
Stereoselective Processes Based on σ-Hole Interactions



Abstract
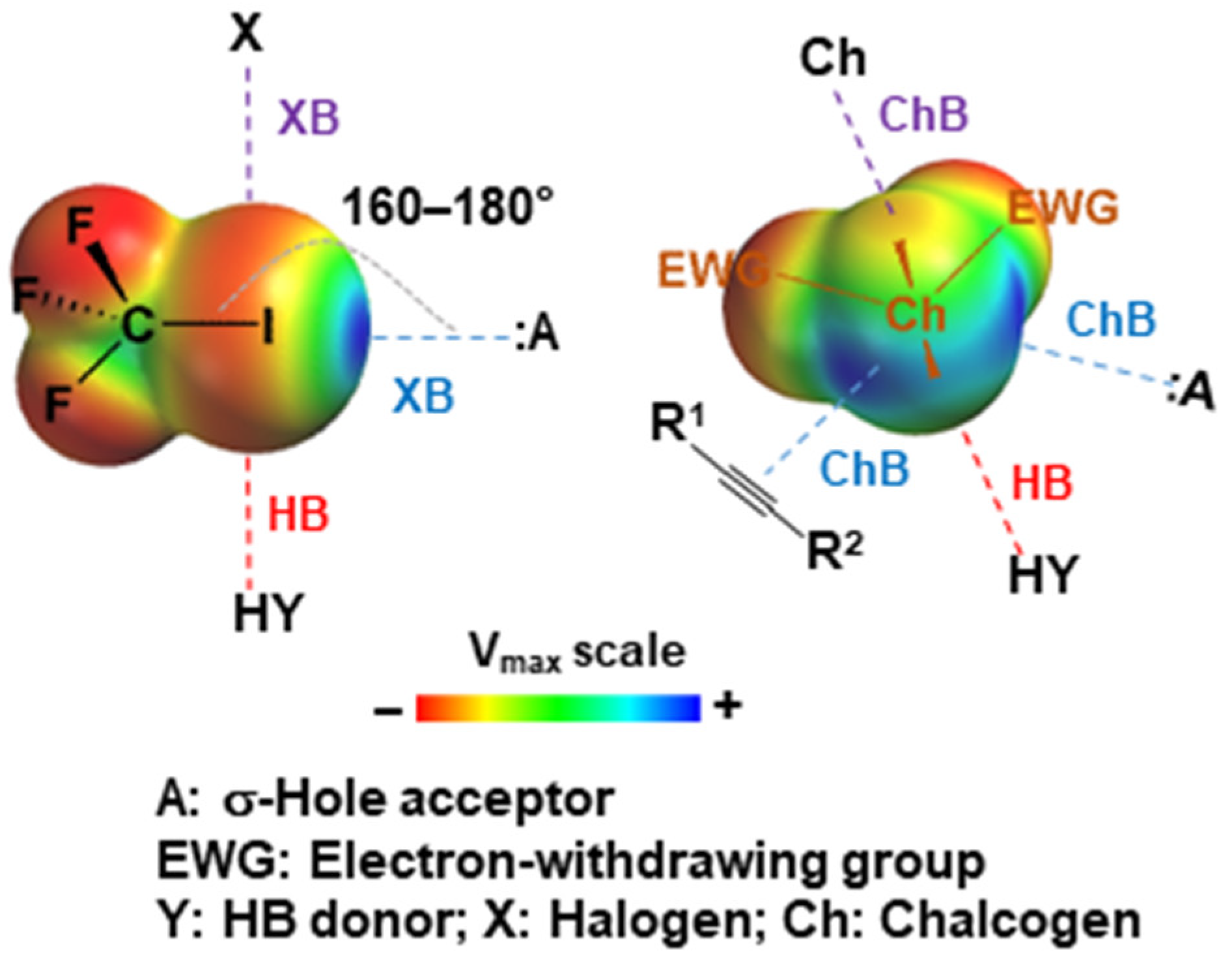
1. Introduction
2. Chiral Supramolecular Assemblies
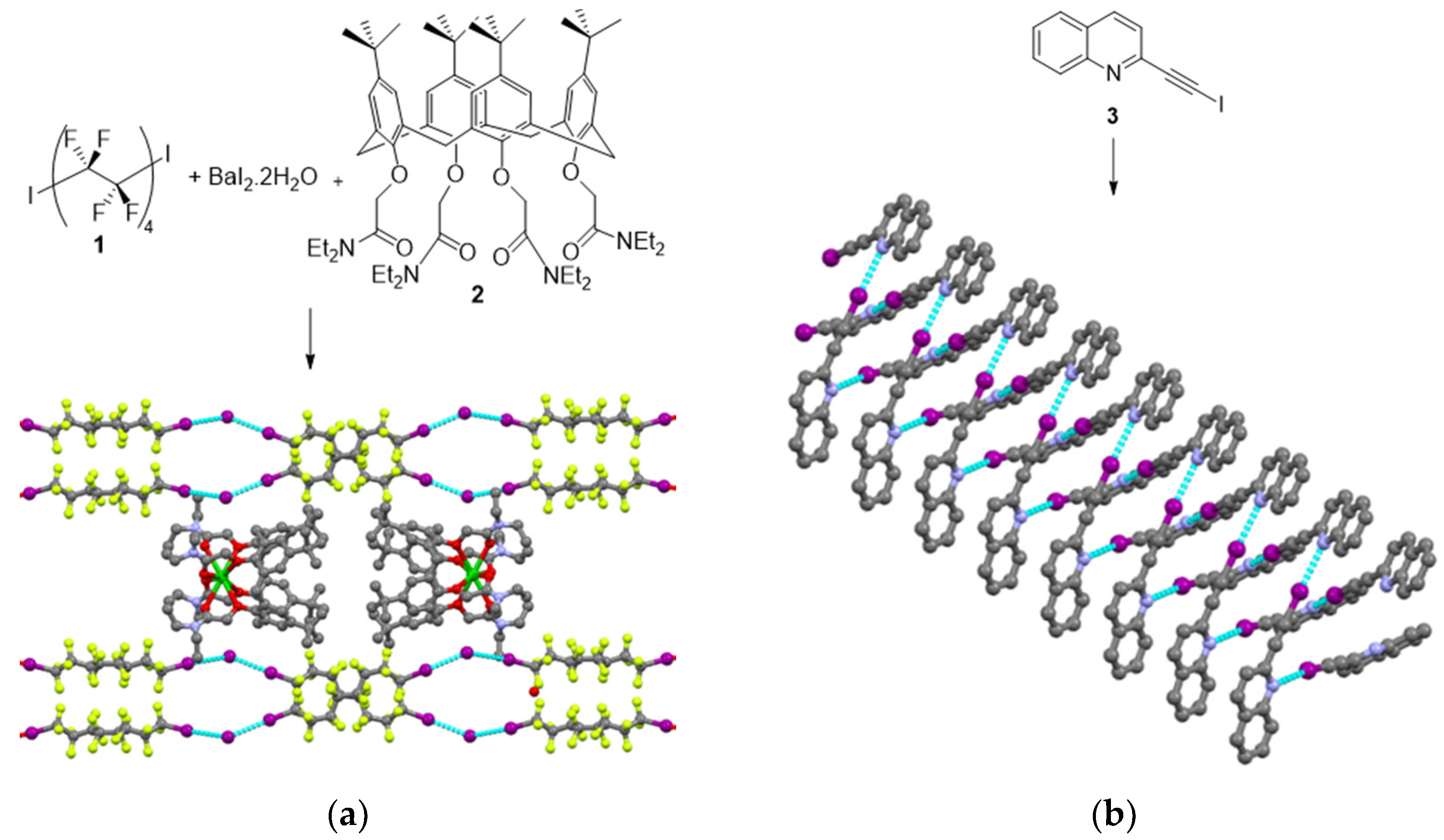
2.1. Spontaneous Resolution
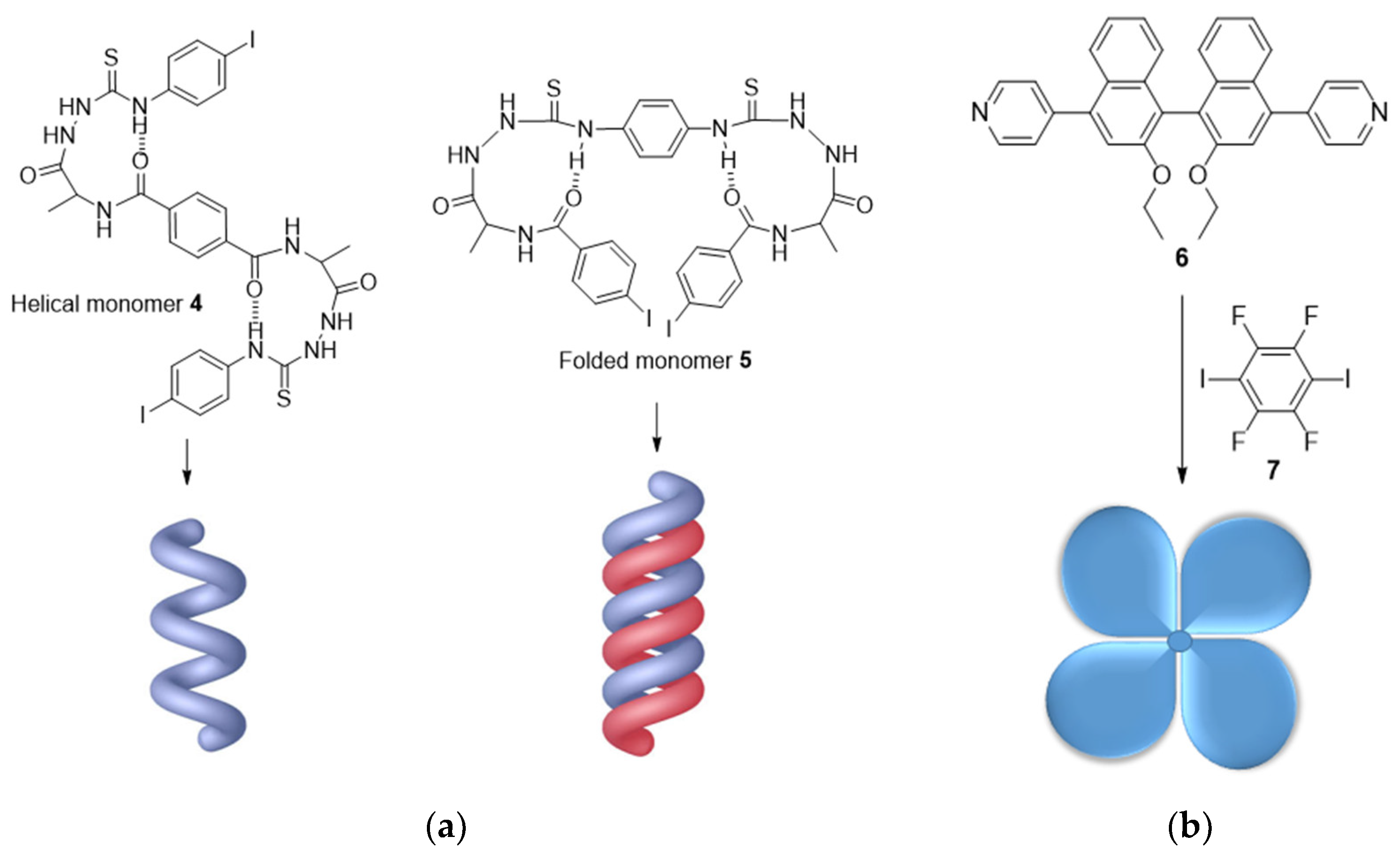
2.2. Chiral Amplification
3. Enantioseparation
3.1. Enantioseparations Based on XB
3.1.1. Initial Observations on the Multiple Role of Halogens in Enantioseparation
