
Supramolecular cis-“Bis(Chelation)” of [M(CN)6]3− (M = CrIII, FeIII, CoIII) by Phloroglucinol (H3PG)

 , , , , , ,
, , , , , ,  and
and 
Abstract
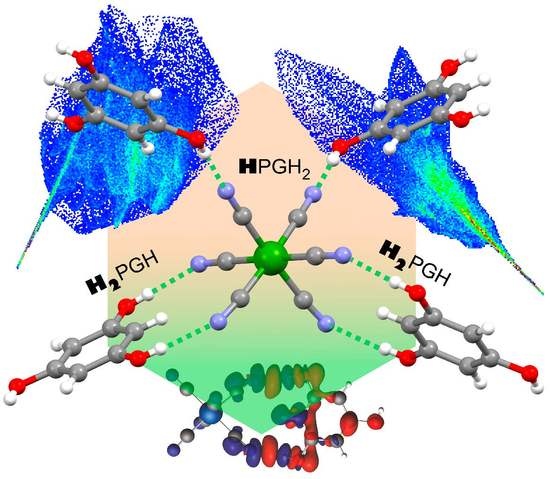
1. Introduction
2. Results and Discussion
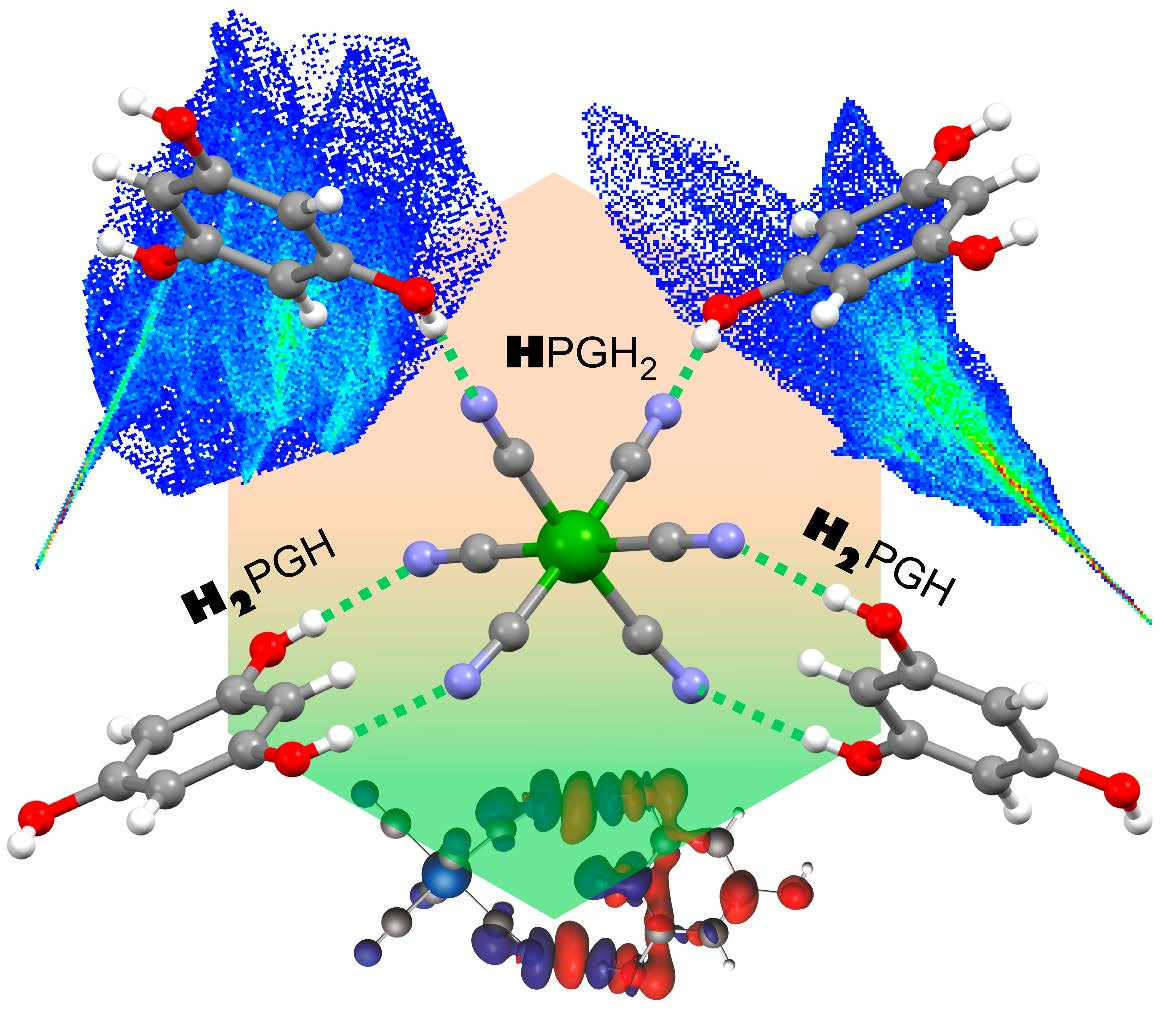
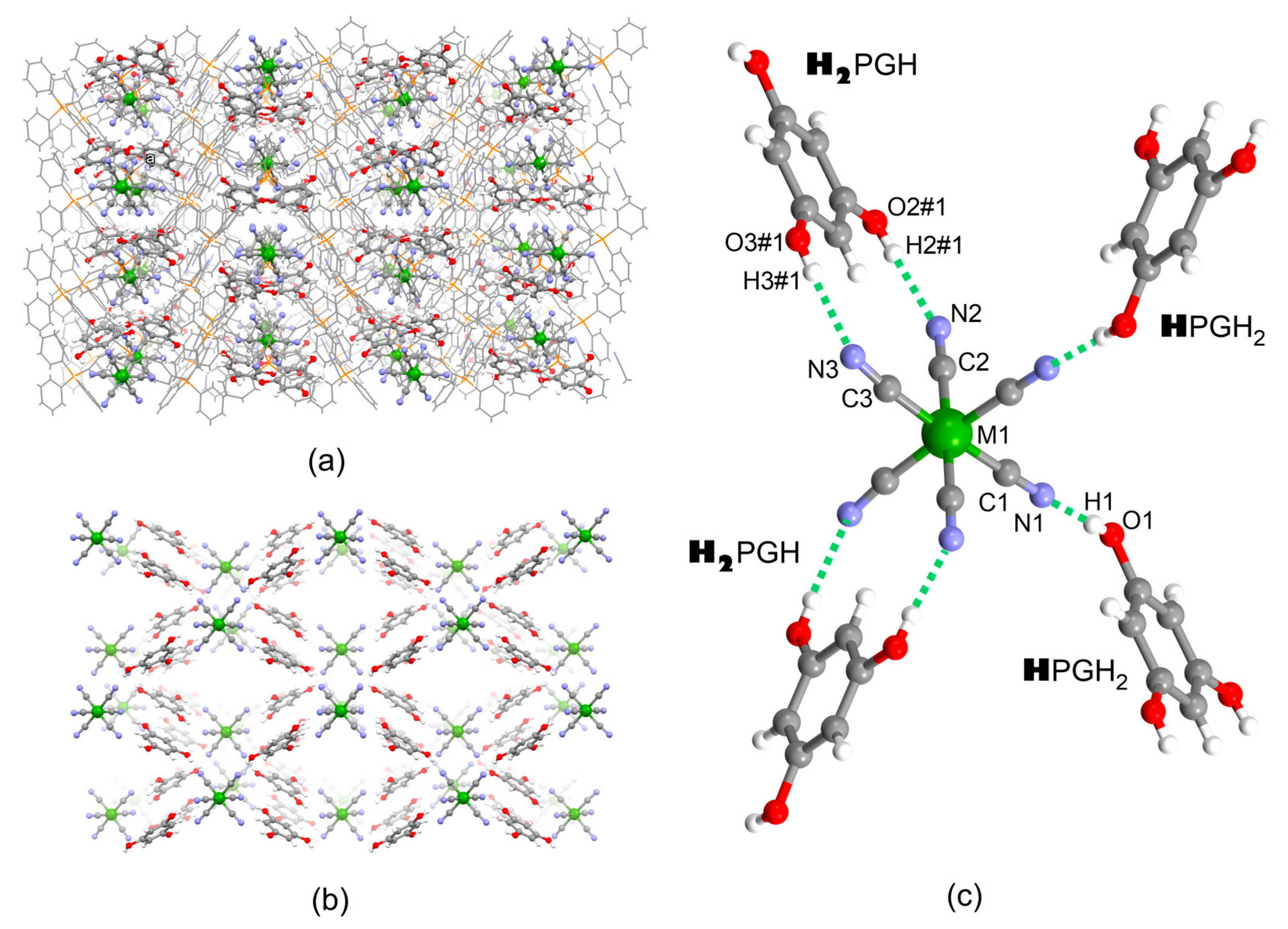
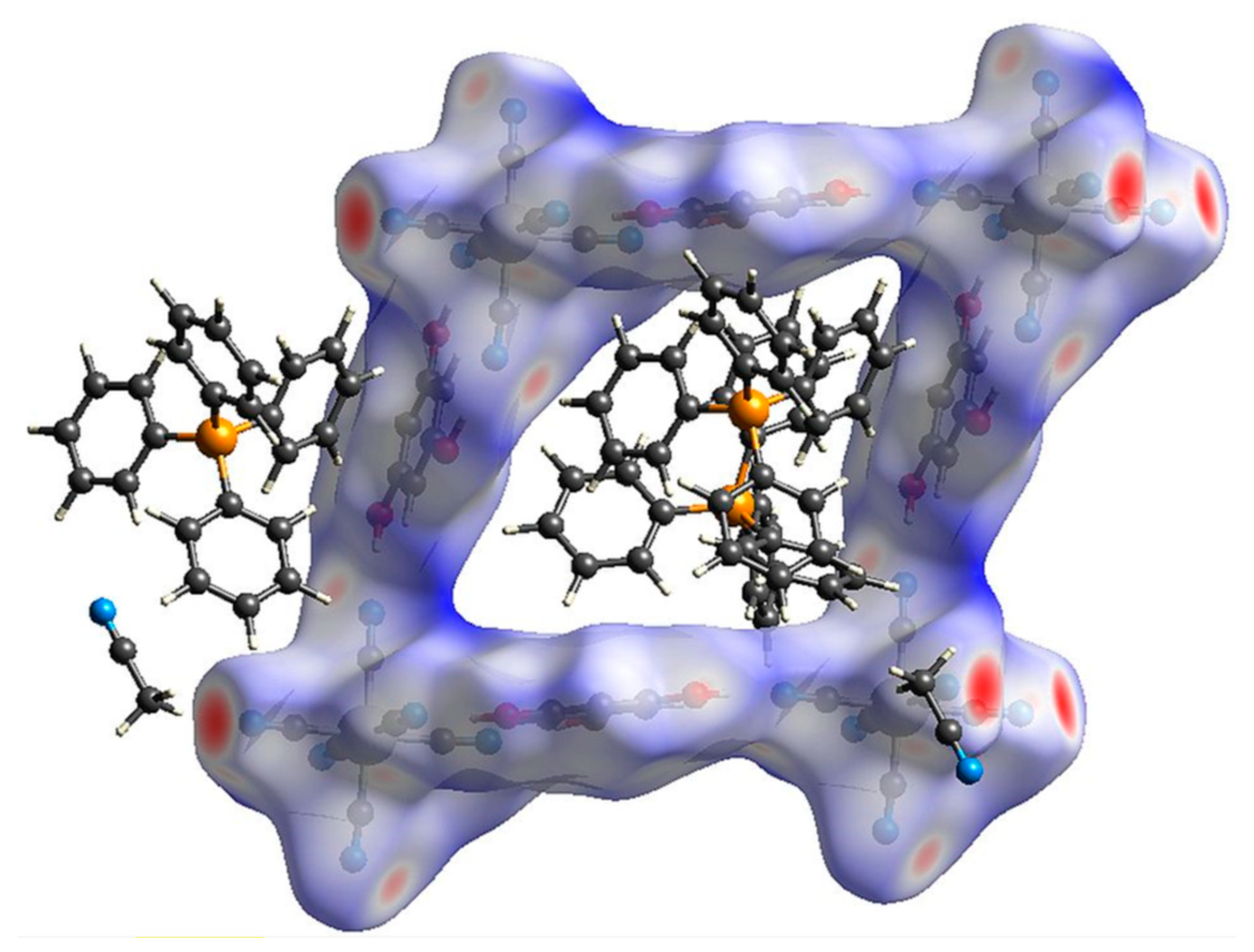
2.1. Structural Studies
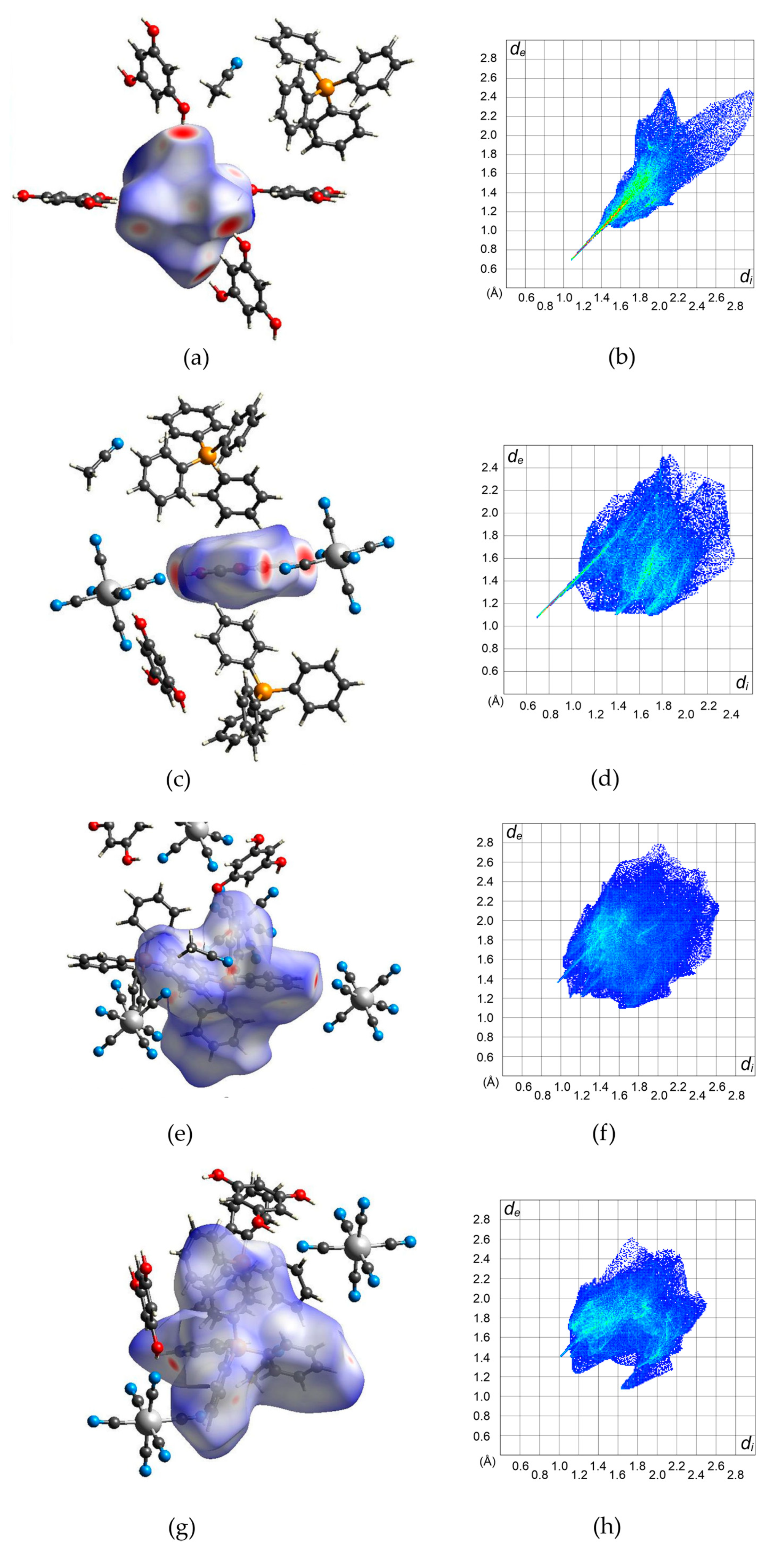
2.2. Hirshfeld Analysis
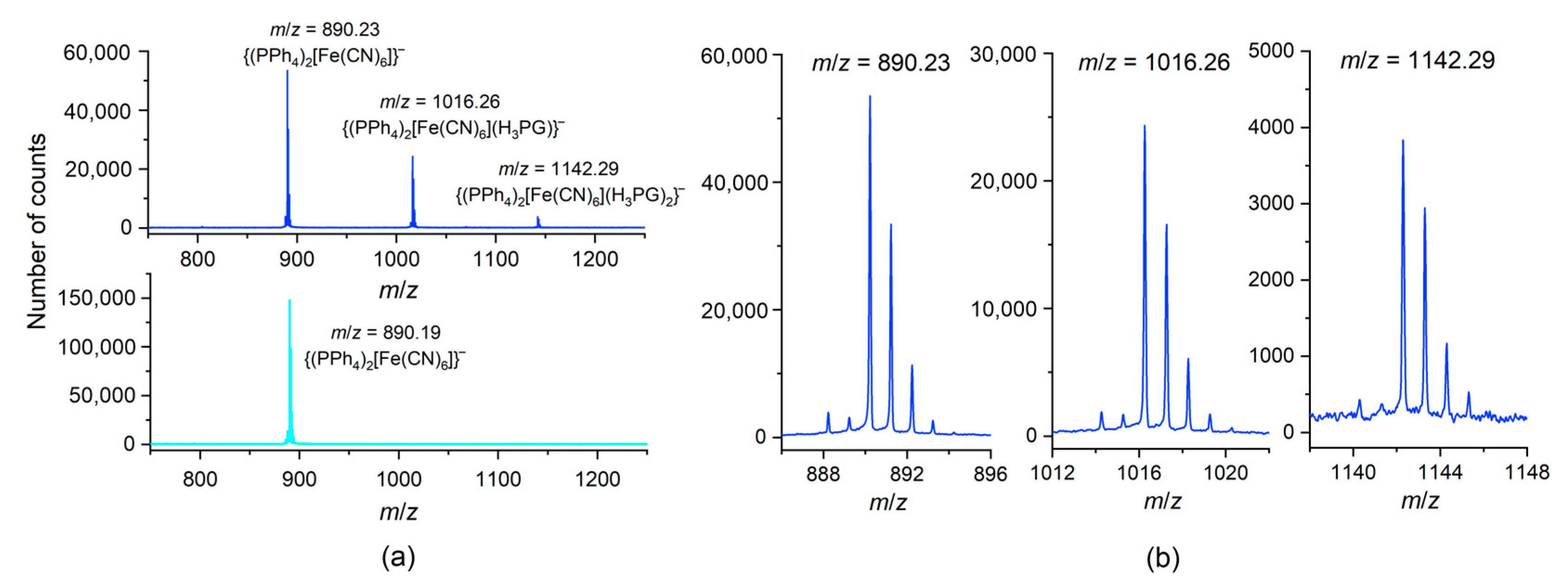
2.3. ESI-MS
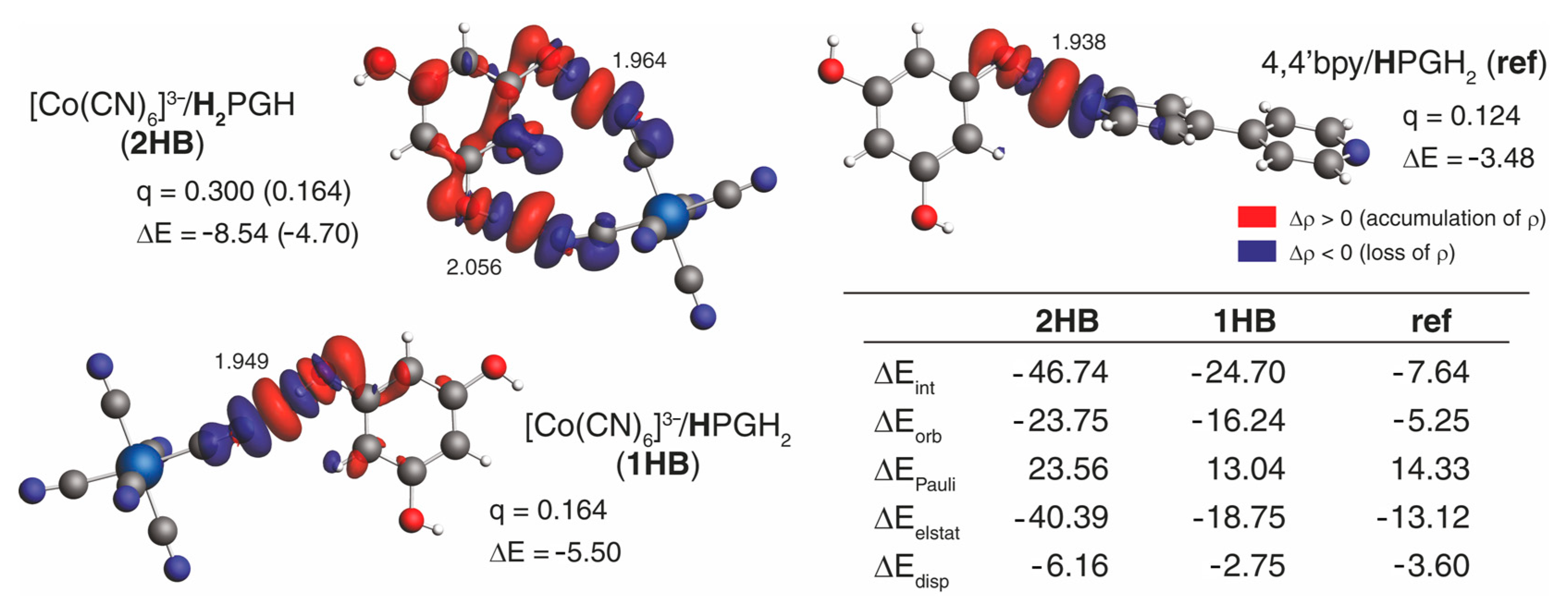
2.4. DFT Calculations
2.5. Spectroscopic Studies
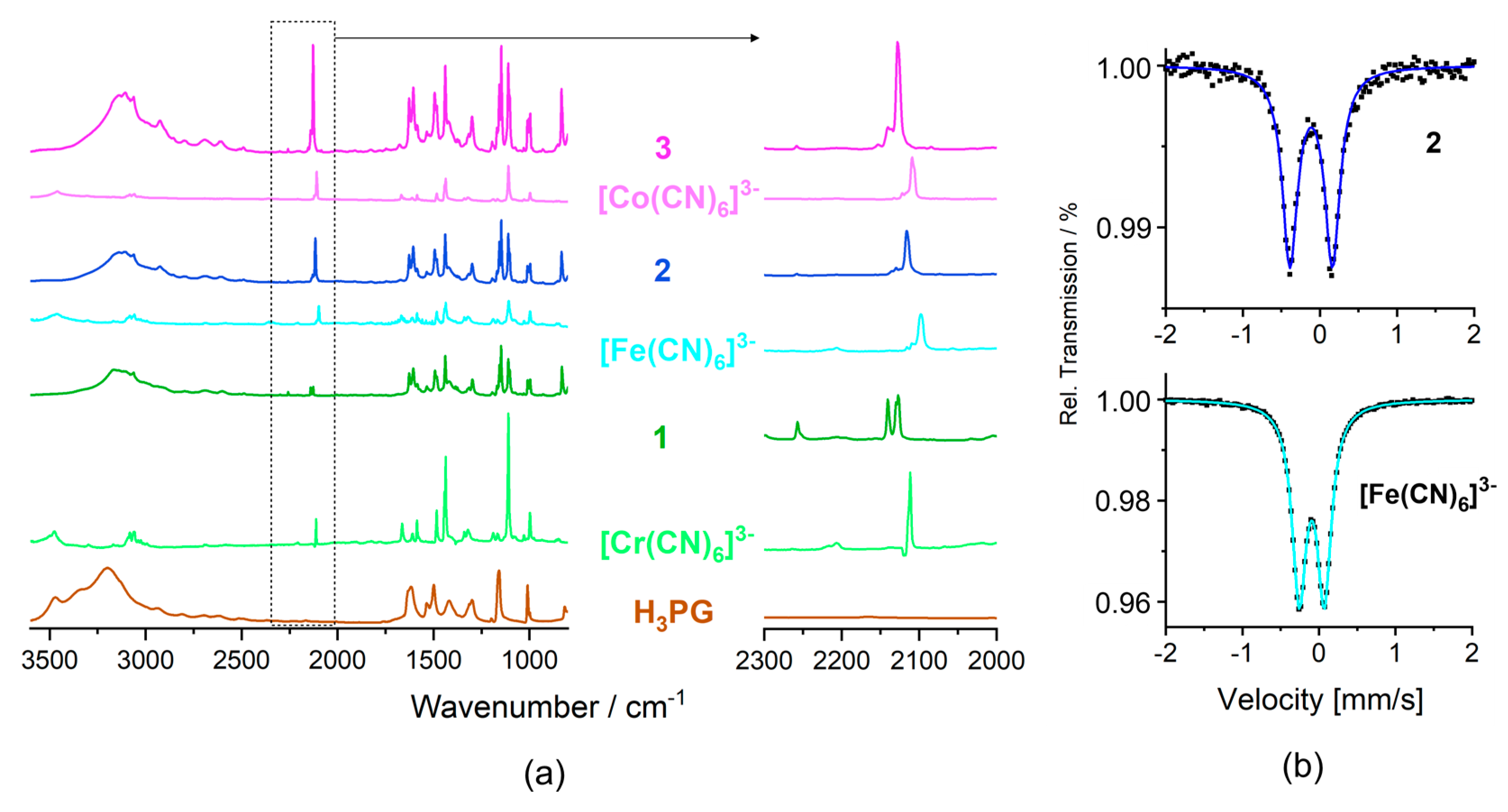
2.5.1. Vibrational Studies
2.5.2. 57Fe Mössbauer Spectra
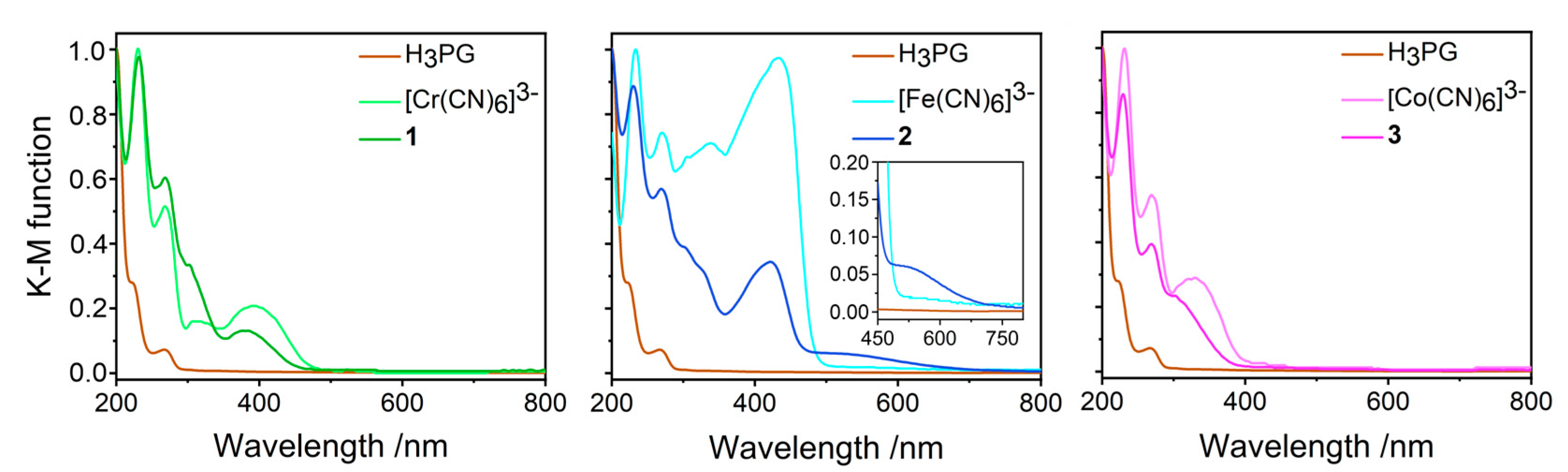
2.5.3. UV-Vis Electronic Absorption Spectra
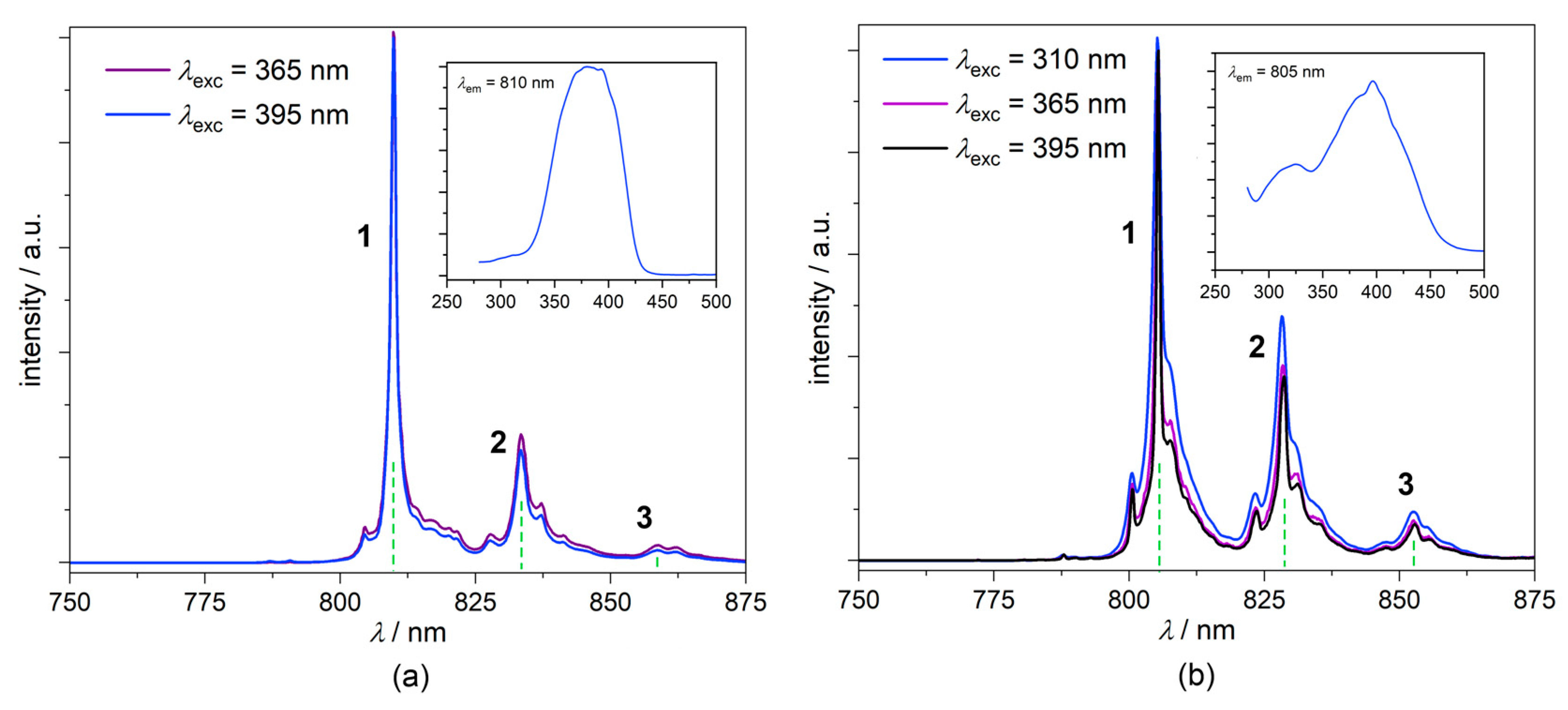
2.5.4. Photoluminescence Studies
3. Conclusions and Perspectives
4. Materials and Methods
4.1. Synthetic Procedures
4.2. X-ray Diffraction Analysis
4.3. Physical Techniques and Calculations
4.4. Hirshfeld Analysis
4.5. Quantum-Chemical Calculations and Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Avilaa, Y.; Acevedo-Peña, P.; Reguera, L.; Reguera, E. Recent progress in transition metal hexacyanometallates: From structure to properties and functionality. Coord. Chem. Rev. 2022, 453, 214274. [Google Scholar] [CrossRef]
- Zakrzewski, J.J.; Liberka, M.; Zychowicz, M.; Chorazy, S. Diverse physical functionalities of rare-earth hexacyanidometallate frameworks and their molecular analogues. Inorg. Chem. Front. 2021, 8, 452–483. [Google Scholar] [CrossRef]
- Chorazy, S.; Zakrzewski, J.J.; Magott, M.; Korzeniak, T.; Nowicka, B.; Pinkowicz, D.; Podgajny, R.; Sieklucka, B. Octacyanidometallates for multifunctional molecule-based materials. Chem. Soc. Rev. 2020, 49, 5945–6001. [Google Scholar] [CrossRef] [PubMed]
- Grothe, E.; Meekes, H.; Vlieg, E.; Ter Horst, J.H.; de Gelder, R. Solvates, salts, and cocrystals: A proposal for a feasible classification system. Cryst. Growth Des. 2016, 16, 3237–3243. [Google Scholar] [CrossRef]
- Zhang, W.; Ye, H.-Y.; Graf, R.; Spiess, H.W.; Yao, Y.-F.; Zhu, R.-Q.; Xiong, R.-G. Tunable and Switchable Dielectric Constant in an Amphidynamic Crystal. J. Am. Chem. Soc. 2013, 135, 5230–5233. [Google Scholar] [CrossRef]
- Qian, K.; Shao, F.; Yan, Z.; Pang, J.; Chen, X.; Yang, C. A perovskite-type cage compound as a temperature-triggered dielectric switchable material. CrystEngComm 2016, 18, 7671–7674. [Google Scholar] [CrossRef]
- Rok, M.; Moskwa, M.; Działowa, M.; Bieńko, A.; Rajnák, C.; Boča, R.; Bator, G. Multifunctional materials based on the double-perovskite organic—Inorganic hybrid (CH3NH3)2[KCr(CN)6] showing switchable dielectric, magnetic, and semiconducting behaviour. Dalton Trans. 2019, 48, 16650–16660. [Google Scholar] [CrossRef]
- Rok, M.; Bator, G.; Medycki, W.; Zamponi, M.; Balčiūnas, S.; Šimėnas, M.; Banys, J. Reorientational dynamics of organic cations in perovskite-like coordination polymers. Dalton Trans. 2018, 47, 17329–17341. [Google Scholar] [CrossRef]
