
Application of Mathematical Modeling and Computational Tools in the Modern Drug Design and Development Process

 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Target Identification
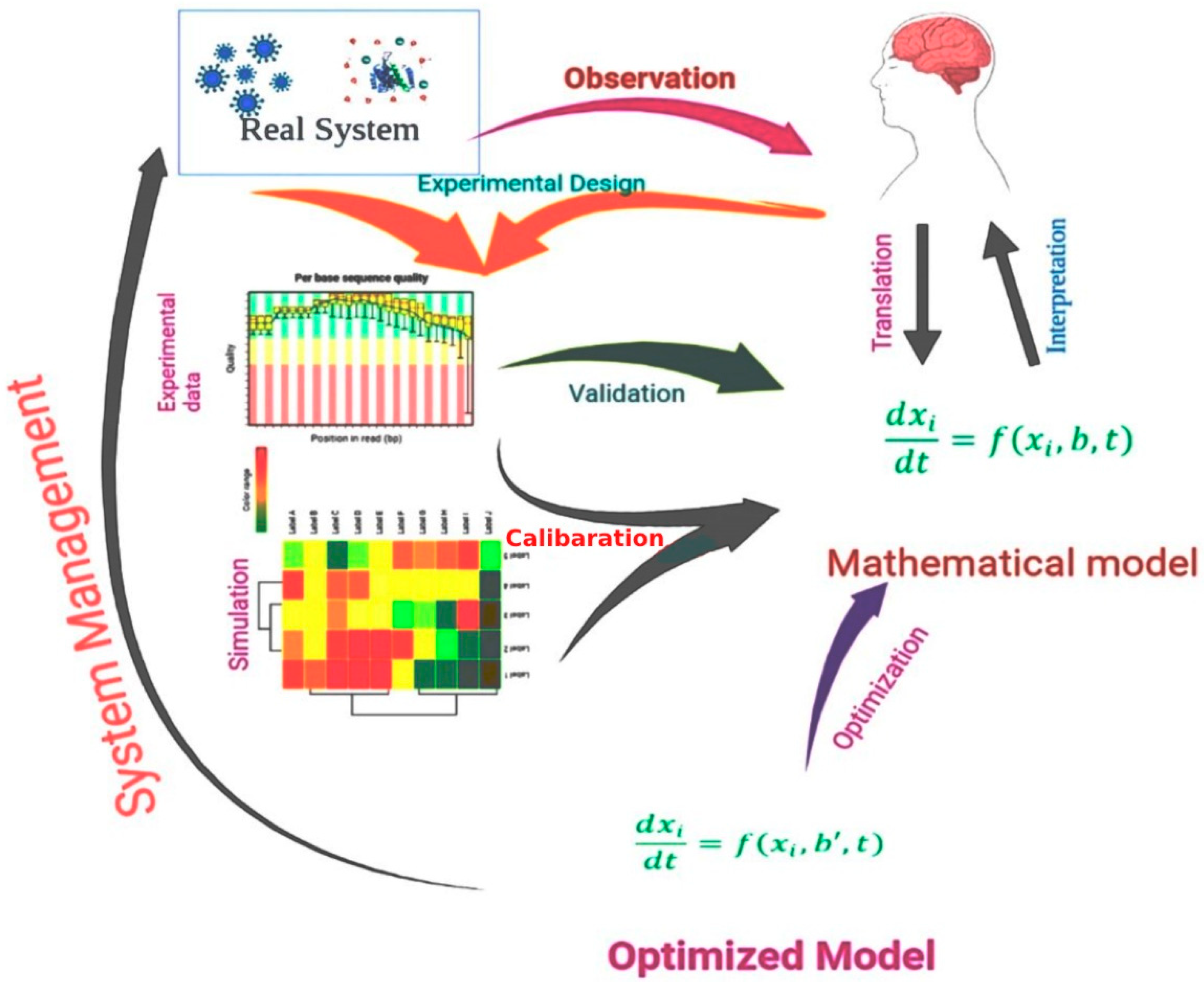
3. Mathematical Models in Drug Design
4. Target Validation
5. Mathematical and Computational Biology Approaches for Target Validation
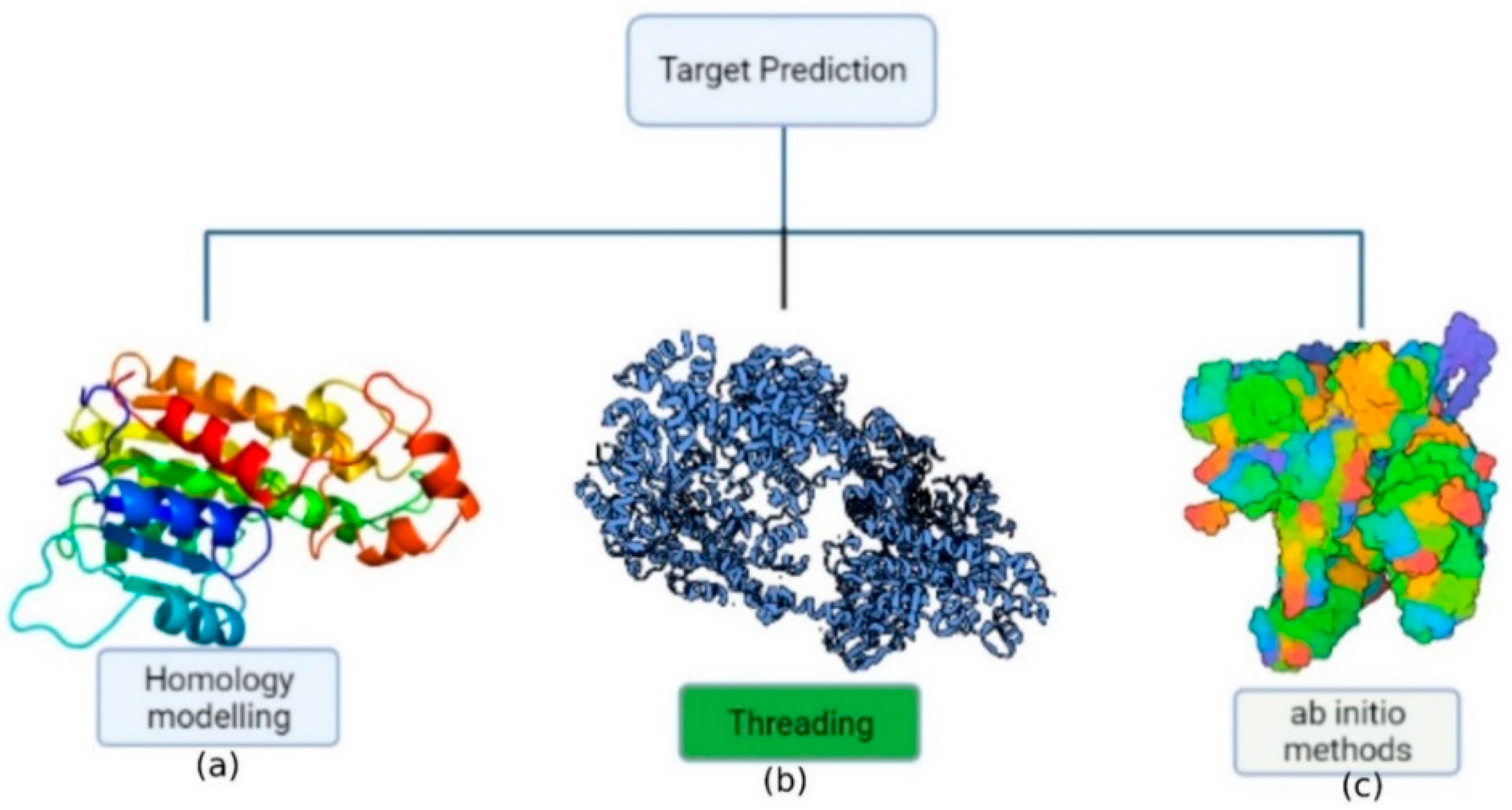
6. Protein Structure Prediction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Name | Application | Availability | Reference |
|---|---|---|---|---|
| 1. | I-TASSER | Reassembling fragment structure via threading | https://zhanggroup.org/I-TASSER/ | [46] |
| 2. | SWISS-MODEL | Segment assembly/local similarity | https://swissmodel.expasy.org/ | [47] |
| 3. | ESyPred3D | 3D modeling, template identification, and alignment | https://www.unamur.be/sciences/biologie/urbm/bioinfo/esypred/ | [48] |
| 4. | HH-suite | Template detection, alignment, 3D modeling | https://arquivo.pt/wayback/20160514083149/http:/toolkit.tuebingen.mpg.de/hhpred | [49] |
| 5. | RaptorX | Protein 3D modeling, remote homology discovery, and binding site prediction | http://raptorx.uchicago.edu/ | [50] |
| 6. | FoldX | Protein design and energy calculations | https://foldxsuite.crg.eu/ | [51] |
| 7. | ROBETTA | Rosetta homology modeling and fragment assembly from scratch with Ginzu domain prediction | http://robetta.bakerlab.org/ | [52] |
| 8. | BHAGEERATH-H | Methods of ab initio folding and homology are combined | http://www.scfbio-iitd.res.in/bhageerath/bhageerath_h.jsp | [53] |
| 9. | Prime | Homology modeling, evaluation, and refining of the produced model using the energy function | https://www.schrodinger.com/prime | [54] |
| 10. | LOMETS | Tertiary structure prediction with a local meta-threading server | https://zhanglab.ccmb.med.umich.edu/ | [55] |
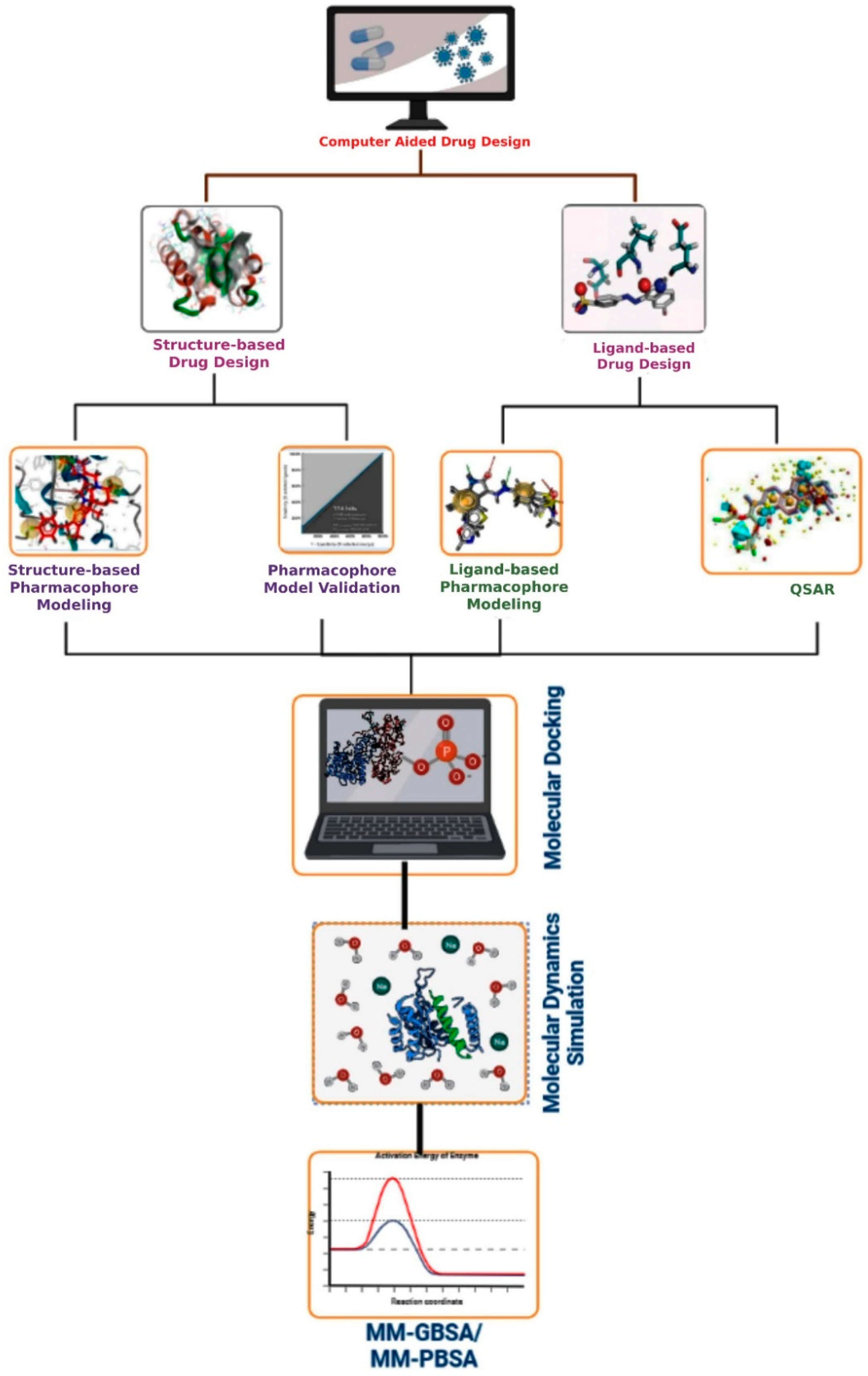
7. Computer-Aided Drug Design
7.1. Structure-Based Drug Design
7.1.1. Structure-Based Pharmacophore Modeling
7.1.2. Pharmacophore Model Validation
Fisher’s Randomization Test
Test Set Prediction
Guner–Henry (GH) Scoring
7.1.3. Virtual Screening
7.1.4. Molecular Docking
7.2. Ligand-Based Drug Design
7.2.1. Quantitative Structure–Activity Relationship (QSAR) Models
7.2.2. Ligand-Based Pharmacophore Modeling
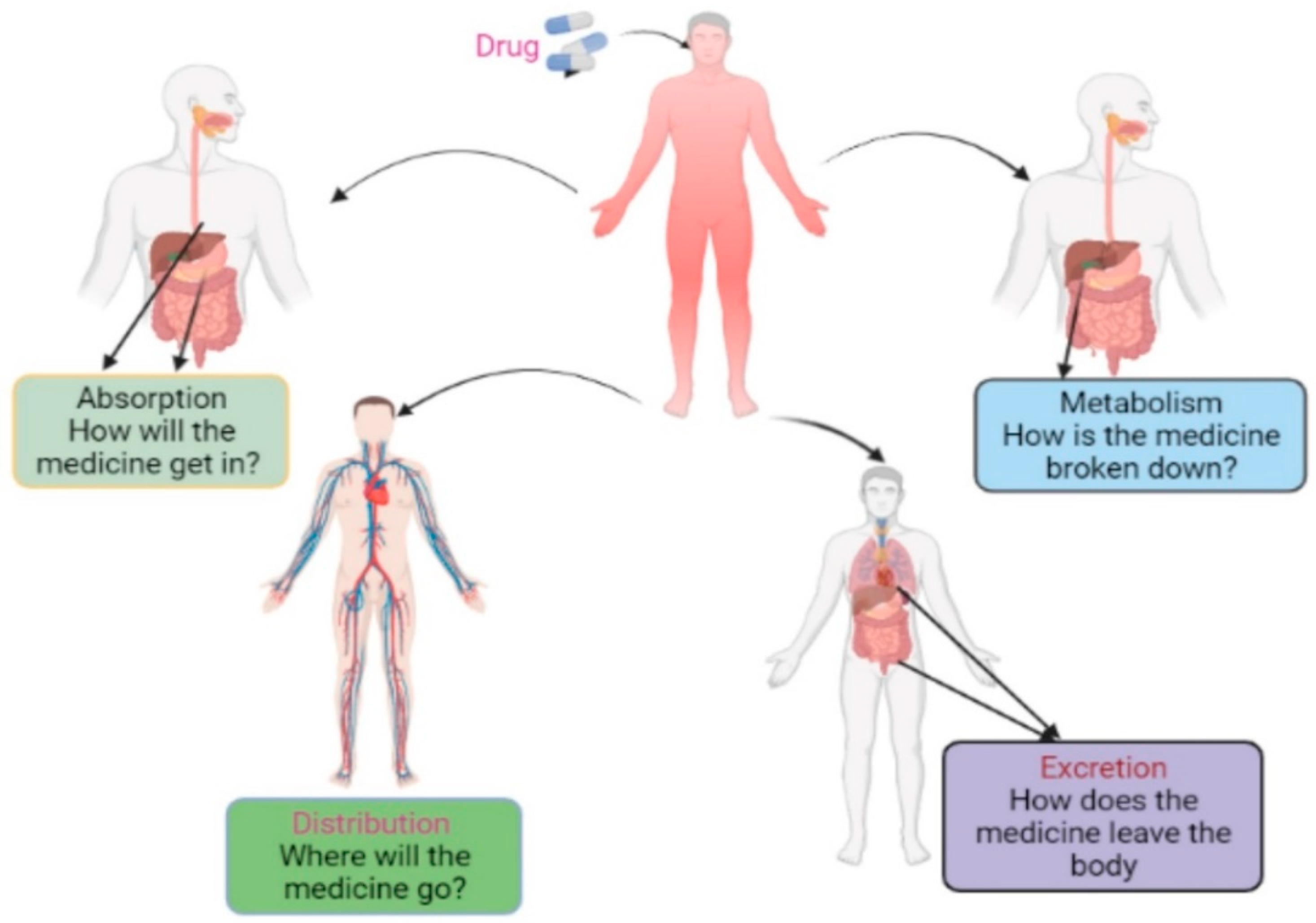
8. Pharmacokinetics Property Analysis
8.1. Absorption
8.2. Distribution
8.3. Metabolism
| S. No. | Program | Description | Accessibility | Reference |
|---|---|---|---|---|
| 1. | ADMETlab | ADMET in a systematic manner utilizing the ADMET database | http://admet.scbdd.com/ | [119] |
| 2. | eMolTox | Molecular toxicity prediction | http://xundrug.cn/moltox | [120] |
| 3. | LIVERTOX | Hepatotoxicity prediction | https://livertox.nih.gov/ | [121] |
| 4. | vNN | ADMET forecasts | https://vnnadmet.bhsai.org | [122] |
| 5. | PreADMET | This online tool calculates the probability of carcinogenicity as well as poisonous potency | https://preadmet.bmdrc.kr/ | [123] |
| 6. | QikProp | Used to forecast ADMET-related features | https://www.schrodinger.com/qikprop | [124] |
| 7. | SwissADME | Estimate physicochemical characteristics and predict ADME | http://www.swissadme.ch/ | [125] |
| 8. | DSSTox | It is a public database of searchable distributed structure toxicity | https://comptox.epa.gov/ | [126] |
| 9. | ChemTree | It is used to forecast ADMETox characteristics. | https://chemtree.kr/ | [127] |
| 10. | Metabase | It is a low-cost radio analytical LIMs in ADME/PK research based on Excel | https://www.metabase.com/ | [128] |
| 11. | TOPKAT | Used in toxicology prediction | https://www.toxit.it/en/services/software/topkat | [129] |
8.4. Excretion
9. Toxicity Analysis
10. Molecular Dynamics Simulation
11. MM-GBSA/MM-PBSA