3.1.2. Enantioseparations Based on Diastereomeric Salt Formation
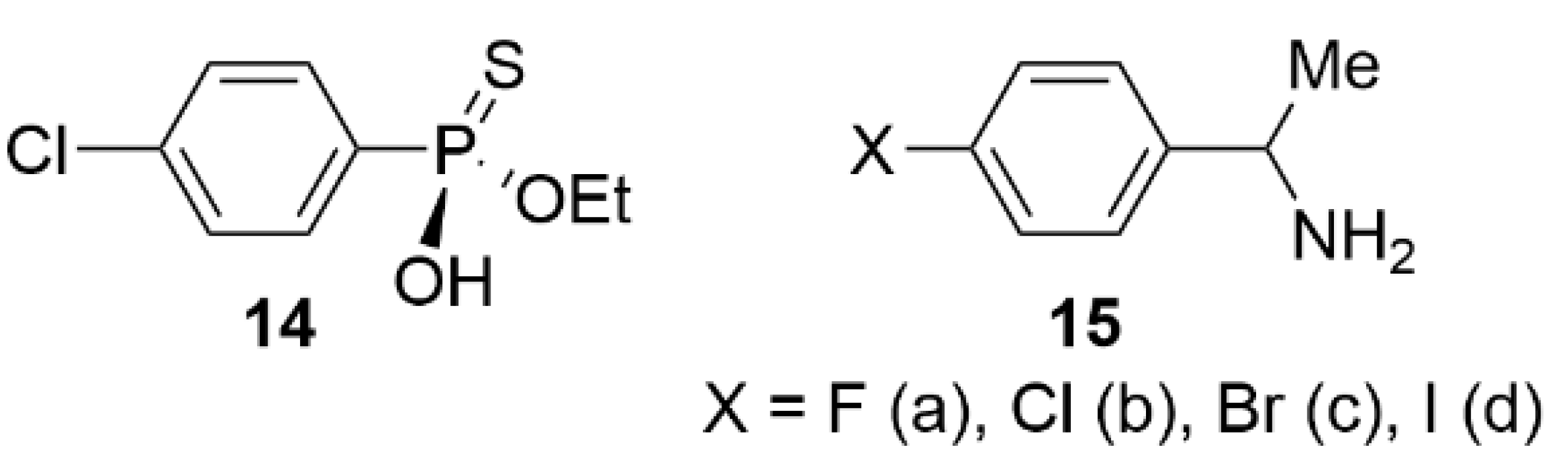
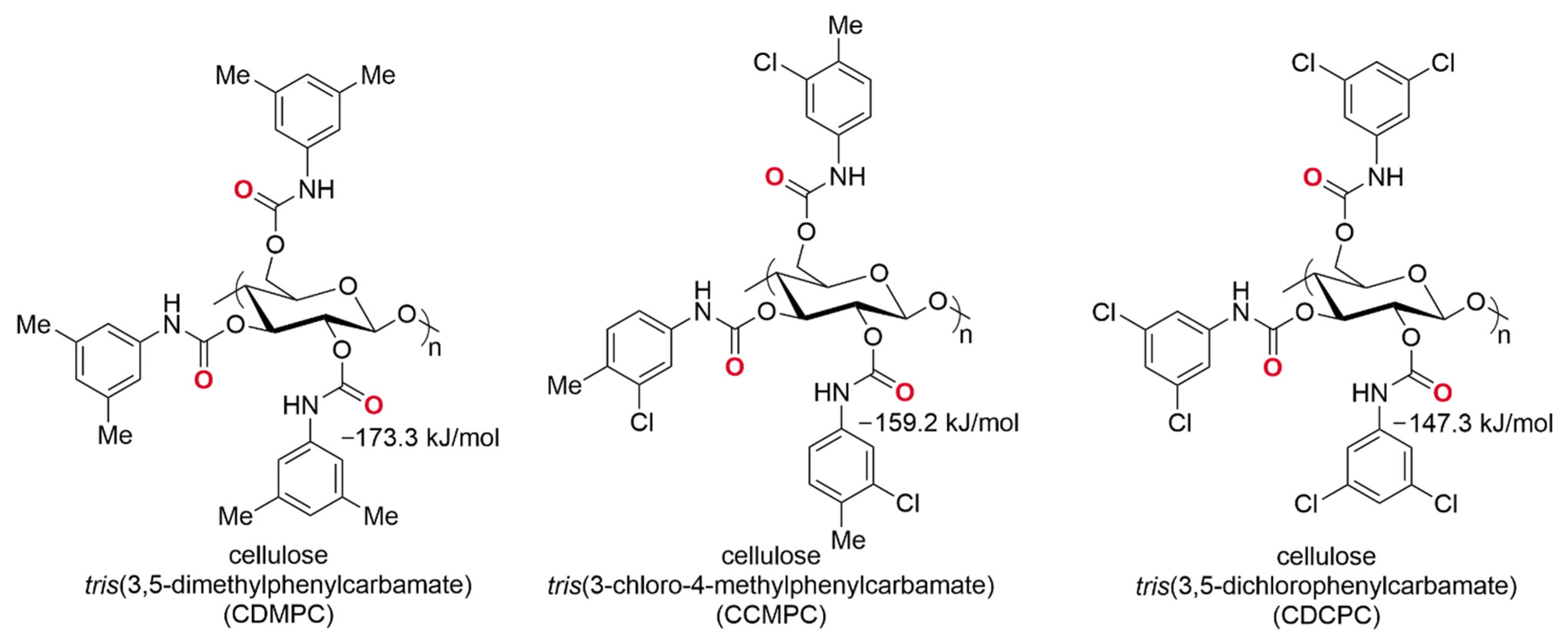
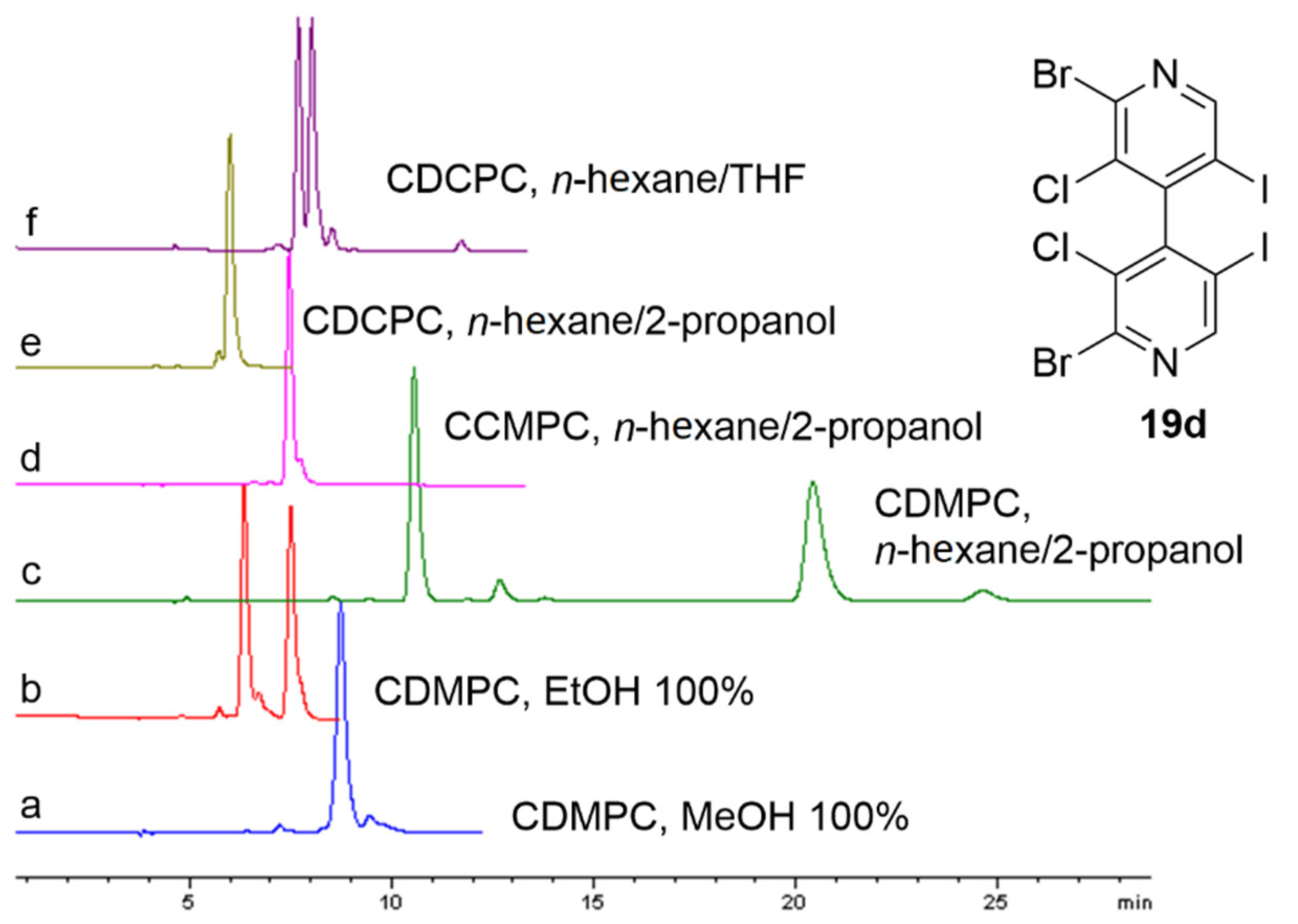
3.1.3. HPLC Enantioseparations
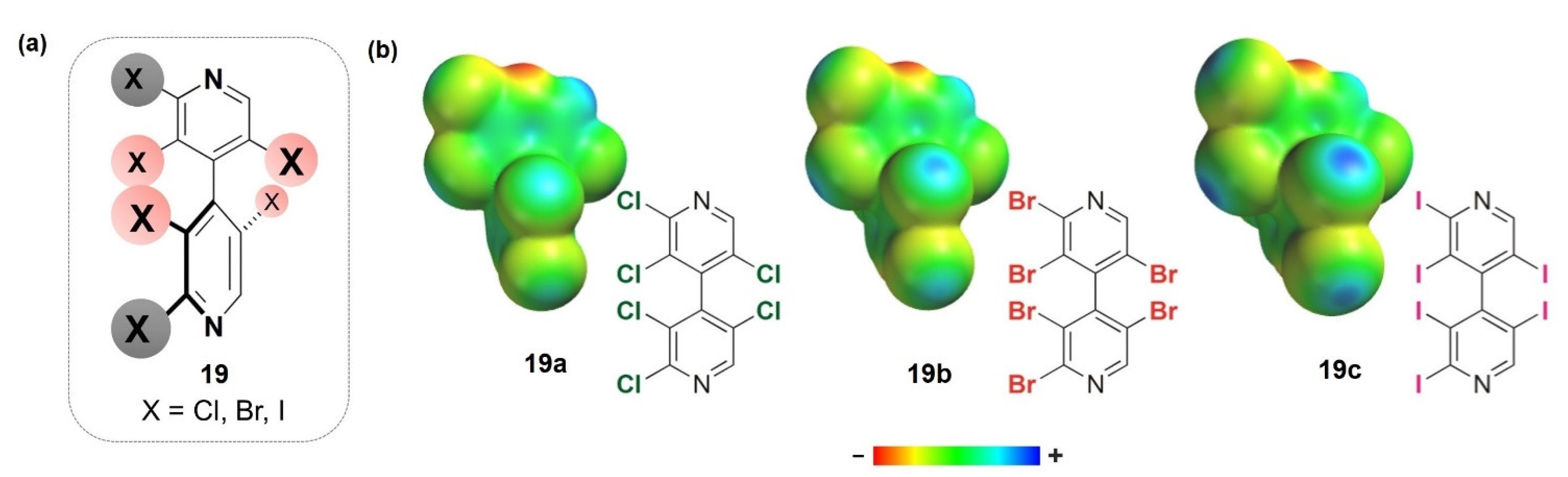
- Enantioseparation of atropisomeric 4,4′-bipyridines based on XB
- 2.
- Enantioseparation of planar chiral ferrocenes based on XB
- 3.
