 2021, 5297162. [Google Scholar] [CrossRef]
- Xu, W.D.; Lin, H.W.; Qiu, F.; Chen, W.S. Chemical constituents of Duchesnea indica Foche. Shenyang Yaokedaxue Xuebao 2007, 24, 402–406. [Google Scholar]
- Wang, Y.Q.; Si, J.Y.; Liu, X.M.; Tang, J.S.; Chang, Q. Isolation, identification and quantitative determination of ellagic acid and brevifolin carboxylic acid in Duchesnea indica by RP-HPLC. Tianran Chanwu Yanjiuyukaifa 2008, 20, 667–671. [Google Scholar]
- Zhen, G.M.; Wei, X.Y.; Xu, L.X.; Xie, H.H.; Wu, P. Chemical constituents from pericarp of longan fruits. Zhongcaoyao 2011, 42, 1485–1489. [Google Scholar]
- Zhao, Y.X.; Yang, D.; Ma, Q.Y.; Li, N.; Cheng, Z.Q.; Zhou, J. Chemical constituents from Osbeckia chinensis. Zhongcaoyao 2011, 42, 1061–1065. [Google Scholar]
- Pfundstein, B.; El Desouky, S.K.; Hull, W.E.; Haubner, R.; Erben, G.; Owen, R.W. Polyphenolic compounds in the fruits of Egyptian medicinal plants (Terminalia bellerica, Terminalia chebula and Terminalia horrida): Characterization, quantitation and determination of antioxidant capacities. Phytochemistry 2010, 71, 1132–1148. [Google Scholar] [CrossRef]
- Rocha, S.H.; Silva, C.C.M.; Caland, N.L.B.; Lopes, J.A.D.; Cito, A.M.; Chaves, M.H. Chemical constituents from bark of Cenostigma macrophyllum: Cholesterol occurrence. Quim. Nova 2007, 30, 1877–1881. [Google Scholar]
- Li, X.Y.; Wang, Y.H.; Wang, H.S.; Shi, Y.N.; Long, C.L. Phenolic derivatives from the leaves of Dipteronia dyeriana. Tianran Chanwu Yanjiuyukaifa 2010, 22, 5–10. [Google Scholar]
- Jossang, A.; Pousset, J.L.; Bodo, B. Comberglutinin, a hydrolysable tannin from Combertum glutinosum. J. Nat. Prod. 1994, 57, 732–737. [Google Scholar] [CrossRef]
- Ito, H.; Li, P.; Koreishi, M.; Nagatomo, A.; Nishida, N.; Yoshida, T. Ellagitannin oligomers and a neolignan from pomegranate arils and their inhibitory effects on the formation of advanced glycation end products. Food Chem. 2014, 152, 323–330. [Google Scholar] [CrossRef]
- Tanaka, T.; Nonaka, G.C.; Itsuo, N. Tannins and related compounds. XLI. Isolation and characterization of novel ellagitannins, punicacorteins A, B, C and D, and punigluconin from the bark of Punica granatum L. Chem. Pharm. Bull. 1986, 34, 656–663. [Google Scholar] [CrossRef]
- Chen, X.W.; Li, C.W.; Cui, C.B.; Hua, W.; Zhu, T.J.; Gu, Q.Q. Nine new and five known polyketides derived from a deep sea-sourced Aspergillus sp. 16-02-1. Mar. Drugs 2014, 12, 3116–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.N.; Su, B.J.; Wang, Y.Q.; Liao, H.B.; Chen, Z.F.; Liang, D. Isolation, absolute configuration, and biological activities of chebulic acid and brevifolincarboxylic acid derivatives from Euphorbia hirta. J. Nat. Prod. 2020, 83, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nonaka, J.; Nishioka, I. Tannins and related compounds. XLII. Isolation and characterization of four new hydrolyzable tannins, terflavins A and B, tergallagin and tercatain from the leaves of Terminalia catappa L. Chem. Pharm. Bull. 1986, 34, 1039–1049. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Tong, H.H.; Xu, Y.M.; Ishimaru, K.; Nonaka, J.; Nishioka, I. Tannins and related compounds. CXVII. Isolation and characterization of three new ellagitannins, lagerstannins A, B and C, having a gluconic acid core, from Lagerstroemia speciosa (L.) Pers. Chem. Pharm. Bull. 1992, 40, 2975–2980. [Google Scholar] [CrossRef] [Green Version]