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and Resupply of Pharmacologically Active Plant-Derived Natural Products: A Review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.Q.; Lei, H.M.; Hu, Q.Y.; Li, G.H.; Zhao, P.J. Recent Advances in the Synthetic Biology of Natural Drugs. Front. Bioeng. Biotechnol. 2021, 9, 640. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Khairnar, S.J.; Jadhav, A.G. Drug Repurposing (DR): An Emerging Approach in Drug Discovery. Drug Repurposing—Hypothesis Mol. Asp. Ther. Appl. 2020. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.S.; Kalindjian, S.B.; Philpott, K.L. Principles of Early Drug Discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimasi, J.A.; Feldman, L.; Seckler, A.; Wilson, A. Trends in Risks Associated with New Drug Development: Success Rates for Investigational Drugs. Clin. Pharmacol. Ther. 2010, 87, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Bajuri, M.R.; Siri, Z.; Abdullah, M.N.S. Mathematical Modeling Research Output Impacting New Technological Development: An Axiomatization to Build Novelty. Axioms 2022, 11, 264. [Google Scholar] [CrossRef]
- Ganusov, V.V. Strong Inference in Mathematical Modeling: A Method for Robust Science in the Twenty-First Century. Front. Microbiol. 2016, 7, 1131. [Google Scholar] [CrossRef] [Green Version]
- Vlachakis, D.; Vlamos, P. Mathematical Multidimensional Modelling and Structural Artificial Intelligence Pipelines Provide Insights for the Designing of Highly Specific AntiSARS-CoV2 Agents. Math. Comput. Sci. 2021. [Google Scholar] [CrossRef]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef]
- Kohen, Z.; Orenstein, D. Mathematical Modeling of Tech-Related Real-World Problems for Secondary School-Level Mathematics. Educ. Stud. Math. 2021, 107, 71–91. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In Silico Pharmacology for Drug Discovery: Methods for Virtual Ligand Screening and Profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.P.F.; Earp, J.C.; Florian, J.; Madabushi, R.; Strauss, D.G.; Wang, Y.; Zhu, H. Quantitative Systems Pharmacology: Landscape Analysis of Regulatory Submissions to the US Food and Drug Administration. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of Computer-Aided Drug Design in Modern Drug Discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, C.; Chandra, I.; Singh, S.K. Artificial Intelligence and Machine Learning Approaches for Drug Design: Challenges and Opportunities for the Pharmaceutical Industries. Mol. Divers. 2021, 1, 1–21. [Google Scholar] [CrossRef]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure Based Pharmacophore Modeling, Virtual Screening, Molecular Docking and ADMET Approaches for Identification of Natural Anti-Cancer Agents Targeting XIAP Protein. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In Silico Methods and Tools for Drug Discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A Practical Guide to Large-Scale Docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef]
- Trabelsi, S.; Issaoui, N.; Brandán, S.A.; Bardak, F.; Roisnel, T.; Atac, A.; Marouani, H. Synthesis and Physic-Chemical Properties of a Novel Chromate Compound with Potential Biological Applications, Bis(2-Phenylethylammonium) Chromate(VI). J. Mol. Struct. 2019, 1185, 168–182. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, M.Z.; Attique, S.A.; Ain, Q.; Alghamdi, H.A.; Bilal, M.; Yan, W.; Zhu, D. Discovery and Characterization of Dual Inhibitors of Human Vanin-1 and Vanin-2 Enzymes through Molecular Docking and Dynamic Simulation-Based Approach. Int. J. Biol. Macromol. 2022, 213, 1088–1097. [Google Scholar] [CrossRef]
- Samad, A.; Ahammad, F.; Nain, Z.; Alam, R.; Imon, R.R.; Hasan, M.; Rahman, M.S. Designing a Multi-Epitope Vaccine against SARS-CoV-2: An Immunoinformatics Approach. J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for Bad Bugs: Confronting the Challenges of Antibacterial Discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Gerry, C.J.; Schreiber, S.L. Chemical Probes and Drug Leads from Advances in Synthetic Planning and Methodology. Nat. Rev. Drug Discov. 2018, 17, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Hotamisligil, G.S. Fatty Acid-Binding Proteins: Role in Metabolic Diseases and Potential as Drug Targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Eckhardt, M.; Hultquist, J.F.; Kaake, R.M.; Hüttenhain, R.; Krogan, N.J. A Systems Approach to Infectious Disease. Nat. Rev. Genet. 2020, 21, 339–354. [Google Scholar] [CrossRef]