- Miscellaneous
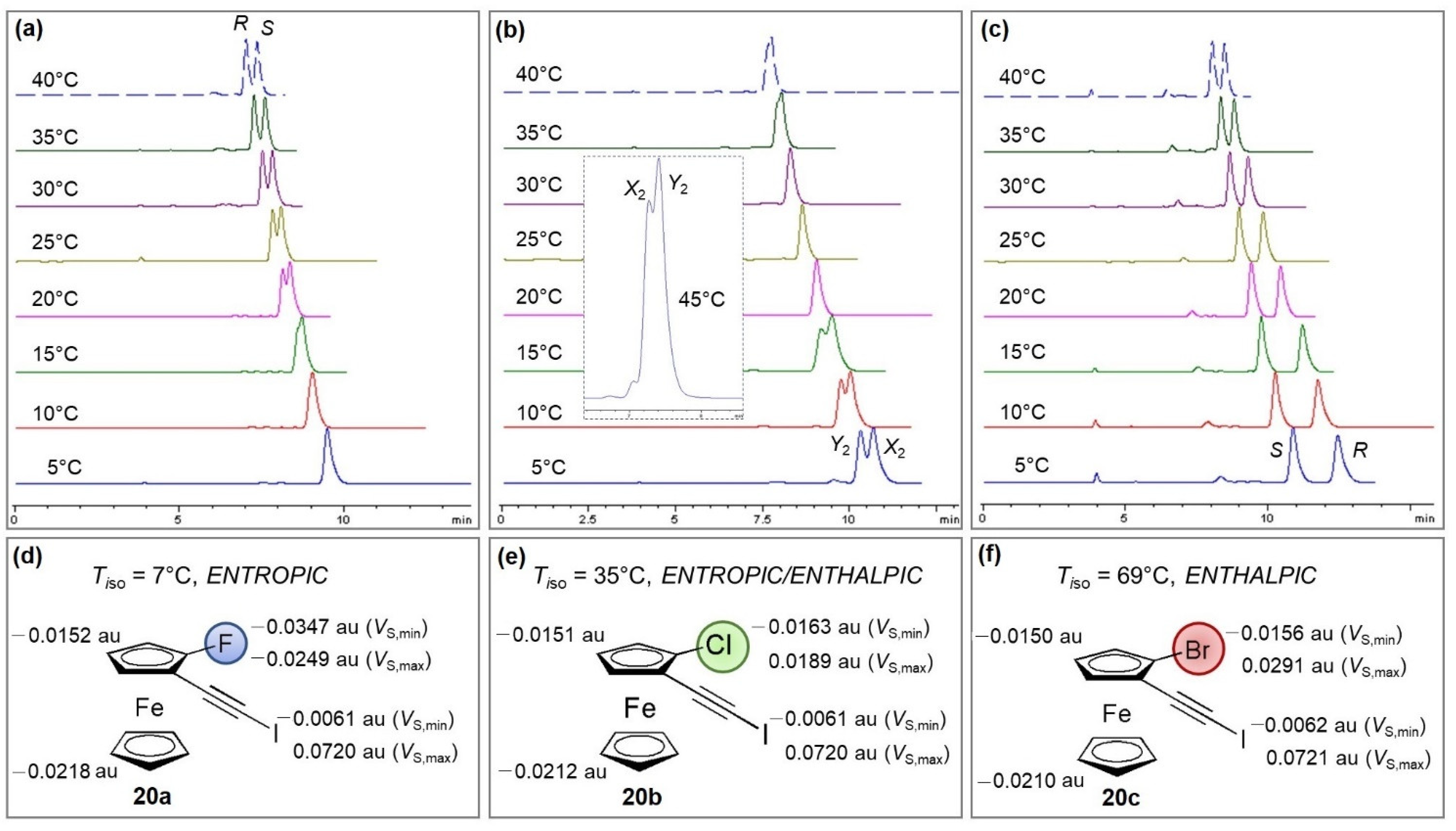
3.1.4. Supercritical Fluid Chromatography (SFC) Enantioseparations
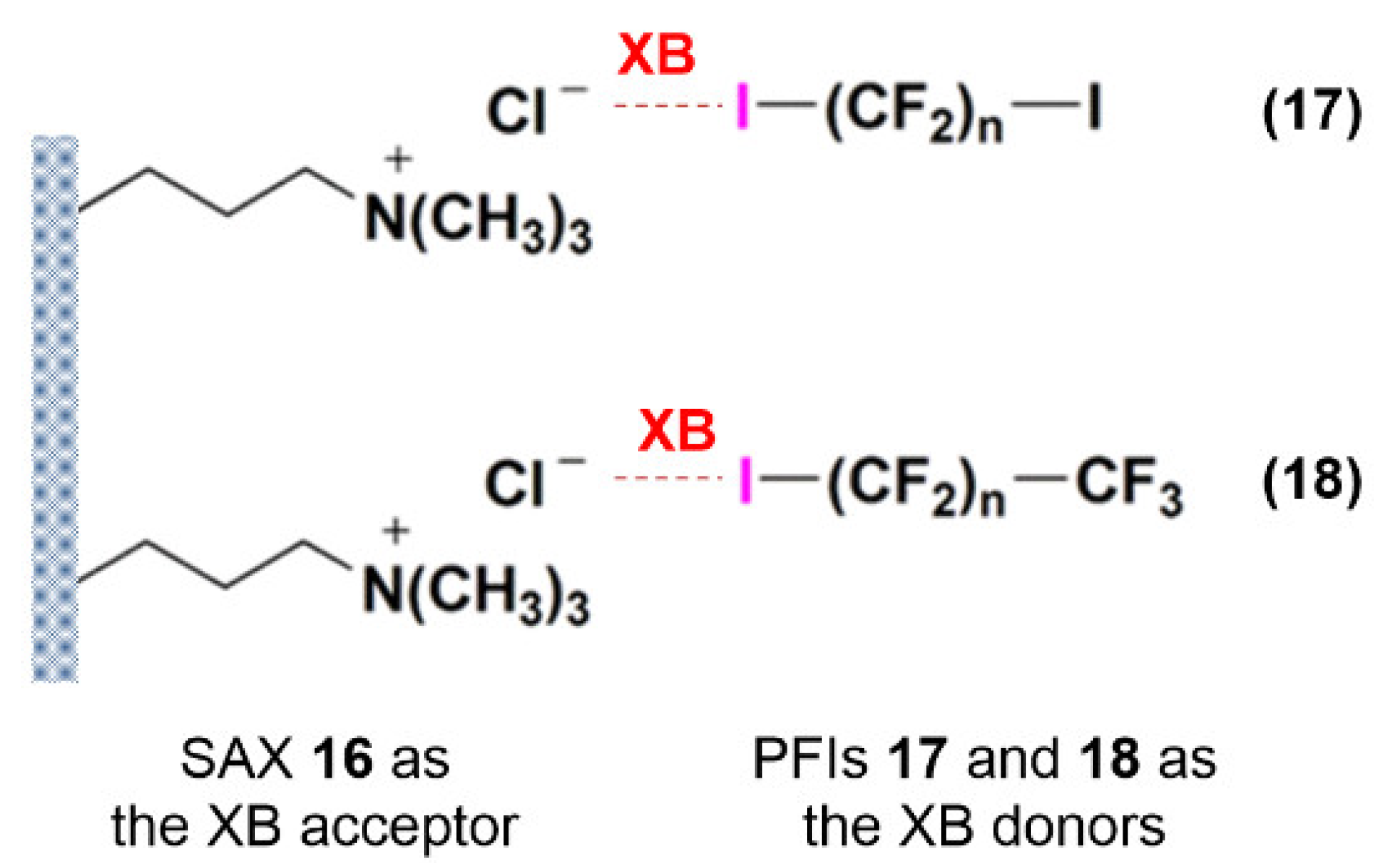
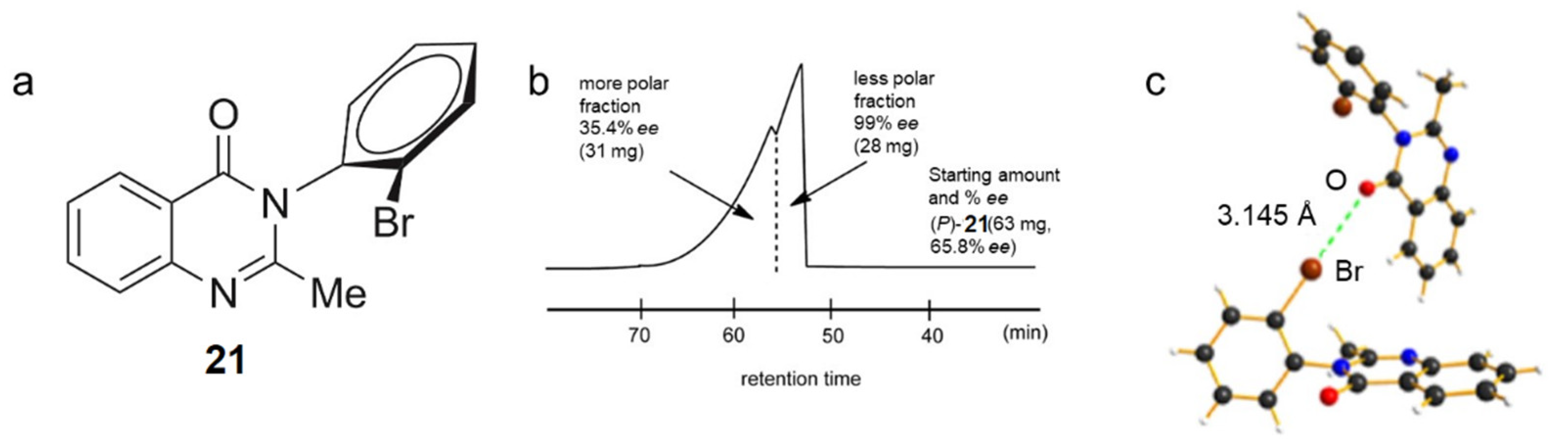
3.1.5. Self-Disproportionation of Enantiomers (SDE)
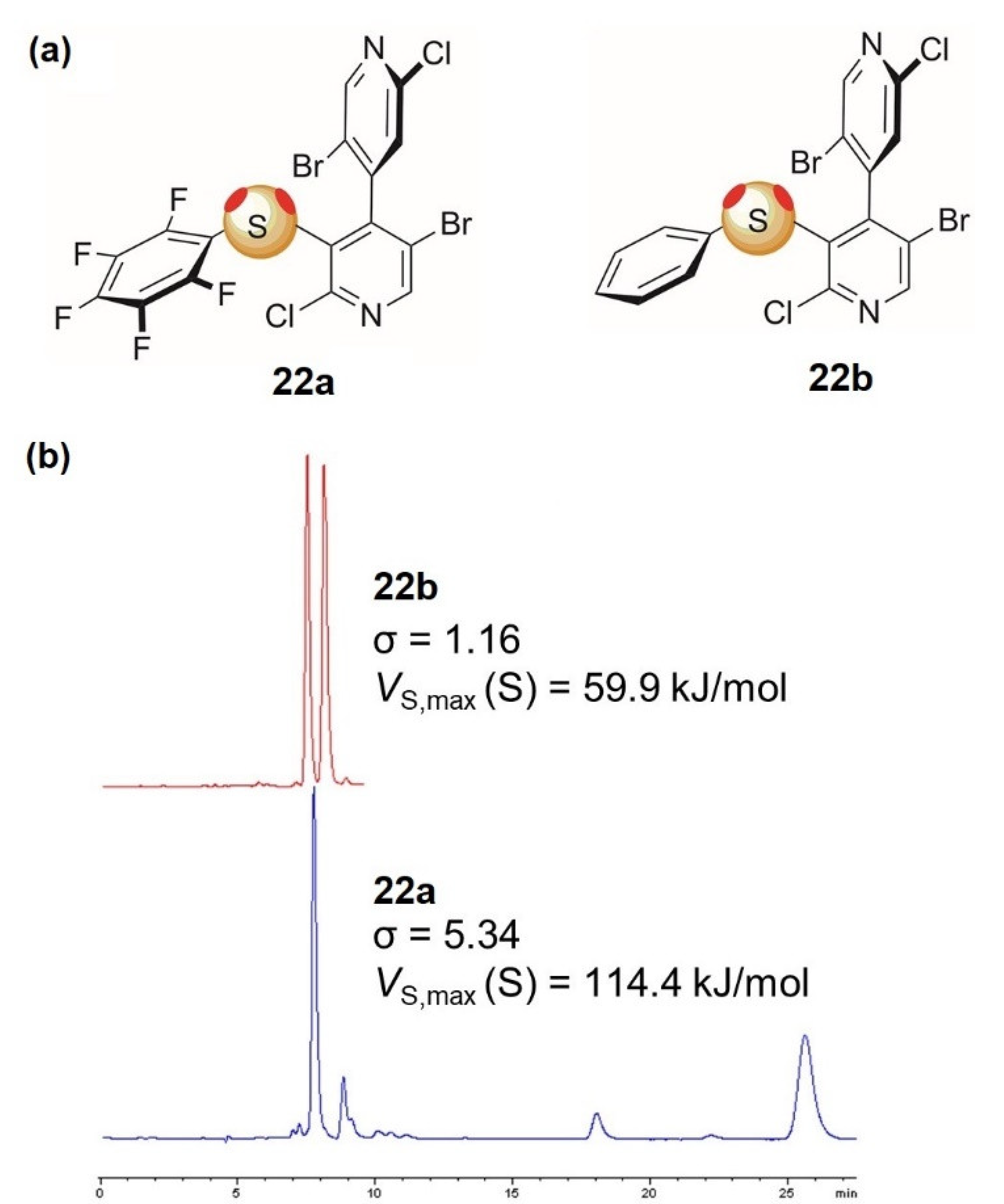
3.2. Enantioseparations Based on ChB
4. Enantioselective Complexation
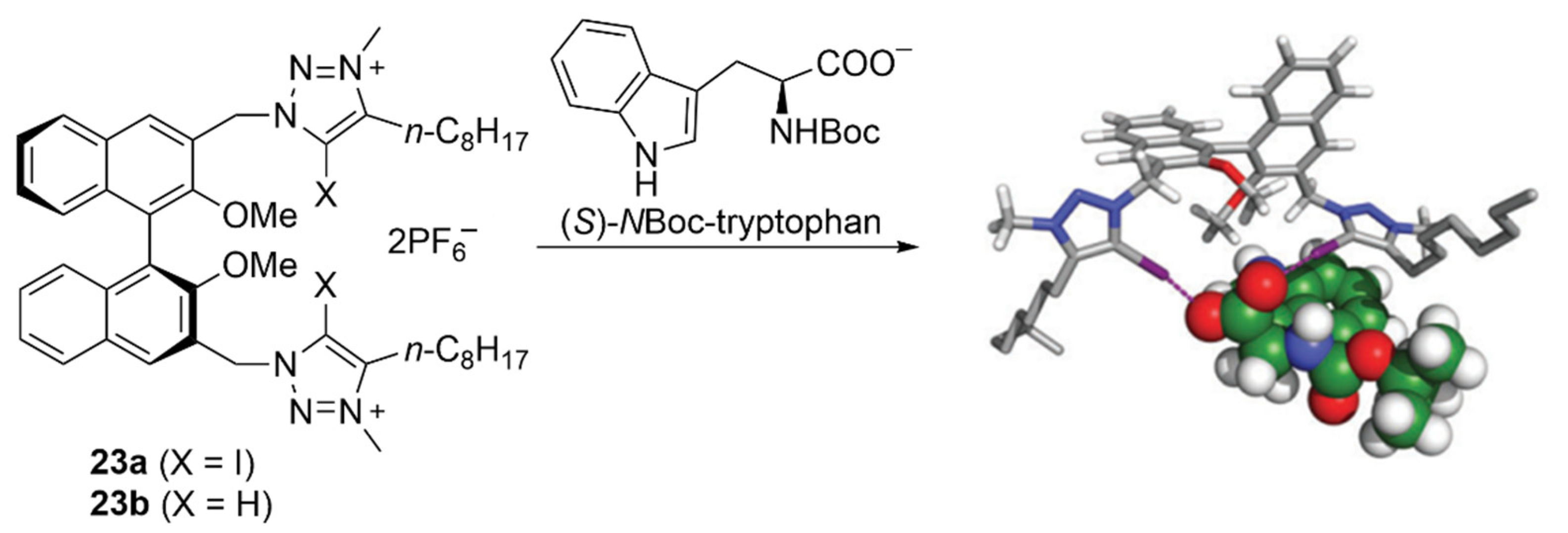
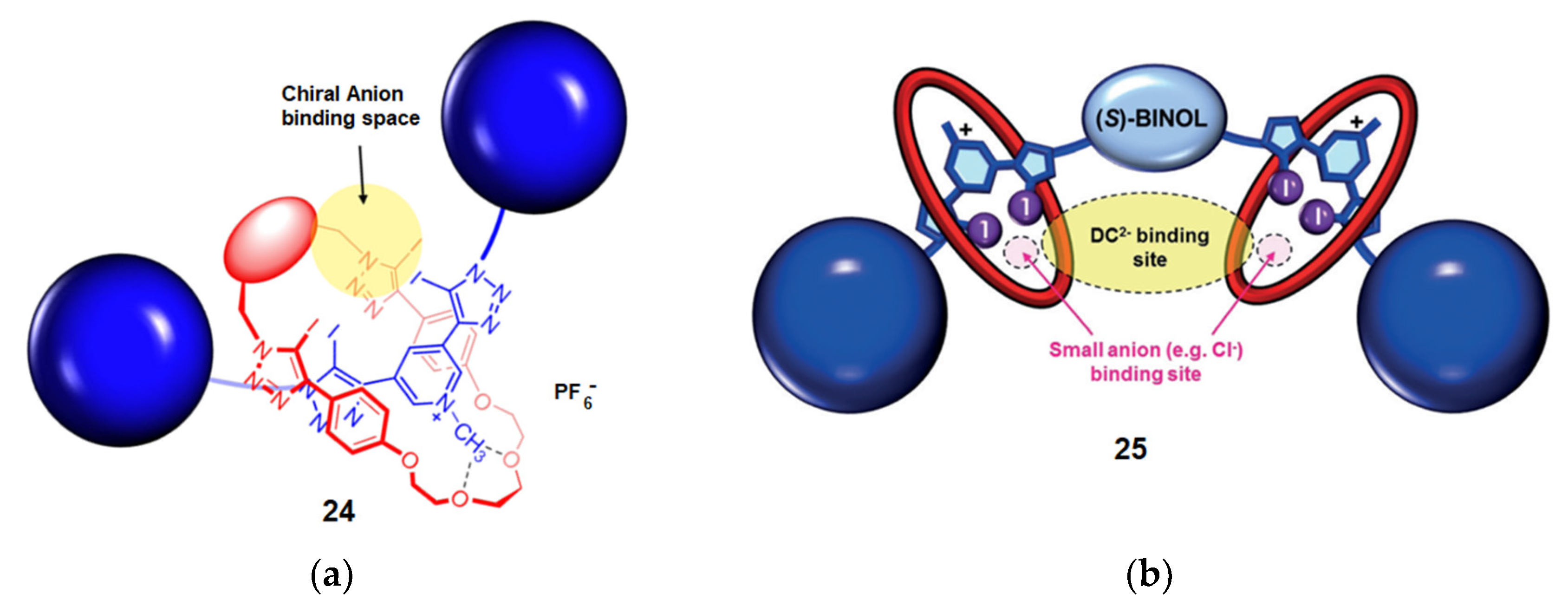
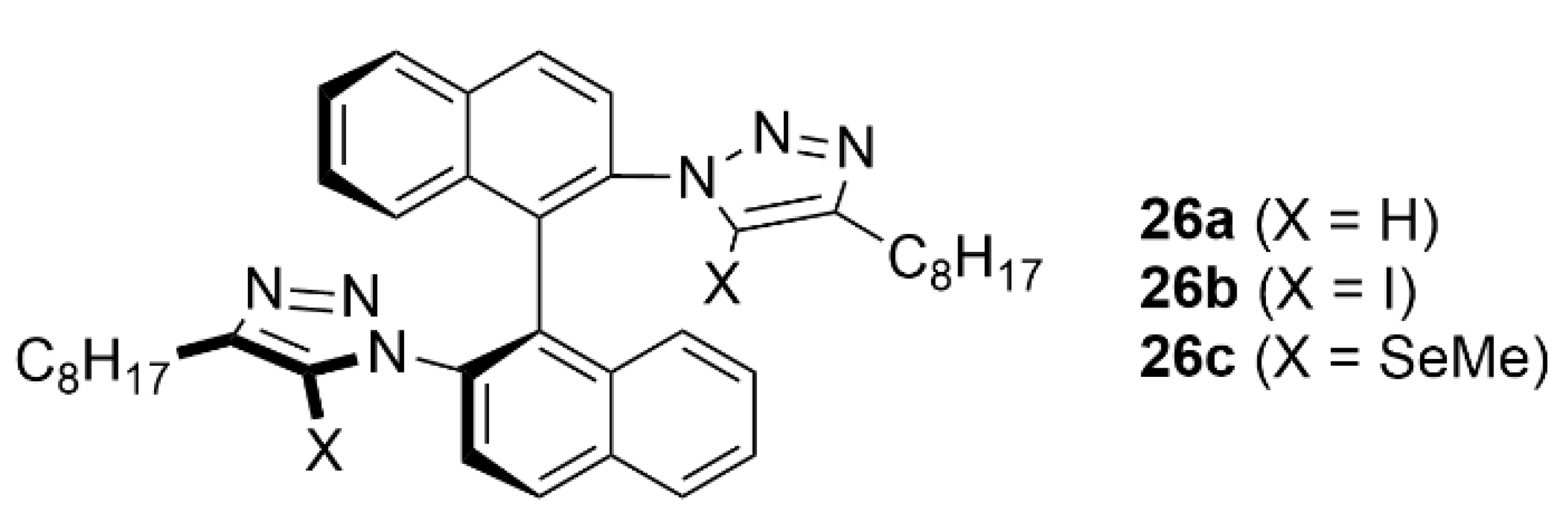
4.1. Recognition of Anionic Guests
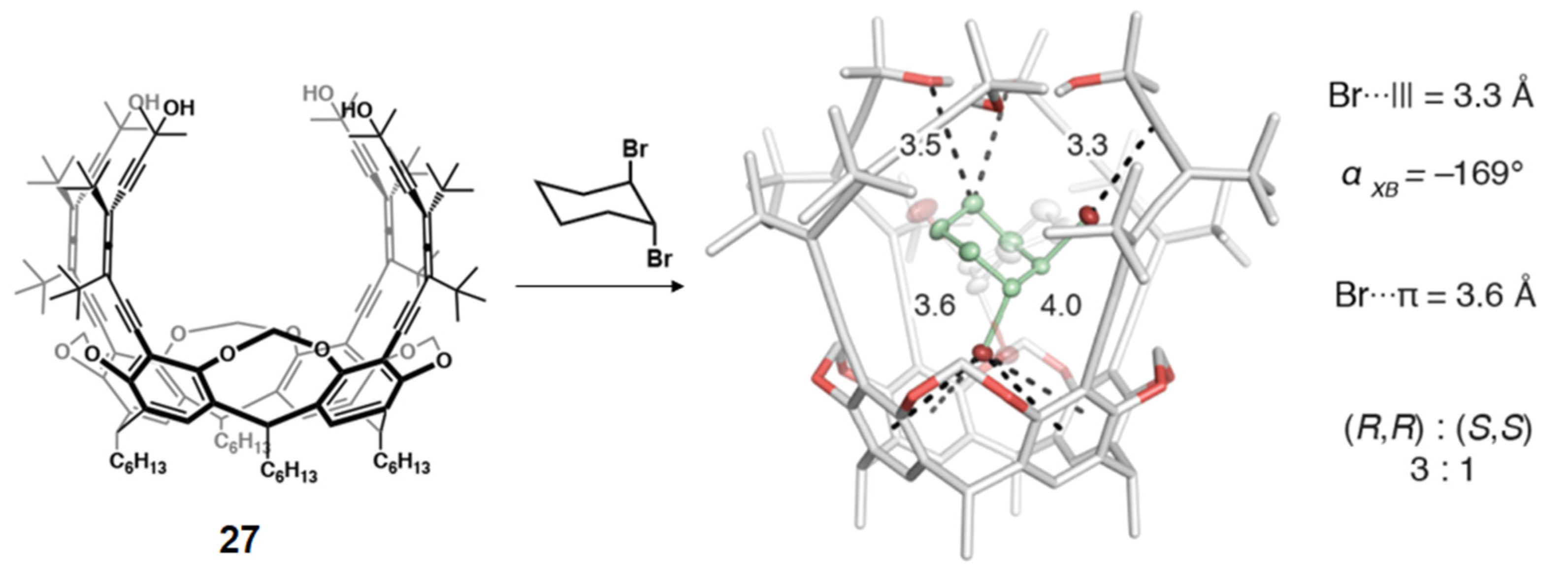
4.2. Recognition of Neutral Guests
5. Asymmetric Catalysis
5.1. XB-Based Catalyzed Asymmetric Reactions
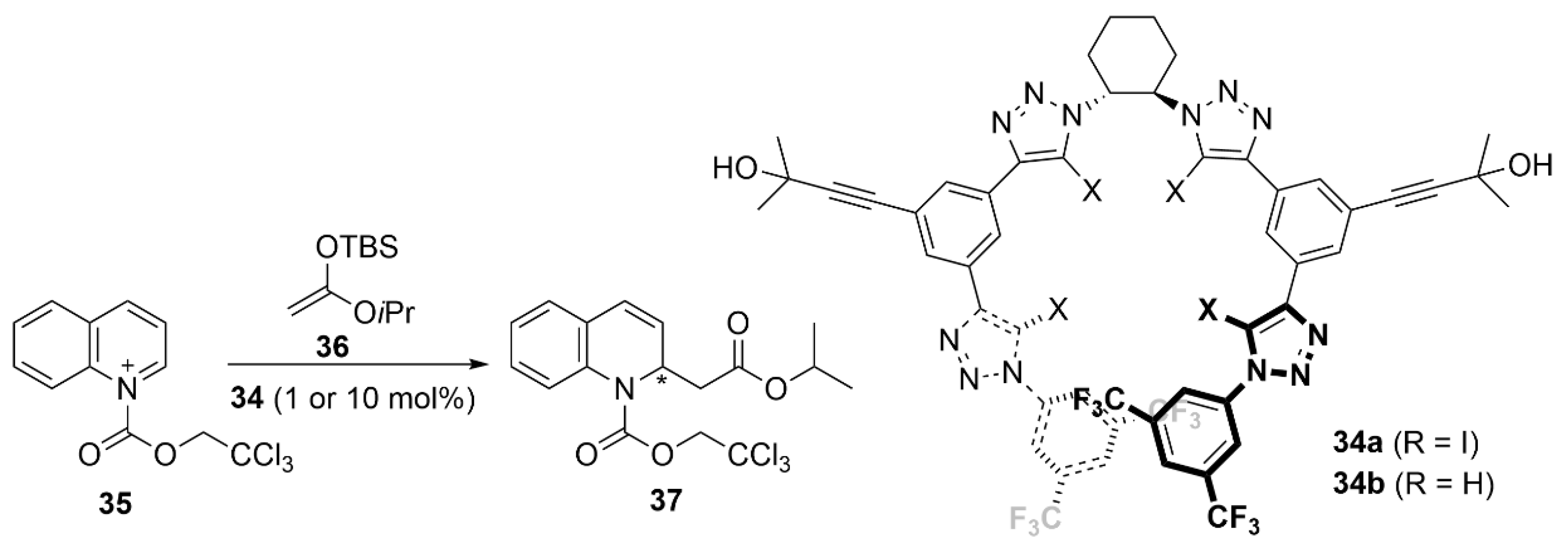
5.2. PnB-Based Catalyzed Asymmetric Reactions
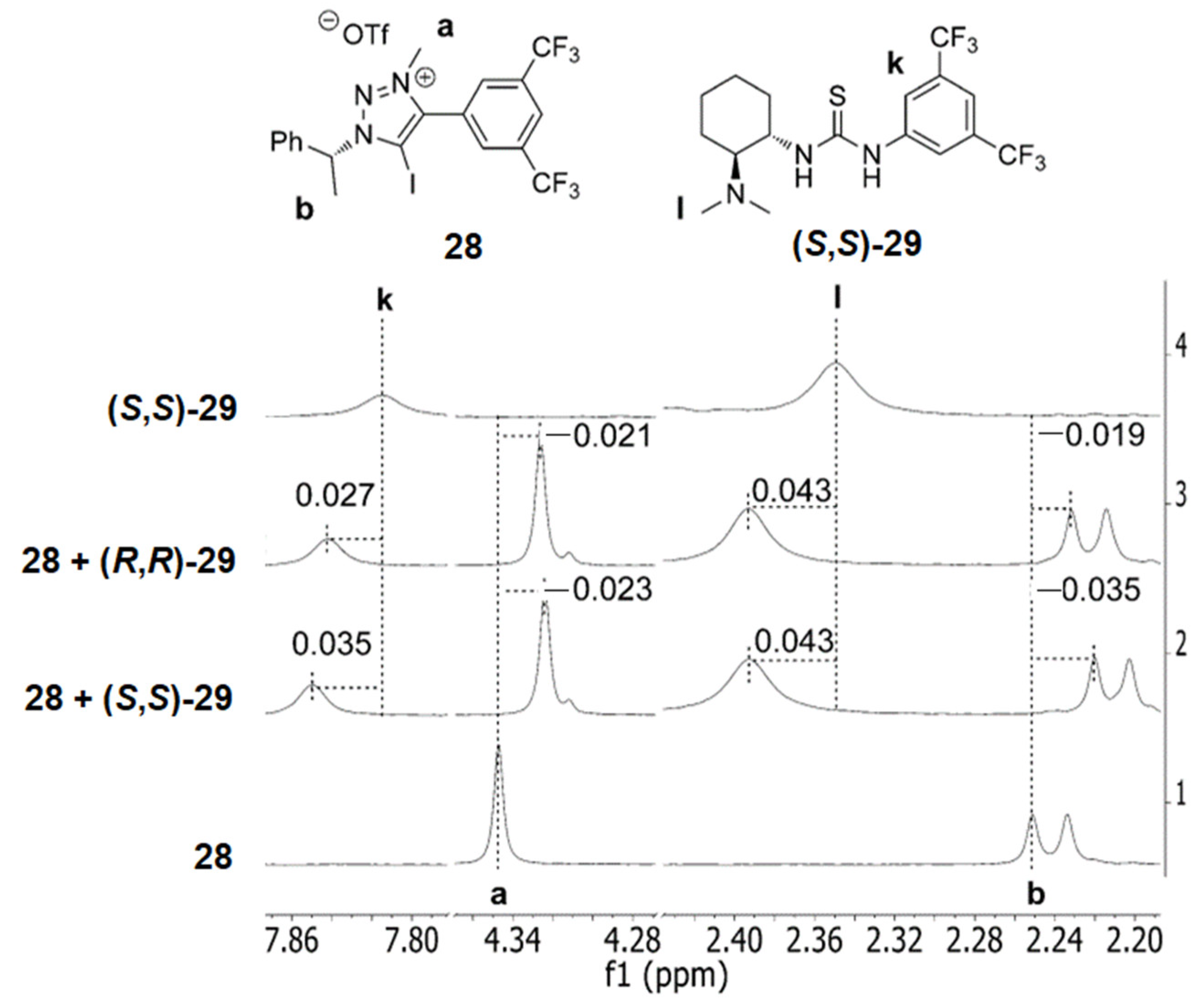
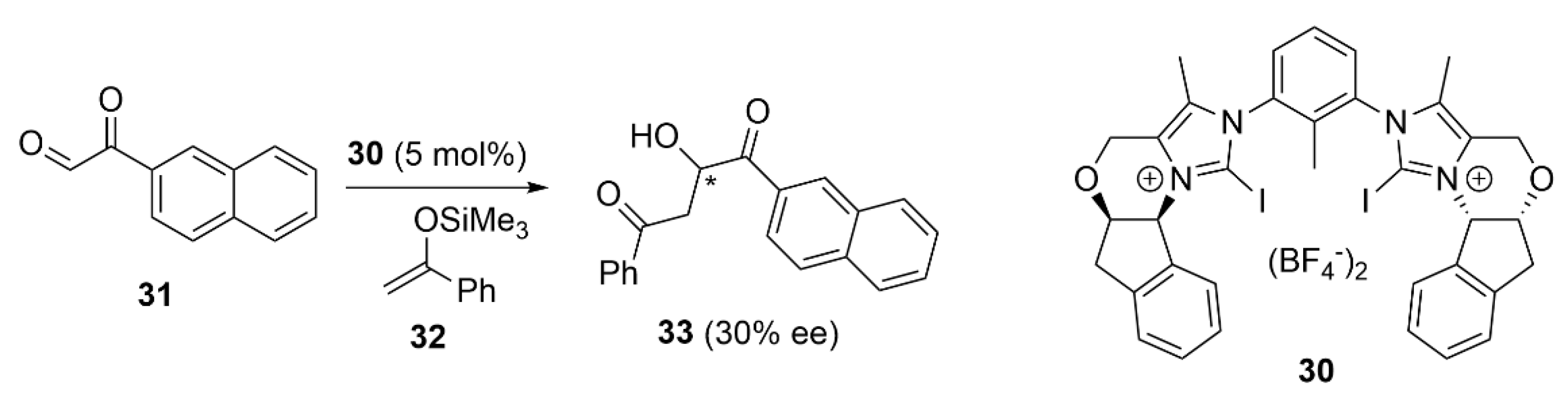
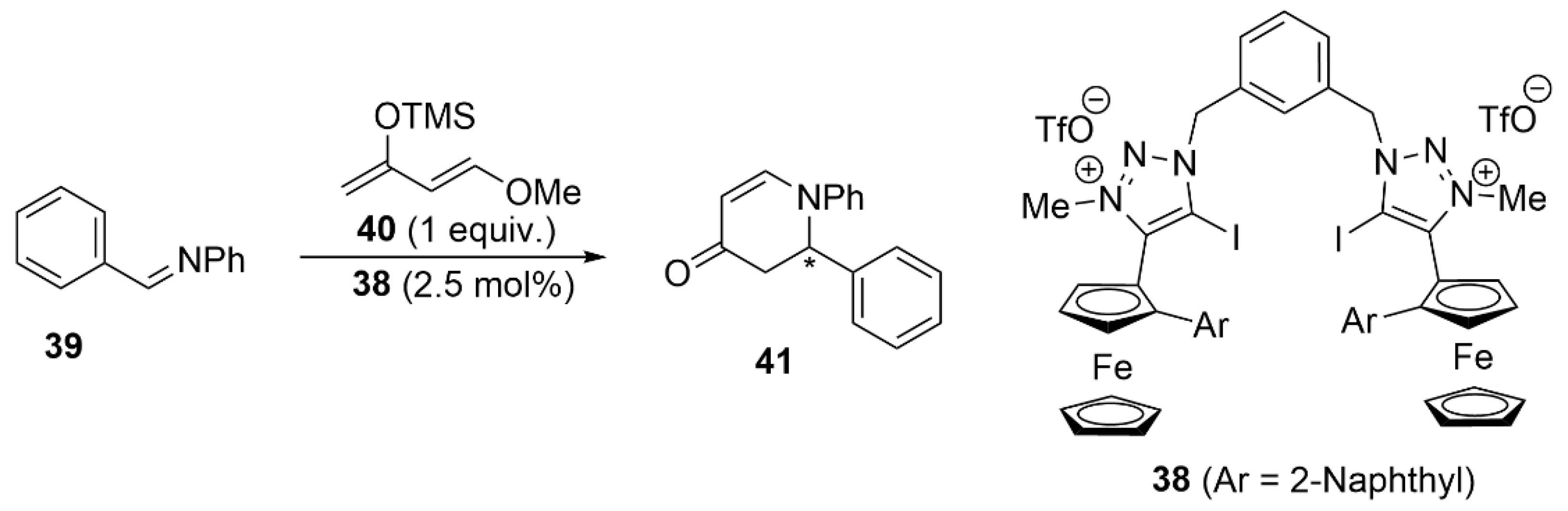
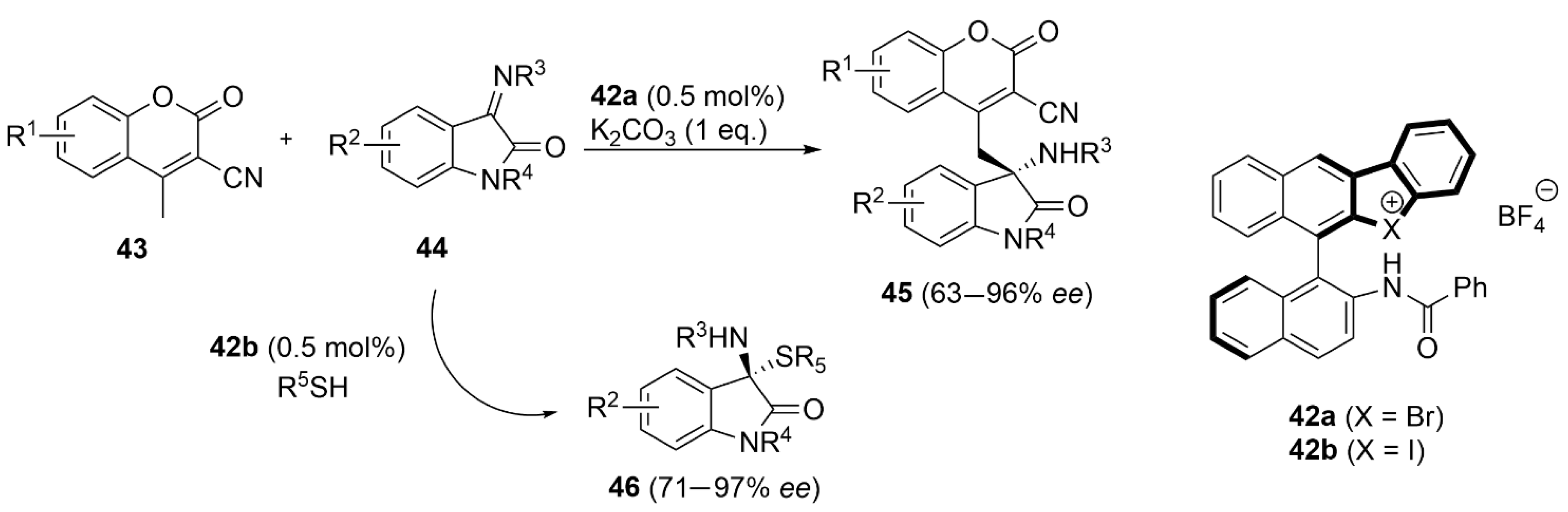
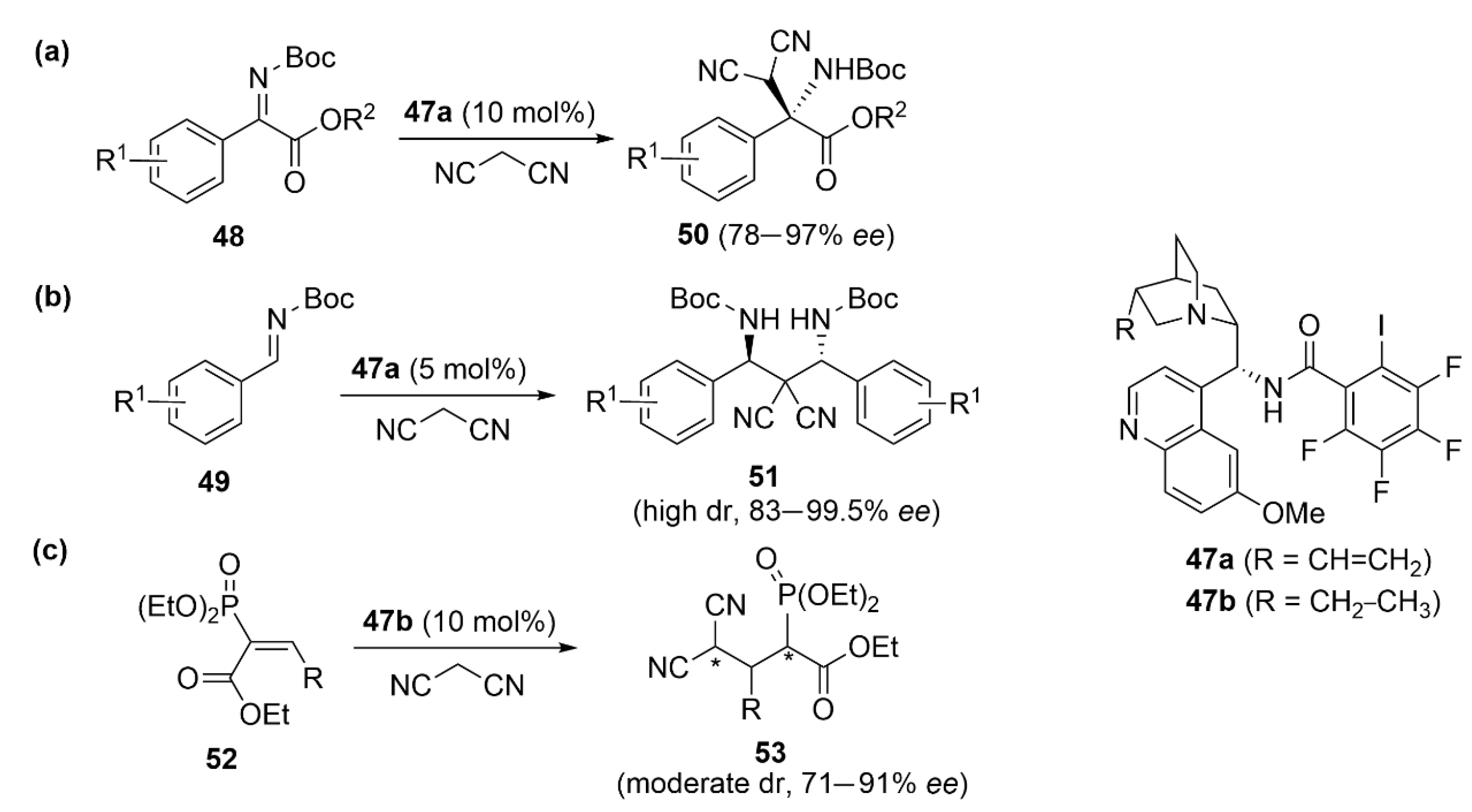
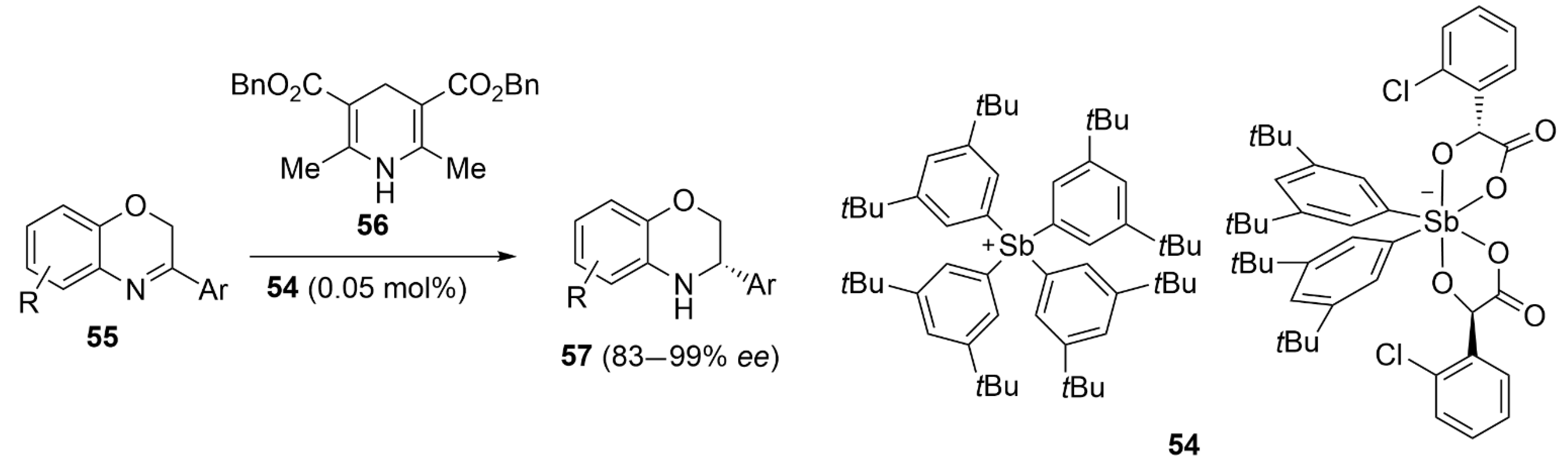
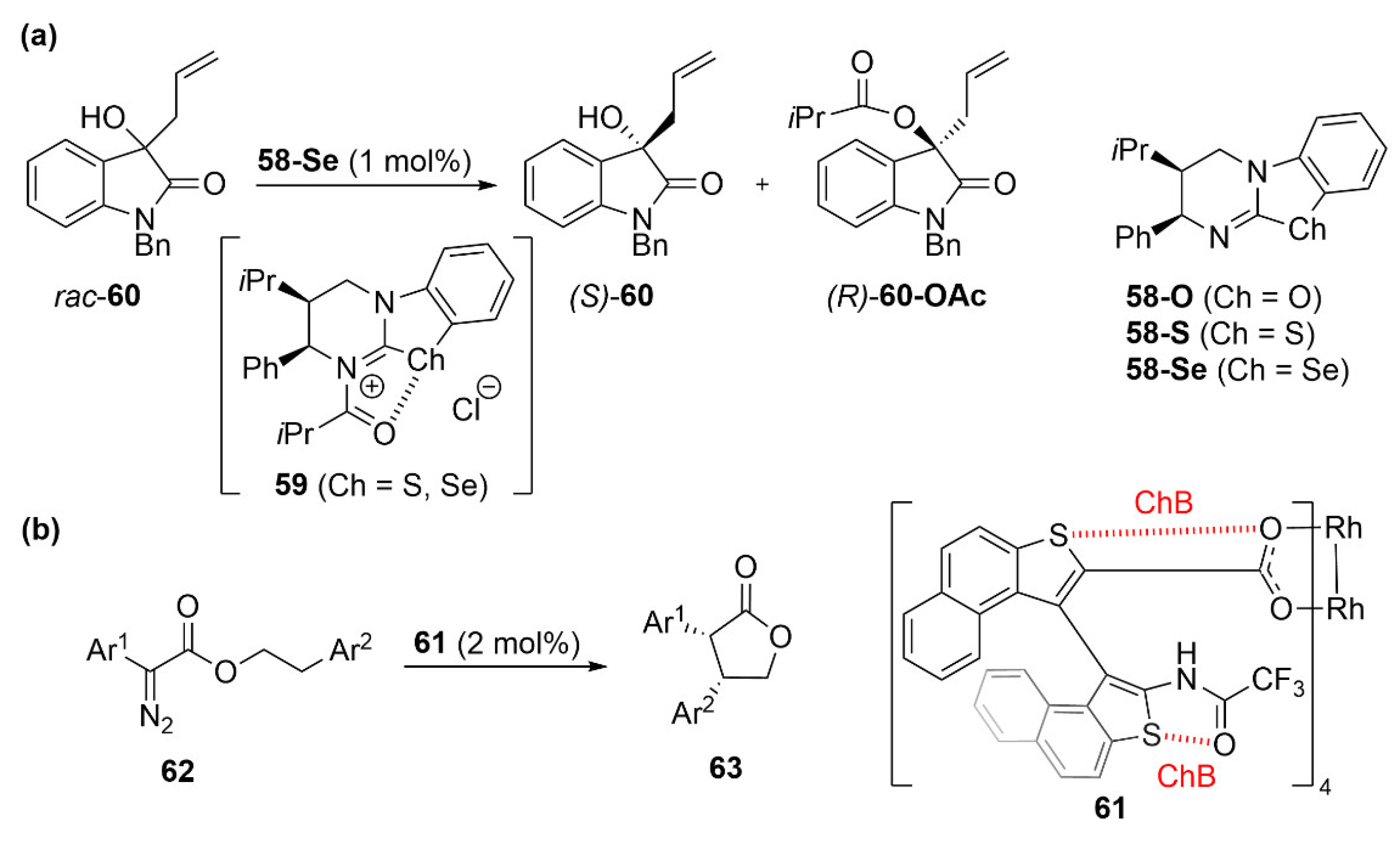
5.3. ChB-Based Catalyzed Asymmetric Reactions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brinck, T.; Murray, J.S.; Politzer, P. Surface Electrostatic Potentials of Halogenated Methanes as Indicators of Directional Intermolecular Interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Clark, T.; Politzer, P. σ-Hole Bonding: Molecules Containing Group VI Atoms. J. Mol. Model. 2007, 13, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-Hole Concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the Chalcogen Bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Scheiner, S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Janjić, G.V.; Zarić, S.D. σ-Hole Interactions of Covalently-Bonded Nitrogen, Phosphorus and Arsenic: A Survey of Crystal Structures. Crystals 2014, 4, 12–31. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Aerogen Bonding Interaction: A New Supramolecular Force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef] [PubMed]
- Stenlid, J.H.; Brinck, T. Extending the σ-Hole Concept to Metals: An Electrostatic Interpretation of the Effects of Nanostructure in Gold and Platinum Catalysis. J. Am. Chem. Soc. 2017, 139, 11012–11015. [Google Scholar] [CrossRef] [PubMed]
- Legon, A.C.; Walker, N.R. What’s in a Name? ‘Coinage-Metal’ Non-covalent Bonds and their Definition. Phys. Chem. Chem. Phys. 2018, 20, 19332–19338. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Alkorta, I.; Elguero, J.; Mooibroek, T.J.; Frontera, A. Spodium Bonds: Noncovalent Interactions Involving Group 12 Elements. Angew. Chem. Int. Ed. 2020, 59, 17482–17487. [Google Scholar] [CrossRef] [PubMed]
- Daolio, A.; Pizzi, A.; Calabrese, M.; Terraneo, G.; Bordignon, S.; Frontera, A.; Resnati, G. Molecular Electrostatic Potential and Noncovalent Interactions in Derivatives of Group 8 Elements. Angew. Chem. Int. Ed. 2021, 60, 20723–20727. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Noncovalent Interactions Involving Group 6 in Biological Systems: The Case of Molybdopterin and Tungstopterin Cofactors. Chem. Eur. J. 2022, e202201660. [Google Scholar] [CrossRef]
- Riel, A.M.S.; Rowe, R.K.; Ho, E.N.; Carlsson, A.-C.C.; Rappé, A.K.; Berryman, O.B.; Ho, P.S. Hydrogen Bond Enhanced Halogen Bonds: A Synergistic Interaction in Chemistry and Biochemistry. Acc. Chem. Res. 2019, 52, 2870–2880. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Type II Halogen…Halogen Contacts are Halogen Bonds. IUCrJ 2014, 1, 5–7. [Google Scholar] [CrossRef]
- Kolb, S.; Oliver, G.A.; Werz, D.B. Chemistry Evolves, Terms Evolve, but Phenomena Do Not Evolve: From Chalcogen–Chalcogen Interactions to Chalcogen Bonding. Angew. Chem. Int. Ed. 2020, 59, 22306–22310. [Google Scholar] [CrossRef]
- Kolár, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Chalcogen Bonding in Synthesis, Catalysis and Design of Materials. Dalton Trans. 2017, 46, 10121–10138. [Google Scholar] [CrossRef] [PubMed]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef] [PubMed]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen Bonding: An Overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The Chalcogen Bond in Crystalline Solids: A World Parallel to Halogen Bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.C.; Beer, P.D. Sigma-Hole Interactions in Anion Recognition. Chem 2018, 4, 731–783. [Google Scholar] [CrossRef]