- Abiodun, O.O.; Rodríguez-Nogales, A.; Algieri, F.; Gomez-Caravaca, A.M.; Segura-Carretero, A.; Utrilla, M.P.; Rodriguez-Cabezas, M.E.; Galvez, J. Antiinflammatory and immunomodulatory activity of an ethanolic extract from the stem bark of Terminalia catappa L. (Combretaceae): In vitro and in vivo evidences. J. Ethnopharmacol. 2016, 192, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, C.; Guo, L.; Chen, Y.; Han, L.; Liu, E.; Gao, X.; Wang, T. Triglyceride accumulation inhibitory effects of phenylpropanoid glycosides from Boschniakia rossica Fedtsch et Flerov. Fitoterapia 2013, 85, 69–75. [Google Scholar] [CrossRef]
- Tanaka, T.; Nonaka, J.; Nishioka, I. Tannins and related compounds. XL. Revision of the structures of punicalin and punicalagin, and isolation and characterization of 2-O-galloylpunicalin from the bark of Punica granatum L. Chem. Pharm. Bull. 1986, 34, 650–655. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.M.; Kim, J.E.; Min, S.K.; Kim, K.H.; Han, S.J.; Yim, J.H.; Park, H.; Kim, J.H.; Kim, I.C. Anti-inflammatory effects of antarctic lichen Umbilicaria antarctica methanol extract in lipopolysaccharide-stimulated RAW 264.7 macrophage cells and zebrafish model. Biomed. Res. Int. 2021, 2021, 8812090. [Google Scholar] [CrossRef]
- Jayakumar, T.; Yang, C.M.; Yen, T.L.; Hsu, C.Y.; Sheu, J.R.; Hsia, C.W.; Manubolu, M.; Huang, W.C.; Hsieh, C.Y.; Hsia, C.H. Anti-inflammatory mechanism of an alkaloid rutaecarpine in LTA-stimulated RAW 264.7 cells: Pivotal role on NF-κB and ERK/p38 signaling molecules. Int. J. Mol. Sci. 2022, 23, 5889. [Google Scholar] [CrossRef]
- Yong, H.Y.; Koh, M.S.; Moon, A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert. Opin. Investig. Drugs 2009, 18, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Corsino, P.; Parker, N.T.; Law, B.K. Identification of a small molecule inhibitor of serine 276 phosphorylation of the p65 subunit of NF-kappaB using in silico molecular docking. Cancer Lett. 2010, 291, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.M.; Kim, Y.S.; Lim, J.Y.; Min, S.J.; Ko, H.C.; Kim, S.J.; Kim, Y. Intestinal anti-inflammatory activity of Sasa quelpaertensis leaf extract by suppressing lipopolysaccharide-stimulated inflammatory mediators in intestinal epithelial Caco-2 cells co-cultured with RAW 264.7 macrophage cells. Nutr. Res. Pract. 2015, 9, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, I.N.; Chang, A.S.; Teng, C.M.; Chen, C.C.; Yang, C.R. Aciculatin inhibits lipopolysaccharide-mediated inducible nitric oxide synthase and cyclooxygenase-2 expression via suppressing NF-κB and JNK/p38 MAPK activation pathways. J. Biomed. Sci. 2011, 18, 28. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-kappaB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596. [Google Scholar] [CrossRef]