- Nogales, C.; Mamdouh, Z.M.; List, M.; Kiel, C.; Casas, A.I.; Schmidt, H.H.H.W. Network Pharmacology: Curing Causal Mechanisms Instead of Treating Symptoms. Trends Pharmacol. Sci. 2022, 43, 136–150. [Google Scholar] [CrossRef]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G Protein-Coupled Receptors: Structure- and Function-Based Drug Discovery. Signal Transduct. Target. Ther. 2021, 6, 1–27. [Google Scholar] [CrossRef]
- Zhao, L.; Zhu, Y.; Wang, J.; Wen, N.; Wang, C.; Cheng, L. A Brief Review of Protein–Ligand Interaction Prediction. Comput. Struct. Biotechnol. J. 2022, 20, 2831–2838. [Google Scholar] [CrossRef]
- Choudhary, K.; DeCost, B.; Chen, C.; Jain, A.; Tavazza, F.; Cohn, R.; Park, C.W.; Choudhary, A.; Agrawal, A.; Billinge, S.J.L.; et al. Recent Advances and Applications of Deep Learning Methods in Materials Science. NPJ Comput. Mater. 2022, 8, 59. [Google Scholar] [CrossRef]
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef] [Green Version]
- Marino, S.; Baxter, N.T.; Huffnagle, G.B.; Petrosino, J.F.; Schloss, P.D. Mathematical Modeling of Primary Succession of Murine Intestinal Microbiota. Proc. Natl. Acad. Sci. USA 2014, 111, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Demers, J.; Robertson, S.L.; Bewick, S.; Fagan, W.F. Implicit versus Explicit Vector Management Strategies in Models for Vector-Borne Disease Epidemiology. J. Math. Biol. 2022, 84, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Kantae, V.; Krekels, E.H.J.; Esdonk, M.J.V.; Lindenburg, P.; Harms, A.C.; Knibbe, C.A.J.; Van der Graaf, P.H.; Hankemeier, T. Integration of Pharmacometabolomics with Pharmacokinetics and Pharmacodynamics: Towards Personalized Drug Therapy. Metabolomics 2017, 13, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Hu, B. Mathematical Modeling and Computational Prediction of Cancer Drug Resistance. Brief. Bioinform. 2017, 19, 1382–1399. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Sarkar, R.; Sinha, S. Mathematical Models of Malaria—A Review. Malar. J. 2011, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Alam, R.; Imon, R.R.; Kabir Talukder, M.E.; Akhter, S.; Hossain, M.A.; Ahammad, F.; Rahman, M.M. GC-MS Analysis of Phytoconstituents FromRuellia ProstrataandSenna Toraand Identification of Potential Anti-Viral Activity against SARS-CoV-2. RSC Adv. 2021, 11, 40120–40135. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Yang, Y.; He, Y.; Wang, X.; Zhang, P.; Li, H.; Liang, S. Synthetic Biology Speeds Up Drug Target Discovery. Front. Pharmacol. 2020, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Kashkooli, F.M.; Abazari, M.A.; Soltani, M.; Ghazani, M.A.; Rahmim, A. A Spatiotemporal Multi-Scale Computational Model for FDG PET Imaging at Different Stages of Tumor Growth and Angiogenesis. Sci. Rep. 2022, 12, 10062. [Google Scholar] [CrossRef]
- Siddiqui, M.R.; AlOthman, Z.A.; Rahman, N. Analytical Techniques in Pharmaceutical Analysis: A Review. Arab. J. Chem. 2017, 10, S1409–S1421. [Google Scholar] [CrossRef] [Green Version]
- Masuo, Y.; Futatsugi, A.; Kato, Y. Experimental Approaches For Studying Drug Transporters. Drug Transp. 2022, 413–431. [Google Scholar] [CrossRef]
- Fischer, H.P. Mathematical Modeling of Complex Biological Systems: From Parts Lists to Understanding Systems Behavior. Alcohol Res. Heal. 2008, 31, 49–59. [Google Scholar]
- Heikkinen, H.A.; Backlund, S.M.; Iwaï, H. Nmr Structure Determinations of Small Proteins Using Only One Fractionally 20%13 C-and Uniformly 100%15 n-Labeled Sample. Molecules 2021, 26, 747. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Marshall, G.R.; Taylor, C.M. Introduction to Computer-Assisted Drug Design—Overview and Perspective for the Future. In Comprehensive Medicinal Chemistry II; Elsevier Ltd.: Amsterdam, The Netherlands, 2006; Volume 4, pp. 13–41. ISBN 9780080450445. [Google Scholar]