- Sutar, R.; Huber, S.M. Catalysis of Organic Reactions through Halogen Bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Bamberger, J.; Ostler, F.; Garcia Mancheño, O. Frontiers in Halogen and Chalcogen-Bond Donor Organocatalysis. ChemCatChem 2019, 11, 5198–5211. [Google Scholar] [CrossRef] [PubMed]
- Biot, N.; Bonifazi, D. Chalcogen-Bond Driven Molecular Recognition at Work. Coord. Chem. Rev. 2020, 413, 213243. [Google Scholar] [CrossRef]
- Taylor, M.S. Anion Recognition Based on Halogen, Chalcogen, Pnictogen and Tetrel Bonding. Coord. Chem. Rev. 2020, 413, 213270. [Google Scholar] [CrossRef]
- Breugst, M.; Koenig, J.J. σ-Hole Interactions in Catalysis. Eur. J. Org. Chem. 2020, 2020, 5473–5487. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. On the Importance of Pnictogen and Chalcogen Bonding Interactions in Supramolecular Catalysis. Int. J. Mol. Sci. 2021, 22, 12550. [Google Scholar] [CrossRef]
- Kaasik, M.; Kanger, T. Supramolecular Halogen Bonds in Asymmetric Catalysis. Front. Chem. 2020, 8, 599064. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, L.; Wang, T. Supramolecular Chirality in Self-Assembled Systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef] [PubMed]
- Adawy, A. Functional Chirality: From Small Molecules to Supramolecular Assemblies. Symmetry 2022, 14, 292. [Google Scholar] [CrossRef]
- Pérez-García, L.; Amabilino, D.B. Spontaneous Resolution under Supramolecular Control. Chem. Soc. Rev. 2002, 31, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Neukirch, H.; Guido, E.; Liantonio, R.; Metrangolo, P.; Pilati, T.; Resnati, G. Spontaneous Resolution in a Halogen Bonded Supramolecular Architecture. Chem. Commun. 2005, 12, 1534–1536. [Google Scholar] [CrossRef]
- Casnati, A.; Liantonio, R.; Metrangolo, P.; Resnati, G.; Ungaro, R.; Ugozzoli, F. Molecular and Supramolecular Homochirality: Enantiopure Perfluorocarbon Rotamers and Halogen-Bonded Fluorous Double Helices. Angew. Chem. Int. Ed. 2006, 45, 1915–1918. [Google Scholar] [CrossRef]
- Muniappan, S.; Lipstman, S.; Goldberg, I. Rational Design of Supramolecular Chirality in Porphyrin Assemblies: The Halogen Bond Case. Chem. Commun. 2008, 15, 1777–1779. [Google Scholar] [CrossRef]
- Ng, C.-F.; Chow, H.-F.; Mak, T.C.W. Halogen-Bond-Mediated Assembly of a Single-Component Supramolecular Triangle and an Enantiomeric Pair of Double Helices from 2-(Iodoethynyl)pyridine Derivatives. Angew. Chem. Int. Ed. 2018, 57, 4986–4990. [Google Scholar] [CrossRef]
- Zong, Z.; Hao, A.; Xing, P. Halogenation Regulates Supramolecular Chirality at Hierarchical Levels of Self-Assembled N-Terminal Aromatic Amino Acids. J. Phys. Chem. Lett. 2021, 12, 1307–1315. [Google Scholar] [CrossRef]
- Yashima, E.; Ousaka, N.; Taura, D.; Shimomura, K.; Ikai, T.; Maeda, K. Supramolecular Helical Systems: Helical Assemblies of Small Molecules, Foldamers, and Polymers with Chiral Amplification and Their Functions. Chem. Rev. 2016, 116, 13752–13990. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yan, X.; He, W.; Li, X.; Li, Z.; Mo, Y.; Liu, M.; Jiang, Y.-B. C−I···π Halogen Bonding Driven Supramolecular Helix of Bilateral N-Amidothioureas Bearing β-Turns. J. Am. Chem. Soc. 2017, 139, 6605–6610. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zou, K.; Cao, J.; Li, X.; Zhao, Z.; Li, Z.; Wu, A.; Liang, W.; Mo, Y.; Jiang, Y. Single-Handed Supramolecular Double Helix of Homochiral Bis(N-Amidothiourea) Supported by Double Crossed C−I···S Halogen Bonds. Nat. Commun. 2019, 10, 3610. [Google Scholar] [CrossRef]
- Xu, Y.; Hao, A.; Xing, P. X···X Halogen Bond-Induced Supramolecular Helices. Angew. Chem. Int. Ed. 2022, 61, e202113786. [Google Scholar]
- An, S.; Hao, A.; Xing, P. Halogen Bonding Mediated Hierarchical Supramolecular Chirality. ACS Nano 2021, 15, 15306–15315. [Google Scholar] [CrossRef]
- Zheng, S.; Han, J.; Jin, X.; Ye, Q.; Zhou, J.; Duan, P.; Liu, M. Halogen Bonded Chiral Emitters: Generation of Chiral Fractal Architecture with Amplified Circularly Polarized Luminescence. Angew. Chem. Int. Ed. 2021, 60, 22711–22716. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Cossu, S. Liquid Chromatography Enantioseparations of Halogenated Compounds on Polysaccharide-Based Chiral Stationary Phases: Role of Halogen Substituents in Molecular Recognition. Chirality 2015, 27, 667–684. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Dessì, A.; Dallocchio, R.; Aubert, E.; Gatti, C.; Mangelings, D.; Cossu, S. Halogen Bond in Separation Science: A Critical Analysis Across Experimental and Theoretical Results. J. Chromatogr. A 2020, 1616, 460788. [Google Scholar] [CrossRef] [PubMed]