- Yan, F.; Cheng, D.; Wang, H.; Gao, M.; Zhang, J.; Cheng, H.; Wang, C.; Zhang, H.; Xiong, H. Corilagin ameliorates con A-induced hepatic injury by restricting M1 macrophage polarization. Front. Immunol. 2022, 13, 807509. [Google Scholar] [CrossRef]
- Shen, R.; Yin, P.; Yao, H.; Chen, L.; Chang, X.; Li, H.; Hou, X. Punicalin ameliorates cell pyroptosis induced by LPS/ATP through suppression of ROS/NLRP3 pathway. J. Inflamm. Res. 2021, 14, 711–718. [Google Scholar] [CrossRef]
- Yan, H.; Gulimire, A.; Zhu, J.; Liang, H.; Gu, Z. Punicalagin suppresses LPS-induced inflammatory responses in murine macrophages via JAK/STAT signaling pathway and zymosan-induced mice paw edema. Indian. J. Pharm. Educ. Res. 2021, 55, 550–555. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | NRC (%) | No. | NRC (%) | No. | NRC (%) |
|---|---|---|---|---|---|
| Normal | 4.0 ± 0.9 | 6 | 85.9 ± 3.2 *** | 14 | 88.2 ± 4.9 *** |
| Control | 100 ± 2.9 | 7 | 84.1 ± 3.0 *** | 15 | 99.2 ± 2.2 |
| Dex | 83.2 ± 2.8 *** | 8 | 80.2 ± 3.6 *** | 16 | 88.7 ± 1.8 *** |
| 1 | 93.4 ± 3.7 * | 9 | 97.6 ± 3.0 | 17 | 90.2 ± 1.4 *** |
| 2 | 101.4 ± 1.3 | 10 | 81.9 ± 4.1 *** | 18 | 74.8 ± 2.8 *** |
| 3 | 94.4 ± 2.5 * | 11 | 91.7 ± 1.8 *** | 19 | 71.0 ± 1.3 *** |
| 4 | 100.1 ± 1.9 | 12 | 100.6 ± 2.5 | 20 | 71.1 ± 2.0 *** |
| 5 | 91.3 ± 3.3 ** | 13 | 101.6 ± 2.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.-M.; Kouye, O.; Yang, D.-S.; Zhang, Y.-Q.; Ruan, J.-Y.; Han, L.-F.; Zhang, Y.; Wang, T. Polyphenols from the Peels of Punica granatum L. and Their Bioactivity of Suppressing Lipopolysaccharide-Stimulated Inflammatory Cytokines and Mediators in RAW 264.7 Cells via Activating p38 MAPK and NF-κB Signaling Pathways. Molecules 2022, 27, 4622. https://doi.org/10.3390/molecules27144622
Li H-M, Kouye O, Yang D-S, Zhang Y-Q, Ruan J-Y, Han L-F, Zhang Y, Wang T. Polyphenols from the Peels of Punica granatum L. and Their Bioactivity of Suppressing Lipopolysaccharide-Stimulated Inflammatory Cytokines and Mediators in RAW 264.7 Cells via Activating p38 MAPK and NF-κB Signaling Pathways. Molecules. 2022; 27(14):4622. https://doi.org/10.3390/molecules27144622
Chicago/Turabian StyleLi, Hui-Min, Ongher Kouye, Ding-Shan Yang, Ya-Qi Zhang, Jing-Ya Ruan, Li-Feng Han, Yi Zhang, and Tao Wang. 2022. "Polyphenols from the Peels of Punica granatum L. and Their Bioactivity of Suppressing Lipopolysaccharide-Stimulated Inflammatory Cytokines and Mediators in RAW 264.7 Cells via Activating p38 MAPK and NF-κB Signaling Pathways" Molecules 27, no. 14: 4622. https://doi.org/10.3390/molecules27144622
APA StyleLi, H.-M., Kouye, O., Yang, D.-S., Zhang, Y.-Q., Ruan, J.-Y., Han, L.-F., Zhang, Y., & Wang, T. (2022). Polyphenols from the Peels of Punica granatum L. and Their Bioactivity of Suppressing Lipopolysaccharide-Stimulated Inflammatory Cytokines and Mediators in RAW 264.7 Cells via Activating p38 MAPK and NF-κB Signaling Pathways. Molecules, 27(14), 4622. https://doi.org/10.3390/molecules27144622