- Dill, K.A.; Ozkan, S.B.; Shell, M.S.; Weikl, T.R. The Protein Folding Problem. Annu. Rev. Biophys. 2008, 37, 289–316. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Lambert, C.; Léonard, N.; De Bolle, X.; Depiereux, E. ESyPred3D: Prediction of Proteins 3D Structures. Bioinformatics 2002, 18, 1250–1256. [Google Scholar] [CrossRef]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-Suite3 for Fast Remote Homology Detection and Deep Protein Annotation. BMC Bioinform. 2019, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-Based Protein Structure Modeling Using the RaptorX Web Server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [Green Version]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX Web Server: An Online Force Field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein Structure Prediction and Analysis Using the Robetta Server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaram, B.; Dhingra, P.; Mishra, A.; Kaushik, R.; Mukherjee, G.; Singh, A.; Shekhar, S. Bhageerath-H: A Homology/Ab Initio Hybrid Server for Predicting Tertiary Structures of Monomeric Soluble Proteins. BMC Bioinform. 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, P.; Li, S.; Xu, C.; Zhang, T.; Zhao, J.; Liu, Y.; Wang, Y.; Xie, Y. PRIME: A Novel Processing-in-Memory Architecture for Neural Network Computation in ReRAM-Based Main Memory. In Proceedings of the Proceedings—2016 43rd International Symposium on Computer Architecture, ISCA 2016, Seoul, Korea, 18–22 June 2016; Volume 44, pp. 27–39. [Google Scholar]
- Wu, S.; Zhang, Y. LOMETS: A Local Meta-Threading-Server for Protein Structure Prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Huang, Z.; Wang, Y.; Wen, C.; Peng, Y.; Ye, Y. Information Extraction from the Text Data on Traditional Chinese Medicine: A Review on Tasks, Challenges, and Methods from 2010 to 2021. Evidence-Based Complement. Altern. Med. 2022, 2022, 1679589. [Google Scholar] [CrossRef] [PubMed]
- Ou-Yang, S.S.; Lu, J.Y.; Kong, X.Q.; Liang, Z.J.; Luo, C.; Jiang, H. Computational Drug Discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahammad, F.; Alam, R.; Mahmud, R.; Akhter, S.; Talukder, E.K.; Tonmoy, A.M.; Fahim, S.; Al-Ghamdi, K.; Samad, A.; Qadri, I. Pharmacoinformatics and Molecular Dynamics Simulation-Based Phytochemical Screening of Neem Plant (Azadiractha Indica) against Human Cancer by Targeting MCM7 Protein. Brief. Bioinform. 2021, 22, bbab098. [Google Scholar] [CrossRef]
- Santos-Filho, A.; Giordano, D.; Biancaniello, C.; Argenio, M.A.; Facchiano, A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharmaceuticals 2022, 15, 646. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-Based Virtual Screening for Drug Discovery: A Problem-Centric Review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Gaurav, A.; Gautam, V. Pharmacophore Based Virtual Screening Approach to Identify Selective PDE4B Inhibitors. Iran. J. Pharm. Res. 2017, 16, 910–923. [Google Scholar]
- Watson, P.F.; Petrie, A. Method Agreement Analysis: A Review of Correct Methodology. Theriogenology 2010, 73, 1167–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.J.; Wang, Y.L.; Wang, Q.H.; Wang, J.; Cheng, M.S. In Silico Design of Human IMPDH Inhibitors Using Pharmacophore Mapping and Molecular Docking Approaches. Comput. Math. Methods Med. 2015, 2015, 418767. [Google Scholar] [CrossRef] [PubMed]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Devnath, H.S.; Medha, M.M.; Biswas, R.P.; Biswas, N.N.; Biswas, B.; Sadhu, S.K. In Silico Profiling of Analgesic, Antidiarrheal and Antihyperglycemic Properties of Tetrastigma Bracteolatum (Wall.) Leaves Extract Supported by in Vivo Studies. Adv. Tradit. Med. 2022, 1–13. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Noureddine, O.; Issaoui, N.; Al-Dossary, O. DFT and Molecular Docking Study of Chloroquine Derivatives as Antiviral to Coronavirus COVID-19. J. King Saud Univ. Sci. 2021, 33, 101248. [Google Scholar] [CrossRef]
- Hernndez-Santoyo, A.; Yair, A.; Altuzar, V.; Vivanco-Cid, H.; Mendoza-Barrer, C. Protein-Protein and Protein-Ligand Docking. In Protein Engineering Technology and Application; IntechOpen: London, UK, 2013; ISBN 978-953-51-1138-2. [Google Scholar]
- Jaishree, V.; Ul Haq, F. Antimicrobial Evaluation and Molecular Docking Studies of Swertiamarin and Quercetin Targeting Dihydropteroate Synthase Enzyme. Adv. Tradit. Med. 2022, 1–7. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [Green Version]