- Siret, L.; Tambuté, A.; Caude, M.; Rosset, R. (S)-Thio-DNBTyr-A and (S)-Thio-DNBTyr-E as Chiral Stationary Phases for Analytical and Preparative Purposes: Application to the Enantiomeric Resolution of Alkyl N-Arylsulphinamoyl Esters. J. Chromatogr. A 1990, 498, 67–79. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Gan, K.Z.; Brice, L.J. The Enhancement of Enantioselectivity by Halogen Substituents. Tetrahedron Asymmetry 1996, 7, 2813–2816. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Gan, K.Z. Facile and Predictable Means of Separating the Enantiomers of 5-Arylhydantoins. J. Chromatogr. A 1997, 790, 65–71. [Google Scholar] [CrossRef]
- Farina, A.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G.; Vecchio, G. Resolution of Racemic 1,2-Dibromohexafluoropropane through Halogen-Bonded Supramolecular Helices. Angew. Chem. Int. Ed. 1999, 38, 2433–2434. [Google Scholar] [CrossRef]
- Messina, M.T.; Metrangolo, P.; Resnati, G. Resolution of Racemic Perfluorocarbons Through Self-Assembly Driven by Electron Donor-Acceptor Intermolecular Recognition. ACS Symp. Ser. 2000, 746, 239–254. [Google Scholar]
- Kobayashi, Y.; Maeda, J.; Ando, T.; Saigo, K. Halogen-Bonding Interaction Stabilizing Cluster-type Diastereomeric Salt Crystals. Cryst. Growth Des. 2010, 10, 685–690. [Google Scholar] [CrossRef]
- Yan, X.Q.; Shen, Q.J.; Zhao, X.R.; Gao, H.Y.; Pang, X.; Jin, W.J. Halogen Bonding: A New Retention Mechanism for the Solid Phase Extraction of Perfluorinated Iodoalkanes. Anal. Chim. Acta 2012, 753, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Mamane, V.; Aubert, E.; Cossu, S. Insights into the Impact of Shape and Electronic Properties on the Enantioseparation of Polyhalogenated 4,4′-Bipyridines on Polysaccharide-Type Selectors. Evidence of Stereoselective Halogen Bonding Interactions. J. Chromatogr. A 2014, 1345, 182–192. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Aubert, E.; Dessì, A.; Dallocchio, R.; Dore, A.; Pale, P.; Cossu, S. Insights into Halogen Bond-Driven Enantioseparations. J. Chromatogr. A 2016, 1467, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Mamane, V.; Dallocchio, R.; Dessì, A.; Villano, R.; Sanna, D.; Aubert, E.; Pale, P.; Cossu, S. Polysaccharide-Based Chiral Stationary Phases as Halogen Bond Acceptors: A Novel Strategy for Detection of Stereoselective σ-Hole Bonds in Solution. J. Sep. Sci. 2018, 41, 1247–1256. [Google Scholar] [CrossRef]
- Mamane, V.; Peluso, P.; Aubert, E.; Cossu, S.; Pale, P. Chiral Hexalogenated 4,4′-Bipyridines. J. Org. Chem. 2016, 81, 4576–4587. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Aubert, E.; Cossu, S. High Performance Liquid Chromatography Enantioseparation of Atropisomeric 4,4′-Bipyridines on Polysaccharide-Type Chiral Stationary Phases: Impact of Substituents and Electronic Properties. J. Chromatogr. A 2012, 1251, 91–100. [Google Scholar] [CrossRef]
- Lu, Y.; Li, H.; Zhu, X.; Zhu, W.; Liu, H. How Does Halogen Bonding Behave in Solution? A Theoretical Study Using Implicit Solvation Model. J. Phys. Chem. A 2011, 115, 4467–4475. [Google Scholar] [CrossRef] [PubMed]
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Resnati, G. The Halogen Bond in the Design of Functional Supramolecular Materials: Recent Advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar] [CrossRef]
- Kolár, M.; Hobza, P.; Bronowska, K. Plugging the Explicit σ-Holes in Molecular Docking. Chem. Commun. 2013, 49, 981–983. [Google Scholar] [CrossRef]
- Dallocchio, R.; Dessì, A.; Solinas, M.; Arras, A.; Cossu, S.; Aubert, E.; Mamane, V.; Peluso, P. Halogen Bond in High-Performance Liquid Chromatography Enantioseparations: Description, features and modelling. J. Chromatogr. A 2018, 1563, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Dallocchio, R.; Sechi, B.; Dessì, A.; Chankvetadze, B.; Cossu, S.; Mamane, V.; Pale, P.; Peluso, P. Enantioseparations of Polyhalogenated 4,4′-Bipyridines on Polysaccharide-Based Chiral Stationary Phases and Molecular Dynamics Simulations of Selector–Selectand Interactions. Electrophoresis 2021, 42, 1853–1863. [Google Scholar] [CrossRef]