- Ma̧czka, M.; Nowok, A.; Zarȩba, J.K.; Stefańska, D.; Ga̧gor, A.; Trzebiatowska, M.; Sieradzki, A. Near-Infrared Phosphorescent Hybrid Organic—Inorganic Perovskite with High-Contrast Dielectric and Third-Order Nonlinear Optical Switching Functionalities. ACS Appl. Mater. Interfaces 2022, 14, 1460–1471. [Google Scholar] [CrossRef]
- Shi, C.; Yu, H.; Wang, Q.-W.; Ye, L.; Gong, Z.-X.; Ma, J.-J.; Jiang, J.-Y.; Hua, M.-M.; Shuai, C.; Zhang, Y.; et al. Hybrid Organic–Inorganic Antiperovskites. Angew. Chem. Int. Ed. 2020, 59, 167–171. [Google Scholar] [CrossRef]
- Ferlay, S.; Félix, O.; Hosseini, M.W.; Planeix, J.-M.; Kyritsakas, N. Second sphere supramolecular chirality: Racemic hybrid H-bonded 2-D molecular networks. Chem. Commun. 2002, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Dechambenoit, P.; Ferlay, S.; Hosseini, M.W.; Planeix, J.-M.; Kyritsakas, N. Molecular tectonics: Control of packing of hybrid 1-D and 2-D H-bonded molecular networks formed between bisamidinium dication and cyanometallate anions. New J. Chem. 2006, 30, 1403–1410. [Google Scholar] [CrossRef]
- Dechambenoit, P.; Ferlay, S.; Kyritsakas, N.; Hosseini, M.W. Molecular Tectonics: Control of Reversible Water Release in Porous Charge-Assisted H-Bonded Networks. J. Am. Chem. Soc. 2008, 130, 17106–17113. [Google Scholar] [CrossRef] [PubMed]
- Dechambenoit, P.; Ferlay, S.; Kyritsakas, N.; Hosseini, M.W. In situ reduction of Fe(III) into Fe(II): An example of post-crystallisation transformation. Chem. Commun. 2009, 6798–6800. [Google Scholar] [CrossRef]
- Olmsted, B.K.; Ferlay, S.; Dechambenoit, P.; Hosseini, M.W.; Ward, M.D. Microscopic Topography of Heterocrystal Interfaces. Cryst. Growth Des. 2009, 9, 2841–2847. [Google Scholar] [CrossRef]
- Catalano, L.; Berthaud, J.; Dushaq, G.; Karothu, D.P.; Rezgui, R.; Rasras, M.; Ferlay, S.; Hosseini, M.W.; Naumov, P. Sequencing and Welding of Molecular Single-Crystal Optical Waveguides. Adv. Funct. Mater. 2020, 30, 2003443. [Google Scholar] [CrossRef]
- Ota, A.; Ouahab, L.; Golhen, S.; Yoshida, Y.; Maesato, M.; Saito, G.; Świetlik, R. Phase Transition from Mott Insulating Phase into the Charge Ordering Phase with Molecular Deformation in Charge-Transfer Salts κ-(ET)4[M(CN)6][N(C2H5)4]·2H2O (M = CoIII and FeIII). Chem. Mater. 2007, 19, 2455–2462. [Google Scholar] [CrossRef]
- Łapiński, A.; Świetlik, R.; Ouahab, L.; Golhen, S. Spectroscopic Studies of the Phase Transition from the Mott Insulator State to the Charge-Ordering State of κ-(ET)4[M(CN)6][N(C2H5)4]·2H2O (M = CoIII and FeIII) Salts. J. Phys. Chem. A 2013, 117, 5241–5250. [Google Scholar] [CrossRef]
- You, M.-H.; Li, M.-H.; Liu, Y.-F.; Li, H.-H.; Lin, M.-J. Unprecedented five-fold interpenetrated donor–acceptor hybrid heterostructure induced by anion–π interactions. CrystEngComm 2019, 21, 6688–6692. [Google Scholar] [CrossRef]
- Kobylarczyk, J.; Pinkowicz, D.; Srebro-Hooper, M.; Hooper, J.; Podgajny, R. Anion–π recognition between [M(CN)6]3− complexes and HAT(CN)6: Structural matching and electronic charge density modification. Dalton Trans. 2017, 46, 3482–3491. [Google Scholar] [CrossRef]