- Noureddine, O.; Issaoui, N.; Medimagh, M.; Al-Dossary, O.; Marouani, H. Quantum Chemical Studies on Molecular Structure, AIM, ELF, RDG and Antiviral Activities of Hybrid Hydroxychloroquine in the Treatment of COVID-19: Molecular Docking and DFT Calculations. J. King Saud Univ. Sci. 2021, 33, 101334. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. Advances and Challenges in Protein-Ligand Docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [Green Version]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated Docking of Flexible Ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Sobolev, V.; Eyal, E.; Gerzon, S.; Potapov, V.; Babor, M.; Prilusky, J.; Edelman, M. SPACE: A Suite of Tools for Protein Structure Prediction and Analysis Based on Complementarity and Environment. Nucleic Acids Res. 2005, 33, W39–W43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for Rigid and Symmetric Docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, D.W. Evaluation of Protein Docking Predictions Using Hex 3.1 in CAPRI Rounds 1 and 2. Proteins Struct. Funct. Genet. 2003, 52, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Gasch, T.; Stahl, M. Binding Site Characteristics in Structure-Based Virtual Screening: Evaluation of Current Docking Tools. J. Mol. Model. 2003, 9, 47–57. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Mottarella, S.E.; Beglov, D.; Beglova, N.; Nugent, M.A.; Kozakov, D.; Vajda, S. Docking Server for the Identification of Heparin Binding Sites on Proteins. J. Chem. Inf. Model. 2014, 54, 2068–2078. [Google Scholar] [CrossRef]
- Bitencourt-Ferreira, G.; de Azevedo, W.F. Docking with SwissDock. Methods Mol. Biol. 2019, 2053, 189–202. [Google Scholar]
- Verdonk, M.L.; Giangreco, I.; Hall, R.J.; Korb, O.; Mortenson, P.N.; Murray, C.W. Docking Performance of Fragments and Druglike Compounds. J. Med. Chem. 2011, 54, 5422–5431. [Google Scholar] [CrossRef]
- Wang, J.; Dokholyan, N.V. MedusaDock 2.0: Efficient and Accurate Protein-Ligand Docking with Constraints. J. Chem. Inf. Model. 2019, 59, 2509–2515. [Google Scholar] [CrossRef]
- Bitencourt-Ferreira, G.; de Azevedo, W.F. Molegro Virtual Docker for Docking. Methods Mol. Biol. 2019, 2053, 149–167. [Google Scholar] [PubMed]
- Paul, D.S.; Gautham, N. MOLS 2.0: Software Package for Peptide Modeling and Protein–Ligand Docking. J. Mol. Model. 2016, 22, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, R.; Pippel, M.; Brandt, F.; Sippl, W.; Baldauf, C. ParaDockS: A Framework for Molecular Docking with Population-Based Metaheuristics. J. Chem. Inf. Model. 2010, 50, 879–889. [Google Scholar] [CrossRef]
- Aparoy, P.; Kumar Reddy, K.; Reddanna, P. Structure and Ligand Based Drug Design Strategies in the Development of Novel 5- LOX Inhibitors. Curr. Med. Chem. 2012, 19, 3763–3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberto, A.V.P.; da Silva Ferreira, N.C.; Soares, R.F.; Alves, L.A. Molecular Modeling Applied to the Discovery of New Lead Compounds for P2 Receptors Based on Natural Sources. Front. Pharmacol. 2020, 11, 1221. [Google Scholar] [CrossRef]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myint, K.Z.; Xie, X.Q. Recent Advances in Fragment-Based QSAR and Multi-Dimensional QSAR Methods. Int. J. Mol. Sci. 2010, 11, 3846–3866. [Google Scholar] [CrossRef]
- Peterson, L. K-Nearest Neighbor. Scholarpedia 2009, 4, 1883. [Google Scholar] [CrossRef]
- Uyanık, G.K.; Güler, N. A Study on Multiple Linear Regression Analysis. Procedia Soc. Behav. Sci. 2013, 106, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Rosipal, R.; Krämer, N. Overview and Recent Advances in Partial Least Squares. In Proceedings of the Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin/Heidelberg, Germany, 2006; Volume 3940 LNCS, pp. 34–51. [Google Scholar]
- Abiodun, O.I.; Jantan, A.; Omolara, A.E.; Dada, K.V.; Mohamed, N.A.E.; Arshad, H. State-of-the-Art in Artificial Neural Network Applications: A Survey. Heliyon 2018, 4, e00938. [Google Scholar] [CrossRef] [Green Version]
- Noble, W.S. What Is a Support Vector Machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Myles, A.J.; Feudale, R.N.; Liu, Y.; Woody, N.A.; Brown, S.D. An Introduction to Decision Tree Modeling. J. Chemom. 2004, 18, 275–285. [Google Scholar] [CrossRef]
- Belgiu, M.; Drăgu, L. Random Forest in Remote Sensing: A Review of Applications and Future Directions. ISPRS J. Photogramm. Remote Sens. 2016, 114, 24–31. [Google Scholar] [CrossRef]
- Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D.; Willett, P. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules 2015, 20, 22799–22832. [Google Scholar] [CrossRef] [Green Version]