- Mamane, V.; Peluso, P.; Aubert, E.; Weiss, R.; Wenger, E.; Cossu, S.; Pale, P. Disubstituted Ferrocenyl Iodo- and Chalcogenoalkynes as Chiral Halogen and Chalcogen Bond Donors. Organometallics 2020, 39, 3936–3950. [Google Scholar] [CrossRef]
- Sechi, B.; Dessì, A.; Gatti, C.; Dallocchio, R.; Chankvetadze, B.; Cosu, S.; Mamane, V.; Pale, P.; Peluso, P. Unravelling Functions of Halogen Substituents in the Enantioseparation of Halogenated Planar Chiral Ferrocenes on Polysaccharide-Based Chiral Stationary Phases: Experimental and Electrostatic Potential Analyses. J. Chromatogr. A 2022, 1673, 463097. [Google Scholar] [CrossRef] [PubMed]
- Gatti, C.; Cargnoni, F.; Bertini, L. Chemical Information from the Source Function. J. Comput. Chem. 2003, 24, 422–436. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.A.V.; Petitbois, J.G.; Vairappan, C.S.; Umezawa, T.; Matsuda, F.; Okino, T. Columbamides D and E: Chlorinated Fatty Acid Amides from the Marine Cyanobacterium Moorea Bouillonii Collected in Malaysia. Org. Lett. 2017, 19, 4231–4234. [Google Scholar] [CrossRef]
- West, C.; Konjaria, M.L.; Shashviashvili, N.; Lemasson, E.; Bonnet, P.; Kakava, R.; Volonterio, A.; Chankvetadze, B. Enantioseparation of Novel Chiral Sulfoxides on Chlorinated Polysaccharide Stationary Phases in Supercritical Fluid Chromatography. J. Chromatogr. A 2017, 1499, 174–182. [Google Scholar] [CrossRef]
- Zhu, B.; Yao, Y.; Deng, M.; Jiang, Z.; Li, Q. Enantioselective Separation of Twelve Pairs of Enantiomers on Polysaccharide-Based Chiral Stationary Phases and Thermodynamic Analysis of Separation Mechanism. Electrophoresis 2018, 39, 2398–2405. [Google Scholar] [CrossRef]
- He, Z.; Wu, F.; Xia, W.; Li, L.; Hu, K.; Kaziem, A.E.; Wang, M. Separation and Detection of Cyproconazole Enantiomers and Its Stereospecific Recognition with Chiral Stationary Phase by High-Performance Liquid Chromatography. Analyst 2019, 144, 5193–5200. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, R.K.; Potlitz, F.; Schulig, L.; Kim, S.; Hasan, M.; Link, A. Subcritical Fluid Chromatography at Sub-ambient Temperatures for the Chiral Resolution of Ketamine Metabolites with Rapid-Onset Antidepressant Effects. Molecules 2019, 24, 1927. [Google Scholar] [CrossRef] [PubMed]
- Bonin, L.; Morvan, A.; Coadou, G.; Furman, C.; Boulanger, E.; Ghinet, A.; Lipka, E. Supercritical Fluid Chromatography for Separation of Chiral Planar Metallocenes. J. Chromatogr. A 2022, 1674, 463115. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, K.; Lesani, M. Does Fluorine Participate in Halogen Bonding? Chem. Eur. J. 2015, 21, 4739–4746. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kawashima, M.; Hatada, K. Chromatographic Resolution. XI. Controlled Chiral Recognition of Cellulose Triphenylcarbamate Derivatives Supported on Silica Gel. J. Chromatogr. A 1986, 363, 173–186. [Google Scholar] [CrossRef]
- Speybrouck, D.; Lipka, E. Preparative Supercritical Fluid Chromatography: A Powerful Tool for Chiral Separations. J. Chromatogr. A 2016, 1467, 33–55. [Google Scholar] [CrossRef]
- Khater, S.; Lozac’h, M.A.; Adam, I.; Francotte, E.; West, C. Comparison of Liquid and Supercritical Fluid Chromatography Mobile Phases for Enantioselective Separations on Polysaccharide Stationary Phases. J. Chromatogr. A 2016, 1467, 463–472. [Google Scholar] [CrossRef]
- Zhu, X.; Lu, Y.; Peng, C.; Hu, J.; Liu, H.; Hu, Y. Halogen Bonding Interactions Between Brominated Ion Pairs and CO2 Molecules: Implications for Design of New and Efficient Ionic Liquids for CO2 Absorption. J. Phys. Chem. B 2011, 115, 3949–3958. [Google Scholar] [CrossRef]
- Terada, S.; Hirai, M.; Honzawa, A.; Kitagawa, O.; Kamizela, A.; Wzorek, A.; Soloshonok, V.A. Possible Case of Halogen Bond-Driven Self-Disproportionation of Enantiomers (SDE) via Achiral Chromatography. Chem. Eur. J. 2017, 23, 14631–14638. [Google Scholar] [CrossRef]
- Soloshonok, V.A. Remarkable Amplification of the Self-Disproportionation of Enantiomers on Achiral-Phase Chromatography Columns. Angew. Chem. Int. Ed. 2006, 45, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Möckel, H.J.; Müller, A.; Diemann, E.; Walberg, H.-J. Retention of Thio- and Selenometalates in Mobile-Phase Ion Chromatography. J. Chromatogr. A 1988, 439, 93–108. [Google Scholar] [CrossRef]
- Vespalec, R.; Corstjens, H.; Billiet, H.A.H.; Frank, J.; Luyben, K.C.A.M. Enantiomeric Separation of Sulfur- and Selenium-Containing Amino Acids by Capillary Electrophoresis Using Vancomycin as a Chiral Selector. Anal. Chem. 1995, 67, 3223–3228. [Google Scholar] [CrossRef]