- Arranz-Mascarós, P.; Bazzicalupi, C.; Bianchi, A.; Giorgi, C.; Godino-Salido, M.-L.; Gutiérrez-Valero, M.-D.; Lopez-Garzón, R.; Savastano, M. Thermodynamics of Anion−π Interactions in Aqueous Solution. J. Am. Chem. Soc. 2013, 135, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Savastano, M.; Arranz-Mascarós, P.; Bazzicalupi, C.; Bianchi, A.; Giorgi, C.; Godino-Salido, M.L.; Gutiérrez-Valero, M.D.; López-Garzón, R. Binding and removal of octahedral, tetrahedral, square planar and linear anions in water by means of activated carbon functionalized with a pyrimidine-based anion receptor. RSC Adv. 2014, 4, 58505–58513. [Google Scholar] [CrossRef]
- Sekine, Y.; Nihei, M.; Oshio, H. Dimensionally Controlled Assembly of an External Stimuli-Responsive [Co2Fe2] Complex into Supramolecular Hydrogen-Bonded Networks. Chem. Eur. J. 2017, 23, 5193–5197. [Google Scholar] [CrossRef] [PubMed]
- Santra, R.; Biradha, K. Solid state double [2 + 2] photochemical reactions in the co-crystal forms of 1,5-bis(4-pyridyl)-1,4-pentadiene-3-one: Establishing mechanism using single crystal X-ray, UV and 1H NMR. CrystEngComm 2011, 13, 3246–3257. [Google Scholar] [CrossRef]
- Dubey, R.; Desiraju, G.R. Combinatorial Crystal Synthesis: Structural Landscape of Phloroglucinol:1,2-bis(4-pyridyl)ethylene and Phloroglucinol:Phenazine. Angew. Chem. Int. Ed. 2014, 53, 13178–13182. [Google Scholar] [CrossRef]
- Thomas, L.H.; Craig, G.A.; Morrison, C.A.; Reilly, A.M.; Wilson, C.C. New Route to Local Order Models for Disordered Crystalline Materials: Diffuse Scattering and Computational Modeling of Phloroglucinol Dihydrate. Cryst. Growth Des. 2011, 11, 2045–2049. [Google Scholar] [CrossRef][Green Version]
- Guo, C.; Zhang, Q.; Zhu, B.; Zhang, Z.; Ma, X.; Dai, W.; Gong, X.; Ren, G.; Mei, X. Drug–Drug Cocrystals Provide Significant Improvements of Drug Properties in Treatment with Progesterone. Cryst. Growth Des. 2020, 20, 3053–3063. [Google Scholar] [CrossRef]
- Pal Singh, M.; Baruah, J.B. Detection of hydroxyaromatics in a superior manner by a water soluble fluorescent iron-complex. Inorg. Chim. Acta 2020, 504, 119467. [Google Scholar] [CrossRef]
- Sarma, B.; Sreenivas Reddy, L.; Nangia, A. The Role of π-Stacking in the Composition of Phloroglucinol and Phenazine Cocrystals. Cryst Growth Des. 2008, 8, 4546–4552. [Google Scholar] [CrossRef]
- Braun, D.E.; Tocher, D.A.; Price, S.L.; Griesser, U.J. The Complexity of Hydration of Phloroglucinol: A Comprehensive Structural and Thermodynamic Characterization. J. Phys. Chem. B 2012, 116, 3961–3972. [Google Scholar] [CrossRef]
- Shanmugaraju, S.; Bar, A.K.; Mukherjee, P.S. Ruthenium−Oxygen Coordination-Driven Self-Assembly of a RuII8 Incomplete Prism: Synthesis, Structure, and Shape-Selective Molecular Recognition Study. Inorg. Chem. 2010, 49, 10235–10237. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Lee, I.S.; Chung, Y.K. Crystal Engineering with Structurally Flexible 1,1′-Substituted Ferrocenes for Nonlinear Optical Materials. Eur. J. Inorg. Chem. 2003, 2003, 2311–2317. [Google Scholar] [CrossRef]
- Arora, K.K.; Talwelkar, M.S.; Pedireddi, V.R. Supramolecular synthesis of some molecular adducts of 4,4′-bipyridine N,N′-dioxide. New J. Chem. 2009, 33, 57–63. [Google Scholar] [CrossRef]
- Shankar, K.; Singh, M.P.; Baruah, J.B. Extent of protonation of 4,4′-bipyridinium cations and nature of host influences the amount of guest intake by cobalt(II) 2,6-pyridinedicarboxylate. Inorg. Chim. Acta 2018, 469, 440–446. [Google Scholar] [CrossRef]