- Anderson, A.C.; O’Neil, R.H.; Surti, T.S.; Stroud, R.M. Approaches to Solving the Rigid Receptor Problem by Identifying a Minimal Set of Flexible Residues during Ligand Docking. Chem. Biol. 2001, 8, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, R.; Jain, P.; Narayan Dikshit, S. Ligand-Based Pharmacophore Detection, Screening of Potential Gliptins and Docking Studies to Get Effective Antidiabetic Agents. Comb. Chem. High Throughput Screen. 2014, 15, 849–876. [Google Scholar] [CrossRef]
- Taminau, J.; Thijs, G.; De Winter, H. Pharao: Pharmacophore Alignment and Optimization. J. Mol. Graph. Model. 2008, 27, 161–169. [Google Scholar] [CrossRef]
- Kurogi, Y.; Guner, O. Pharmacophore Modeling and Three-Dimensional Database Searching for Drug Design Using Catalyst. Curr. Med. Chem. 2012, 8, 1035–1055. [Google Scholar] [CrossRef]
- Chen, I.J.; Foloppe, N. Conformational Sampling of Druglike Molecules with MOE and Catalyst: Implications for Pharmacophore Modeling and Virtual Screening. J. Chem. Inf. Model. 2008, 48, 1773–1791. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Mallik, B.; Morìkis, D. Development of a Quasi-Dynamic Pharmacophore Model for Anti-Complement Peptide Analogues. J. Am. Chem. Soc. 2005, 127, 10967–10976. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Camacho, C.J. Pharmer: Efficient and Exact Pharmacophore Search. J. Chem. Inf. Model. 2011, 51, 1307–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosco, P.; Balle, T. Open3DQSAR: A New Open-Source Software Aimed at High-Throughput Chemometric Analysis of Molecular Interaction Fields. J. Mol. Model. 2011, 17, 201–208. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A Webserver for Ligand-Based Pharmacophore Detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. GRID-Based Three-Dimensional Pharmacophores I: FLAPpharm, a Novel Approach for Pharmacophore Elucidation. J. Chem. Inf. Model. 2012, 52, 2587–2598. [Google Scholar] [CrossRef]
- Bocci, G.; Carosati, E.; Vayer, P.; Arrault, A.; Lozano, S.; Cruciani, G. ADME-Space: A New Tool for Medicinal Chemists to Explore ADME Properties. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Li, A.P. Screening for Human ADME/Tox Drug Properties in Drug Discovery. Drug Discov. Today 2001, 6, 357–366. [Google Scholar] [CrossRef]
- Flynn, E. Drug Bioavailability. In xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands; Boston, MA, USA, 2007; pp. 1–2. ISBN 9780080552323. [Google Scholar]
- Zhao, Y.; Roy, K.; Vidossich, P.; Cancedda, L.; De Vivo, M.; Forbush, B.; Cao, E. Structural Basis for Inhibition of the Cation-Chloride Cotransporter NKCC1 by the Diuretic Drug Bumetanide. Nat. Commun. 2022, 13, 1–12. [Google Scholar] [CrossRef]
- Shen, Q.; Wang, L.; Zhou, H.; Jiang, H.-D.; Yu, L.S.; Zeng, S. Stereoselective Binding of Chiral Drugs to Plasma Proteins. Acta Pharmacol. Sin. 2013, 34, 998–1006. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Wei, T.; Goldberg, H.; Wang, W.; Cullion, K.; Kohane, D.S. Getting Drugs Across Biological Barriers. Adv. Mater. 2017, 29, 1606596. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Krämer, S.D. The Biochemistry of Drug Metabolism—An Introduction Part 1. Principles and Overview. Chem. Biodivers. 2006, 3, 1053–1101. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy Metabolism in the Liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Wang, N.N.; Yao, Z.J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.P.; Cao, D.S. Admetlab: A Platform for Systematic ADMET Evaluation Based on a Comprehensively Collected ADMET Database. J. Cheminform. 2018, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Svensson, F.; Zoufir, A.; Bender, A. EMolTox: Prediction of Molecular Toxicity with Confidence. Bioinformatics 2018, 34, 2508–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoofnagle, J.H. LiverTox: A Website on Drug-Induced Liver Injury. In Drug-Induced Liver Disease; Academic Press: Cambridge, MA, USA, 2013; pp. 725–732. ISBN 9780123878175. [Google Scholar]
- Schyman, P.; Liu, R.; Desai, V.; Wallqvist, A. VNN Web Server for ADMET Predictions. Front. Pharmacol. 2017, 8, 889. [Google Scholar] [CrossRef] [Green Version]