- Hou, J.-G.; Han, X.-Q.; Liu, H.-T.; Wang, Y.-L.; Gao, J.-Z. Chiral Separation of Glycidyl Selenide and Glycidyl Sulfide Racemates on Cellulose-Based Chiral Stationary Phases. J. Chromatogr. Sci. 2001, 39, 388–392. [Google Scholar] [CrossRef][Green Version]
- Weiss, R.; Aubert, E.; Peluso, P.; Cossu, S.; Pale, P.; Mamane, V. Chiral Chalcogen Bond Donors Based on the 4,4′-bipyridine scaffold. Molecules 2019, 24, 4484. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Gatti, C.; Dessì, A.; Dallocchio, R.; Weiss, R.; Aubert, E.; Pale, P.; Cossu, S.; Mamane, V. Enantioseparation of Fluorinated 3-Arylthio-4,4′-Bipyridines: Insights into Chalcogen and π-Hole Bonds in High-Performance Liquid Chromatography. J. Chromatogr. A 2018, 1567, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Gatti, C.; Dessì, A.; Dallocchio, R.; Mamane, V.; Cossu, S.; Weiss, R.; Pale, P.; Aubert, E.; Peluso, P. Factors Impacting σ- and π-Hole Regions as Revealed by the Electrostatic Potential and Its Source Function Reconstruction: The Case of 4,4′-Bipyridine Derivatives. Molecules 2020, 25, 4409. [Google Scholar] [CrossRef]
- Peluso, P.; Dessì, A.; Dallocchio, R.; Sechi, B.; Gatti, C.; Chankvetadze, B.; Mamane, V.; Weiss, R.; Pale, P.; Aubert, E.; et al. Enantioseparation of 5,5′-Dibromo-2,2′-Dichloro-3-Selanyl-4,4′-Bipyridines on Polysaccharide-based Chiral Stationary Phases: Exploring Chalcogen Bonds in Liquid-phase Chromatography. Molecules 2021, 26, 221. [Google Scholar] [CrossRef]
- Gropp, C.; Quigley, B.L.; Diederich, F. Molecular Recognition with Resorcin[4]arene Cavitands: Switching, Halogen-Bonded Capsules, and Enantioselective Complexation. J. Am. Chem. Soc. 2018, 140, 2705–2717. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Marques, I.; Ferreira, L.; Félix, V.; Beer, P.D. Enhancing the Enantioselective Recognition and Sensing of Chiral Anions by Halogen Bonding. Chem. Commun. 2016, 52, 5527–5530. [Google Scholar] [CrossRef]
- Borissov, A.; Lim, J.Y.C.; Brown, A.; Christensen, K.E.; Thompson, A.L.; Smith, M.D.; Beer, P.D. Neutral Iodotriazole Foldamers as Tetradentate Halogen Bonding Anion Receptors. Chem. Commun. 2017, 53, 2483–2486. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.C.; Marques, I.; Félix, V.; Beer, P.D. Enantioselective Anion Recognition by Chiral Halogen-Bonding [2]Rotaxanes. J. Am. Chem. Soc. 2017, 139, 12228–12239. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.C.; Marques, I.; Félix, V.; Beer, P.D. A Chiral Halogen-Bonding [3]Rotaxane for the Recognition and Sensing of Biologically Relevant Dicarboxylate Anions. Angew. Chem. Int. Ed. 2018, 57, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.C.; Marques, I.; Félix, V.; Beer, P.D. Chiral Halogen and Chalcogen Bonding Receptors for Discrimination of Stereo- and Geometric Dicarboxylate Isomers in Aqueous Media. Chem. Commun. 2018, 54, 10851–10854. [Google Scholar] [CrossRef]
- Gropp, C.; Husch, T.; Trapp, N.; Reiher, M.; Diederich, F. Dispersion and Halogen-Bonding Interactions: Binding of the Axial Conformers of Monohalo- and (±)-trans-1,2-Dihalocyclohexanes in Enantiopure Alleno-Acetylenic Cages. J. Am. Chem. Soc. 2017, 139, 12190–12200. [Google Scholar] [CrossRef]
- Kaasik, M.; Kaabel, S.; Kriis, K.; Järving, I.; Aav, R.; Rissanen, K.; Kanger, T. Synthesis and Characterisation of Chiral Triazole-Based Halogen-Bond Donors: Halogen Bonds in the Solid State and in Solution. Chem. Eur. J. 2017, 23, 7337–7344. [Google Scholar] [CrossRef]
- Sutar, R.L.; Engelage, E.; Stoll, R.; Huber, S.M. Bidentate Chiral Bis(imidazolium)-Based Halogen-Bond Donors: Synthesis and Applications in Enantioselective Recognition and Catalysis. Angew. Chem. Int. Ed. 2020, 59, 6806–6810. [Google Scholar] [CrossRef]
- Ostler, F.; Piekarski, D.G.; Danelzik, T.; Taylor, M.S.; García Mancheño, O. Neutral Chiral Tetrakis-Iodo-Triazole Halogen-Bond Donor for Chiral Recognition and Enantioselective Catalysis. Chem. Eur. J. 2021, 27, 2315–2320. [Google Scholar] [CrossRef]
- Aubert, E.; Doudouh, A.; Wenger, E.; Sechi, B.; Peluso, P.; Pale, P.; Mamane, V. Chiral Ferrocenyl−Iodotriazoles and −Iodotriazoliums as Halogen Bond Donors. Synthesis, Solid State Analysis and Catalytic Properties. Eur. J. Inorg. Chem. 2022, 2022, e202100927. [Google Scholar] [CrossRef]