- Pfletscher, M.; Wölper, C.; Gutmann, J.S.; Mezger, M.; Giese, M. A modular approach towards functional supramolecular aggregates—Subtle structural differences inducing liquid crystallinity. Chem. Commun. 2016, 52, 8549–8552. [Google Scholar] [CrossRef] [PubMed]
- Giese, M.; Krappitz, T.; Dong, R.Y.; Michal, C.A.; Hamad, W.Y.; Patrick, B.O.; MacLachlan, M.J. Tuning the photonic properties of chiral nematic mesoporous organosilica with hydrogen-bonded liquid-crystalline assemblies. J. Mater. Chem. C 2015, 3, 1537–1545. [Google Scholar] [CrossRef]
- Pfletscher, M.; Hölscher, S.; Wölper, C.; Mezger, M.; Giese, M. Structure–Property Relationships in Hydrogen-Bonded Liquid Crystals. Chem. Mater. 2017, 29, 8462–8471. [Google Scholar] [CrossRef]
- Kobylarczyk, J.; Pakulski, P.; Potępa, I.; Podgajny, R. Binary and Ternary Core–Shell Crystals of Polynuclear Coordination Clusters via Epitaxial Growth. under revision.
- Shanmugaraju, S.; Bar, A.K.; Joshi, S.A.; Tatil, P.Y.P.; Mukherjee, P.S. Constructions of 2D-Metallamacrocycles Using Half-Sandwich RuII2 Precursors: Synthesis, Molecular Structures, and Self-Selection for a Single Linkage Isomer. Organometallics 2011, 30, 1951–1960. [Google Scholar] [CrossRef]
- Cho, Y.L.; Uh, H.; Chang, S.-Y.; Chang, H.-Y.; Choi, M.-G.; Shin, I.; Jeong, K.-S. A Double-Walled Hexagonal Supermolecule Assembled by Guest Binding. J. Am. Chem. Soc. 2001, 123, 1258–1259. [Google Scholar] [CrossRef]
- Sekiya, R.; Nishikiori, S.; Ogura, K. Crystalline Inclusion Compounds Constructed through Self-Assembly of Isonicotinic Acid and Thiocyanato Coordination Bridges. J. Am. Chem. Soc. 2004, 126, 16587–16600. [Google Scholar] [CrossRef]
- Trokowski, R.; Akine, S.; Nabeshima, T. Synthesis, characterization and molecular recognition of a bis-platinum terpyridine dimer. Chem. Commun. 2008, 889–890. [Google Scholar] [CrossRef] [PubMed]
- Shankar, K.; Kirillov, A.M.; Baruah, J.B. A modular approach for molecular recognition by zinc dipicolinate complexes. Dalton Trans. 2015, 44, 14411–14423. [Google Scholar] [CrossRef] [PubMed]
- Blanco, V.; Abella, D.; Rama, T.; Alvarino, C.; García, M.D.; Peinador, C.; Quintela, J.M. Guest-induced stereoselective self-assembly of quinoline-containing PdII and PtII metallacycles. RSC Adv. 2016, 6, 80181–80192. [Google Scholar] [CrossRef]
- Singh, M.P.; Baruah, J.B. Stable host–guest complexes of bis-2,6-pyridinedicarboxylate iron(III) with dihydroxybenzenes. Polyhedron 2017, 138, 103–108. [Google Scholar] [CrossRef]
- Shankar, K.; Baruah, J.B. Inclusion of dihydroxyaromatics by a lanthanum(III) 2,6-dipicolinate complex. Polyhedron 2017, 126, 262–267. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Paul, M.; Desiraju, G.R. From a Binary to a Quaternary Cocrystal: An Unusual Supramolecular Synthon. Angew. Chem. Int. Ed. 2019, 58, 12027–12031. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Alvarez, S.; Alemany, P.; Casanova, D.; Cirera, J.; Llunell, M.; Avnir, D. Shape maps and polyhedral interconversion paths in transition metal chemistry. Coord. Chem. Rev. 2005, 249, 1693–1708. [Google Scholar] [CrossRef]