- Viana Nunes, A.M.; das Chagas Pereira de Andrade, F.; Filgueiras, L.A.; de Carvalho Maia, O.A.; Cunha, R.L.O.R.; Rodezno, S.V.A.; Maia Filho, A.L.M.; de Amorim Carvalho, F.A.; Braz, D.C.; Mendes, A.N. PreADMET Analysis and Clinical Aspects of Dogs Treated with the Organotellurium Compound RF07: A Possible Control for Canine Visceral Leishmaniasis? Environ. Toxicol. Pharmacol. 2020, 80, 103470. [Google Scholar] [CrossRef]
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Richard, A.M.; Williams, C.L.R. Distributed Structure-Searchable Toxicity (DSSTox) Public Database Network: A Proposal. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2002, 499, 27–52. [Google Scholar] [CrossRef]
- Ekins, S. In Silico Approaches to Predicting Drug Metabolism, Toxicology and Beyond. Biochem. Soc. Trans. 2003, 31, 611–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesharwani, R.K.; Vishwakarma, V.K.; Keservani, R.K.; Singh, P.; Katiyar, N.; Tripathi, S. Role of ADMET Tools in Current Scenario: Application and Limitations. In Computer-Aided Drug Design; Springer: Singapore, 2020; pp. 71–87. ISBN 10.1007/9789811. [Google Scholar]
- Mamadalieva, N.Z.; Youssef, F.S.; Hussain, H.; Zengin, G.; Mollica, A.; Musayeib, N.M.A.; Ashour, M.L.; Westermann, B.; Wessjohann, L.A. Validation of the Antioxidant and Enzyme Inhibitory Potential of Selected Triterpenes Using in Vitro and in Silico Studies, and the Evaluation of Their Admet Properties. Molecules 2021, 26, 6331. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, R.; Friend, S.H. Toxicogenomics and Drug Discovery: Will New Technologies Help Us Produce Better Drugs? Nat. Rev. Drug Discov. 2002, 1, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ciallella, H.L.; Aleksunes, L.M.; Zhu, H. Advancing Computer-Aided Drug Discovery (CADD) by Big Data and Data-Driven Machine Learning Modeling. Drug Discov. Today 2020, 25, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; kumar, R.; Sulabh, G.; Singh, P.; Kumar, J.; Singh, V.K.; Ojha, K.K. In Silico ADMET, Molecular Docking and Molecular Simulation-Based Study of Glabridin’s Natural and Semisynthetic Derivatives as Potential Tyrosinase Inhibitors. Adv. Tradit. Med. 2022, 1–19. [Google Scholar] [CrossRef]
- Petraccone, L.; Garbett, N.C.; Chaires, J.B.; Trent, J.O. An Integrated Molecular Dynamics (MD) and Experimental Study of Higher Order Human Telomeric Quadruplexes. Biopolymers 2010, 93, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Aljahdali, M.O.; Molla, M.H.R.; Ahammad, F. Compounds Identified from Marine Mangrove Plant (Avicennia Alba) as Potential Antiviral Drug Candidates against WDSV, an In-Silico Approach. Mar. Drugs 2021, 19, 253. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Forouzesh, N.; Mishra, N. An Effective MM/GBSA Protocol for Absolute Binding Free Energy Calculations: A Case Study on SARS-CoV-2 Spike Protein and the Human ACE2 Receptor. Molecules 2021, 26, 2383. [Google Scholar] [CrossRef]
- Jha, P.; Chaturvedi, S.; Bhat, R.; Jain, N.; Mishra, A.K. Insights of Ligand Binding in Modeled H5-HT1A Receptor: Homology Modeling, Docking, MM-GBSA, Screening and Molecular Dynamics. J. Biomol. Struct. Dyn. 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sargolzaei, M. Effect of Nelfinavir Stereoisomers on Coronavirus Main Protease: Molecular Docking, Molecular Dynamics Simulation and MM/GBSA Study. J. Mol. Graph. Model. 2021, 103, 107803. [Google Scholar] [CrossRef] [PubMed]
| No. | Programs | Application | Accessibility | Reference |
|---|---|---|---|---|
| 1. | AutoDock | It is employed in molecular docking. It predicts the binding capacity of a tiny chemical and assigns a target protein to a 3D structure | https://autodock.scripps.edu/ | [74] |
| 2. | LPCCSU | Based on a comprehensive investigation of interatomic interactions and interface complementarity | https://oca.weizmann.ac.il/oca-bin/lpccsu | [75] |
| 3. | PatchDock | The method performs rigid docking, with surface variability | https://bioinfo3d.cs.tau.ac.il/PatchDock/php.php | [76] |
| 4. | Hex | For docking studies | http://hex.loria.fr/ | [77] |
| 5. | Glide Schrodinger | Comprehensive molecular modeling and computer-aided drug development (CADD) tool | https://www.schrodinger.com/ | [78] |
| 6. | Molecular operating environment | Comprehensive molecular modeling and computer-aided drug development (CADD) tool | https://www.chemcomp.com/ | [79] |