- Yoshida, Y.; Mino, T.; Sakamoto, M. Chiral Hypervalent Bromine(III) (Bromonium Salt): Hydrogen- and Halogen-Bonding Bifunctional Asymmetric Catalysis by Diaryl-λ3-bromanes. ACS Catal. 2021, 11, 13028–13033. [Google Scholar] [CrossRef]
- Yoshida, Y.; Fujimura, T.; Mino, T.; Sakamoto, M. Chiral Binaphthyl-Based Iodonium Salt (Hypervalent Iodine(III)) as Hydrogen- and Halogen-Bonding Bifunctional Catalyst: Insight into Abnormal Counteranion Effect and Asymmetric Synthesis of N,S-Acetals. Adv. Synth. Catal. 2022, 364, 1091–1097. [Google Scholar] [CrossRef]
- Kuwano, S.; Nishida, Y.; Suzuki, T.; Arai, T. Catalytic Asymmetric Mannich-Type Reaction of Malononitrile with N-Boc α-Ketiminoesters Using Chiral Organic Base Catalyst with Halogen Bond Donor Functionality. Adv. Synth. Catal. 2020, 362, 1674–1678. [Google Scholar] [CrossRef]
- Kuwano, S.; Ogino, E.; Arai, T. Enantio- and Diastereoselective Double Mannich Reaction of Malononitrile with N-Boc Imines using Quinine-Derived Bifunctional Organoiodine Catalyst. Org. Biomol. Chem. 2021, 19, 6969–6973. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, M.; Martonova, J.; Erkman, K.; Metsala, A.; Järving, I.; Kanger, T. Enantioselective Michael Addition to Vinyl Phosphonates via Hydrogen Bond-Enhanced Halogen Bond Catalysis. Chem. Sci. 2021, 12, 7561–7568. [Google Scholar] [CrossRef] [PubMed]
- Kriis, K.; Martõnov, H.; Miller, A.; Erkman, K.; Järving, I.; Kaasik, M.; Kanger, T. Multifunctional Catalysts in the Asymmetric Mannich Reaction of Malononitrile with N-Phosphinoylimines: Coactivation by Halogen Bonding versus Hydrogen Bonding. J. Org. Chem. 2022, 87, 7422–7435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wei, J.; Ding, W.-Y.; Li, S.; Xiang, S.-H.; Tan, B. Asymmetric Pnictogen-Bonding Catalysis: Transfer Hydrogenation by a Chiral Antimony(V) Cation/Anion Pair. J. Am. Chem. Soc. 2021, 143, 6382–6387. [Google Scholar] [CrossRef] [PubMed]
- Young, C.M.; Elmi, A.; Pascoe, D.J.; Morris, R.K.; McLaughlin, C.; Woods, A.M.; Frost, A.B.; de la Houpliere, A.; Ling, K.B.; Smith, T.K.; et al. The Importance of 1,5-Oxygen···Chalcogen Interactions in Enantioselective Isochalcogenourea Catalysis. Angew. Chem. Int. Ed. 2020, 59, 3705–3710. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Young, C.M.; Watts, A.A.; Slawin, A.M.Z.; Boyce, G.R.; Bühl, M.; Smith, A.D. Isothiourea-Catalyzed Enantioselective Michael Addition of Malonates to α,β-Unsaturated Aryl Esters. Org. Lett. 2022, 24, 4040–4045. [Google Scholar] [CrossRef] [PubMed]
- Murai, T.; Lu, W.; Kuribayashi, T.; Morisaki, K.; Ueda, Y.; Hamada, S.; Kobayashi, Y.; Sasamori, T.; Tokitoh, N.; Kawabata, T.; et al. Conformational Control in Dirhodium(II) Paddlewheel Catalysts Supported by Chalcogen-Bonding Interactions for Stereoselective Intramolecular C−H Insertion Reactions. ACS Catal. 2021, 11, 568–578. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | X | R1 | R2 | k11 | α 2 |
|---|---|---|---|---|---|
| 8 | H | - | - | 3.72 | 3.17 |
| 8 | p-I | - | - | 4.10 | 5.12 |
| 9 | H | - | - | 1.39 | 6.74 |
| 9 | p-F | - | - | 1.17 | 7.29 |
| 9 | p-Cl | - | - | 1.48 | 11.6 |
| 9 | p-Br | - | - | 1.61 | 12.8 |
| 9 | p-I | - | - | 1.75 | 13.7 |
| 9 | m-Br | - | - | 1.61 | 13.1 |
| 10 | H | - | - | 0.87 | 1.29 |
| 10 | p-F | - | - | 0.83 | 1.39 |
| 10 | p-Cl | - | - | 0.84 | 1.55 |
| 10 | p-Br | - | - | 0.86 | 1.66 |
| 11 | F | H | H | 0.62 | 2.06 |
| 11 | F | Me | H | 1.17 | 2.52 |
| 11 | F | Me | Me | 3.59 | 3.40 |
| 11 | Cl | H | H | 0.62 | 2.61 |
| 11 | Cl | Me | H | 1.22 | 3.31 |
| 11 | Cl | Me | Me | 3.81 | 4.96 |
| 11 | Br | H | H | 0.66 | 2.82 |
| 11 | Br | Me | H | 1.31 | 3.58 |
| 11 | Br | Me | Me | 3.97 | 5.20 |
| 11 | I | H | H | 0.69 | 2.97 |
| 11 | I | Me | H | 1.41 | 3.90 |
| 11 | I | Me | Me | 4.29 | 5.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peluso, P.; Mamane, V. Stereoselective Processes Based on σ-Hole Interactions. Molecules 2022, 27, 4625. https://doi.org/10.3390/molecules27144625
Peluso P, Mamane V. Stereoselective Processes Based on σ-Hole Interactions. Molecules. 2022; 27(14):4625. https://doi.org/10.3390/molecules27144625
Chicago/Turabian StylePeluso, Paola, and Victor Mamane. 2022. "Stereoselective Processes Based on σ-Hole Interactions" Molecules 27, no. 14: 4625. https://doi.org/10.3390/molecules27144625
APA StylePeluso, P., & Mamane, V. (2022). Stereoselective Processes Based on σ-Hole Interactions. Molecules, 27(14), 4625. https://doi.org/10.3390/molecules27144625