- Dance, I.; Scudder, M. Supramolecular Motifs: Concerted Multiple Phenyl Embraces between Ph4P+ Cations Are Attractive and Ubiquitous. Chem.-Eur. J. 1996, 2, 481–486. [Google Scholar] [CrossRef]
- Kuzniak, E.; Hooper, J.; Srebro-Hooper, M.; Kobylarczyk, J.; Dziurka, M.; Musielak, B.; Pinkowicz, D.; Raya, J.; Ferlay, S.; Podgajny, R. A concerted evolution of supramolecular interactions in a {cation; metal complex; π-acid;solvent} anion-π system. Inorg. Chem. Front. 2020, 7, 1851–1863. [Google Scholar] [CrossRef]
- Parker, R.J.; Spiccia, L.; Batten, S.R.; Cashion, J.D.; Fallon, G.D. The Encapsulation of Ferrocyanide by Copper(II) Complexes of Tripodal Tetradentate Ligands. Novel H-Bonding Networks Incorporating Heptanuclear and Pentanuclear Heterometallic Assemblies. Inorg. Chem. 2001, 40, 4696–4704. [Google Scholar] [CrossRef] [PubMed]
- Bencini, A.; Bianchi, A.; Garcia-Espana, E.; Giusti, M.; Mangani, S.; Micheloni, M.; Orioli, P.; Paoletti, P. Anion coordination chemistry. 2. Electrochemical, thermodynamic, and structural studies on supercomplex formation between large polyammonium cycloalkanes and the two complex anions hexacyanoferrate(II) and hexacyanocobaltate(III). Inorg. Chem. 1987, 26, 3902–3907. [Google Scholar] [CrossRef]
- Brorson, M.; Galsbøl, F.; Simonsen, K.; Skov, L.K.; Søtofte, I. Preparation and Characterization of cis- and trans-[Ir(tn)2Cl2]CF3SO3 and of [Ir(tn)3]Cl3 (tn=propane-1,3-diamine). Acta Chem. Scand. 1998, 52, 1017–1023. [Google Scholar] [CrossRef][Green Version]
- Kou, H.Z.; Jiang, Y.-B.; Zhou, B.C.; Wang, R.J. Cyano-Bridged 2D CuII−CrIII Coordination Polymers: Structural Evidence for Formation of a Polymeric Macrocyclic Metallic Compound. Inorg. Chem. 2004, 43, 3271–3276. [Google Scholar] [CrossRef]
- Cvrtila, I.; Stilinović, V. New Tricks by Old Anions: Hydrogen Bonded Hexacyanoferrous Anionic Networks. Cryst. Growth Des. 2017, 17, 6793–6800. [Google Scholar] [CrossRef]
- Alborés, P.; Slep, L.D.; Weyhermüller, T.; Rentschler, E.; Baraldo, L.M. Exchange coupling across the cyanide bridge: Structural and DFT interpretation of the magnetic properties of a binuclear chromium(III) complex. Dalton Trans. 2006, 948–954. [Google Scholar] [CrossRef]
- Graham, M.J.; Zadrozny, J.M.; Shiddiq, M.; Anderson, J.S.; Fataftah, M.S.; Hill, S.; Freedman, D.E. Influence of Electronic Spin and Spin–Orbit Coupling on Decoherence in Mononuclear Transition Metal Complexes. J. Am. Chem. Soc. 2014, 136, 7623–7626. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef] [PubMed]
- Coupar, P.I.; Ferguson, G.; Glidewell, C. Hydrogen-Bonded Chains in 4,4′-Dihydroxybenzophenone-4,4′-Bipyridyl (1/1) and Chains of Rings in 1,3,5-Trihydroxybenzene-4,4′-Bipyridyl (2/3). Acta Cryst. 1996, C52, 2524–2528. [Google Scholar] [CrossRef]
- Goerigk, L.; Mehta, N. Trip to the Density Functional Theory Zoo: Warnings and Recommendations for the User. Aust. J. Chem. 2019, 72, 563–573. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef]
- van der Lubbe, S.C.C.; Guerra, C.F. The Nature of Hydrogen Bonds: A Delineation of the Role of Different Energy Components on Hydrogen Bond Strengths and Lengths. Chem.-Asian J. 2019, 14, 2760–2769. [Google Scholar]