| 7. | DockingServer | A user-friendly web-based interface that manages all elements of molecular docking. | https://www.dockingserver.com/web | [80] |
| 8. | SwissDock | A web service for predicting a protein’s association with a small molecule ligand. | http://www.swissdock.ch/ | [81] |
| 9. | LeDock | A molecular docking program for docking ligands with protein targets | http://www.lephar.com/software.htm | [82] |
| 10. | MedusaDock 2.0 | Fast flexible docking with a discrete rotamer library of ligands | https://dokhlab.med.psu.edu/cpi/#/MedusaDock | [83] |
| 11. | Molegro Virtual Docker | Based on a novel heuristic search method that integrates differential evolution and a cavity prediction algorithm | http://molexus.io/molegro-virtual-docker/ | [84] |
| 12. | MOLS 2.0 | Using mutually orthogonal Latin squares, induced-fit peptide–protein, and small molecule–protein docking | https://sourceforge.net/projects/mols2-0/ | [85] |
| 13. | ParaDockS | Metaheuristics for population-based molecular docking | http://www.paradocks.at/ | [86] |
| No | Techniques | Equation | Activity | Reference |
|---|---|---|---|---|
| 1. | K-nearest neighbor | Linear | Simple | [91] |
| 2. | Multiple linear regression | Linear | Simple | [92] |
| 3. | Partial least squares | Linear | Performs effectively on data including a big dataset | [93] |
| 4. | Artificial neural network | Nonlinear | Works well with nonlinear data | [94] |
| 5. | Support vector machine | Nonlinear | A most effective approach for classification and regression | [95] |
| 6. | Decision tree | Nonlinear | Extremely interpretable | [96] |
| 7. | Random forest | Nonlinear | A better and more reliable estimate | [97] |
| No. | Programs | Application | Accessibility | Reference |
|---|---|---|---|---|
| 1. | Align-it | Pharmacophore alignment | http://silicos-it.be/ | [101] |
| 2. | Catalyst | Pharmacophore modeling | http://accelrys.com/products/discovery-studio/pharmacophore.html | [102] |
| 3. | MOE | Pharmacophore modeling | http://www.chemcomp.com/MOE-Pharmacophore_Discovery.htm | [103] |
| 4. | LigandScout | Pharmacophore modeling | http://www.inteligand.com/ligandscout/ | [104] |
| 5. | Phase | Pharmacophore modeling | http://www.schrodinger.com/Phase/ | [105] |
| 6. | Quasi | Pharmacophore modeling | http://www.denovopharma.com/page2.asp?PageID=485 | [106] |
| 7. | Pharmer | Pharmacophore search | http://smoothdock.ccbb.pitt.edu/pharmer/ | [107] |
| 8. | Open3DQSAR | Exploration of pharmacophores using high-throughput chemometric analysis | http://open3dqsar.sourceforge.net/ | [108] |
| 9. | Pharmagist | A website for the discovery of ligand-based pharmacophores | https://bioinfo3d.cs.tau.ac.il/PharmaGist/ | [109] |
| 10. | FLAP | The fingerprints are characterized by pharmacophoric properties | https://www.moldiscovery.com/software/flap/ | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, M.R.; Alsaiari, A.A.; Fakhurji, B.Z.; Molla, M.H.R.; Asseri, A.H.; Sumon, M.A.A.; Park, M.N.; Ahammad, F.; Kim, B. Application of Mathematical Modeling and Computational Tools in the Modern Drug Design and Development Process. Molecules 2022, 27, 4169. https://doi.org/10.3390/molecules27134169
Hasan MR, Alsaiari AA, Fakhurji BZ, Molla MHR, Asseri AH, Sumon MAA, Park MN, Ahammad F, Kim B. Application of Mathematical Modeling and Computational Tools in the Modern Drug Design and Development Process. Molecules. 2022; 27(13):4169. https://doi.org/10.3390/molecules27134169
Chicago/Turabian StyleHasan, Md Rifat, Ahad Amer Alsaiari, Burhan Zain Fakhurji, Mohammad Habibur Rahman Molla, Amer H. Asseri, Md Afsar Ahmed Sumon, Moon Nyeo Park, Foysal Ahammad, and Bonglee Kim. 2022. "Application of Mathematical Modeling and Computational Tools in the Modern Drug Design and Development Process" Molecules 27, no. 13: 4169. https://doi.org/10.3390/molecules27134169
APA StyleHasan, M. R., Alsaiari, A. A., Fakhurji, B. Z., Molla, M. H. R., Asseri, A. H., Sumon, M. A. A., Park, M. N., Ahammad, F., & Kim, B. (2022). Application of Mathematical Modeling and Computational Tools in the Modern Drug Design and Development Process. Molecules, 27(13), 4169. https://doi.org/10.3390/molecules27134169