- Perruchas, S.; Boubekeur, K.; Molinié, P. Radical cation salts of TTF(CH2OH)4 with cyanide anions: Synthesis, crystal structure and magnetic properties of [TTF(CH2OH)4][TCNQF4] and [TTF(CH2OH)4]3[18-crown-6 ⊂ K][M(CN)6]·DMF (M = Fe, Co). Polyhedron 2005, 24, 1555–1564. [Google Scholar] [CrossRef]
- Hanuza, J.; Strek, W.; Hermanowicz, K.; Jezowska-Trzebiatowska, B.; Trabjerg, I. Spectroscopic behaviour of Cr(CN)63− ion isolated in KCl host. J. Mol. Struct. 1986, 144, 141–153. [Google Scholar] [CrossRef]
- Jain, S.C.; Warrier, A.V.R.; Sehgal, H.K. Vibrational and electronic properties of Co(CN)63− doped in alkali halides. J. Phys. C Sol. St. Phys. 1972, 5, 1511–1518. [Google Scholar] [CrossRef]
- Ross, M.; Andersen, A.; Fox, Z.W.; Zhang, Y.; Hong, K.; Lee, J.-H.; Cordones, A.; March, A.M.; Doumy, G.; Southworth, S.H.; et al. Comprehensive Experimental and Computational Spectroscopic Study of Hexacyanoferrate Complexes in Water: From Infrared to X-ray Wavelengths. J. Phys. Chem. B 2018, 122, 5075–5086. [Google Scholar] [CrossRef]
- Kuzniak, E.; Pinkowicz, D.; Hooper, J.; Srebro-Hooper, M.; Hetmańczyk, Ł.; Podgajny, R. Molecular Deformation, Charge Flow, and Spongelike Behavior in Anion–π {[M(CN)4]2–;[HAT(CN)6]}∞ (M=Ni, Pd, Pt) Supramolecular Stacks. Chem. Eur. J. 2018, 24, 16302–16314. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, R.; Knobel, M.; Reguera, E. Modification of the magnetic properties in molecular magnets based on Prussian blue analogues through adsorbed species. J. Phys. Condens. Matter 2006, 18, 11243–11254. [Google Scholar] [CrossRef]
- Hendrickx, M.F.A.; Mironov, V.S.; Chibotaru, L.F.; Ceulemans, A. An Ab Initio Study of the Ligand Field and Charge-Transfer Transitions of Cr(CN)63− and Mo(CN)63−. J. Am. Chem. Soc. 2003, 125, 3694–3695. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hanuza, J.; Strek, W.; Hermanowicz, K.; Jezowska-Trzebiatowska, B.; Trabjerg, I. Spectroscopic properties of Cr(CN)63− doped in a KBr crystal. Chem. Phys. 1984, 86, 137–145. [Google Scholar] [CrossRef]
- Mitsuru, S.; Hirohiko, A.; Hideo, Y. The DV-Xα MO Study of the Electronic Structures of [M(CN)6]3− (M = Cr, Mn, Fe, and Co) and [Fe(CN)6]4−. Bull. Chem. Soc. Jap. 1981, 54, 2898–2903. [Google Scholar]
- Alexander, J.J.; Gray, H.B. Electronic structures of hexacyanometalate complexes. J. Am. Chem. Soc. 1968, 90, 4260–4271. [Google Scholar] [CrossRef]
- Sreekantan, S.; Lalithambika, N.; Atak, K.; Seidel, R.; Neubauer, A.; Brandenburg, T.; Xiao, J.; Winter, B.; Aziz, E.F. Chemical bonding in aqueous hexacyano cobaltate from photon- and electron-detection perspectives. Sci. Rep. 2017, 7, 40811. [Google Scholar]
- Sano, M.; Hatano, Y.; Kashiwagi, H.; Yamatera, H. An Ab Initio Calculations of the Electronic Structure of the [Co(CN)6]3− Ion. Bull. Chem. Soc. Jap. 1981, 54, 1523–1530. [Google Scholar] [CrossRef]
- Hahn, A.W.; Van Kuiken, B.E.; Chilkuri, V.G.; Levin, N.; Bill, E.; Weyhermüller, T.; Nicolaou, A.; Miyawaki, J.; Harada, Y.; DeBeer, S. Probing the Valence Electronic Structure of Low-Spin Ferrous and Ferric Complexes Using 2p3d Resonant Inelastic X-ray Scattering (RIXS). Inorg. Chem. 2018, 57, 9515–9530. [Google Scholar] [CrossRef]
- Ojeda, J.; Arrell, C.A.; Longetti, L.; Chergui, M.; Helbing, J. Charge-transfer and impulsive electronic-to-vibrational energy conversion in ferricyanide: Ultrafast photoelectron and transient infrared studies. Phys. Chem. Chem. Phys. 2017, 19, 17052–17062. [Google Scholar] [CrossRef]
- Yu, P.; Yang, F.; Zhao, J.; Wang, J. Hydration Dynamics of Cyanoferrate Anions Examined by Ultrafast Infrared Spectroscopy. J. Phys. Chem. B 2014, 118, 3104–3114. [Google Scholar] [CrossRef]
- Zhang, W.; Ji, M.; Sun, Z.; Gaffney, K.J. Dynamics of Solvent-Mediated Electron Localization in Electronically Excited Hexacyanoferrate(III). J. Am. Chem. Soc. 2012, 134, 2581–2588. [Google Scholar] [CrossRef] [PubMed]
- Chorazy, S.; Sieklucka, B.; Ohkoshi, S. Near-Infrared Photoluminescence in Hexacyanido-Bridged Nd–Cr Layered Ferromagnet. Cryst. Growth Des. 2016, 16, 4918–4925. [Google Scholar] [CrossRef]
- Kitzmann, W.R.; Moll, J.; Heinze, K. Spin-fip luminescence. Photochem. Photobiol. Sci. 2022. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liang, Y.; Ma, P.; Li, S.; Wang, J.; Niu, J. Self assembly of carboxylate/alcoholate functionalized ring-shape phosphomolybdates. CrystEngComm 2014, 16, 8041–8046. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Liu, J.-L.; Yang, H.-M.; Jia, A.-Q.; Zhang, Q.-F. Inclusion of ferrocene within the deepened cavities formed from the hydrogen bonding self-assembly of calix[4]resorcinarenes with bis-pyridines. J. Incl. Phenom. Macro. 2016, 85, 105–110. [Google Scholar] [CrossRef]
- Papaefstathiou, G.S.; Darrow, B.G.; MacGillivray, L.R. Crystal and molecular structure of [Cu2(3,5-dihydroxybenzoate)4 (acetonitrile)2]·8H2O. J. Chem. Crystallogr. 2002, 32, 191–195. [Google Scholar] [CrossRef]
- Feng, L.; Chen, Z.; Zeller, M.; Luck, R.L. Dicopper moieties stabilized by Fréchet-type dendrons: Syntheses and structural characterizations. Polyhedron 2014, 80, 206–215. [Google Scholar] [CrossRef]
- Chaumont, C.; Mobian, P.; Kyritsakas, N.; Henry, M. Synthesis, topology and energy analysis of crystalline resorcinol-based oligophenylene molecules with various symmetries. CrystEngComm 2013, 15, 6845–6862. [Google Scholar] [CrossRef]
- Dan, M.; Cheetham, A.K.; Rao, C.N.R. Diverse Structures and Dimensionalities in Hybrid Frameworks of Strontium and Lanthanum with Isomeric Dihydroxybenzoates. Inorg. Chem. 2006, 45, 8227–8238. [Google Scholar]
- Liwporncharoenvong, T.; Luck, R.L. Quadruply Bonded Dimolybdenum Atoms Surrounded by Dendrons: Preparation, Characterization, and Electrochemistry. J. Am. Chem. Soc. 2001, 123, 3615–3616. [Google Scholar] [CrossRef] [PubMed]
- Bhyrappa, P.; Wilson, S.R.; Suslick, K.S. View Author Information Hydrogen-Bonded Porphyrinic Solids: Supramolecular Networks of Octahydroxy Porphyrins. J. Am. Chem. Soc. 1997, 119, 8492–8502. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF 2019.304, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: https://www.scm.com (accessed on 21 April 2022).
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Santra, G.; Sylvetsky, N.; Martin, J.M.L. Minimally Empirical Double-Hybrid Functionals Trained against the GMTKN55 Database: RevDSD-PBEP86-D4, RevDOD-PBE-D4, and DOD-SCAN-D4. J. Phys. Chem. A 2019, 123, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Regular Two-Component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Total Energy Using Regular Approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Mitoraj, M.; Michalak, A. Natural Orbitals for Chemical Valence as Descriptors of Chemical Bonding in Transition Metal Complexes. J. Mol. Model. 2007, 13, 347–355. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Formula | double hydrogen bonds | ||
| N2⋅⋅⋅O2#1 a | 2.769 | 2.777 | 2.787 |
| N2⋅⋅⋅H2#1 | 1.959 | 1.940 | 1.964 |
| N2⋅⋅⋅H2#1-O2#1 | 176.89 | 173.77 | 178.21 |
| N3⋅⋅⋅O3#1 | 2.815 | 2.827 | 2.835 |
| N3⋅⋅⋅H3#1 | 2.016 | 1.987 | 2.056 |
| N3⋅⋅⋅H3#1-O3#1 | 179.53 | 178.09 | 177.10 |
| related angles in [M(CN)6]3− | |||
| M1-N2-C2 | 176.70 | 177.62 | 177.89 |
| M1-N3-C3 | 171.31 | 173.53 | 173.52 |
| C2-M1-C3 | 84.87 | 85.19 | 85.87 |
| N2⋅⋅⋅M1⋅⋅⋅N3 | 80.82 | 82.01 | 82.75 |
| single hydrogen bonds | |||
| N1⋅⋅⋅O1 | 2.733 | 2.751 | 2.762 |
| N1⋅⋅⋅H1 | 1.944 | 1.916 | 1.949 |
| N1⋅⋅⋅H1-O1 | 174.67 | 172.81 | 173.99 |
| related angles in [M(CN)6]3− | |||
| M1-C1-N1 | 176.72 | 177.35 | 177.63 |
| C1-M1-C b | 88.39 | 89.24 | 89.27 |
| 90.18 | 91.13 | 91.08 | |
| 91.37 | 91.02 | 91.38 | |
| 88.56 | 88.55 | 88.81 | |
| N1⋅⋅⋅M1⋅⋅⋅N b | 88.37 | 89.14 | 89.19 |
| 90.47 | 91.47 | 91.62 | |
| 92.26 | 91.89 | 92.06 | |
| 87.60 | 87.87 | 88.00 | |
| [Cr(CN)6]3− | [Fe(CN)6]3− | [Co(CN)6]3− | 4,4′bpy | |
|---|---|---|---|---|
| BLYP + D4//TZP a | ||||
| (H2PGH)2 (HPGH2)2 | −125.33 | −133.58 | −131.42 | – |
| H2PGH | −44.98 | −48.44 | −46.74 | – |
| HPGH2 | † | −27.30 | −24.70 | −7.64 |
| B3LYP + D4//TZP a | ||||
| (H2PGH)2 (HPGH2)2 | −124.87 | −133.38 | −130.88 | – |
| H2PGH | −44.65 | −47.98 | −46.40 | – |
| HPGH2 | −22.30 | −24.29 | −23.99 | −7.70 |
| rev-DOD-BLYP + D4//TZ2P a | ||||
| (H2PGH)2 (HPGH2)2 | −134.00 | −142.66 | −138.84 | – |
| H2PGH | −47.28 | −51.02 | −48.77 | – |
| HPGH2 | −23.99 | −26.60 | −26.46 | −9.50 |
| Compound | 1 | (PPh4)3[Cr(CN)6]⋅2H2O |
|---|---|---|
| Band 1 range | 802–822 (12,469–12,165) | 797–817 (12,547–12,240) |
| Band 1 max. | 810 (12,346) | 805 (12,422) |
| Band 2 range | 825–850 (12,121–11,765) | 820–842 (12,195–11,876) |
| Band 2 max. | 833 (12,005) | 828.5 (12,070) |
| Band 3 range | 855–875 (11,696–11,428) | 844–868 (11,848–11,521) |
| Band 3 max. | 858 (11,655) | 852.5 (11,730) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jędrzejowska, K.; Kobylarczyk, J.; Glosz, D.; Kuzniak-Glanowska, E.; Tabor, D.; Srebro-Hooper, M.; Zakrzewski, J.J.; Dziedzic-Kocurek, K.; Muzioł, T.M.; Podgajny, R. Supramolecular cis-“Bis(Chelation)” of [M(CN)6]3− (M = CrIII, FeIII, CoIII) by Phloroglucinol (H3PG). Molecules 2022, 27, 4111. https://doi.org/10.3390/molecules27134111
Jędrzejowska K, Kobylarczyk J, Glosz D, Kuzniak-Glanowska E, Tabor D, Srebro-Hooper M, Zakrzewski JJ, Dziedzic-Kocurek K, Muzioł TM, Podgajny R. Supramolecular cis-“Bis(Chelation)” of [M(CN)6]3− (M = CrIII, FeIII, CoIII) by Phloroglucinol (H3PG). Molecules. 2022; 27(13):4111. https://doi.org/10.3390/molecules27134111
Chicago/Turabian StyleJędrzejowska, Katarzyna, Jedrzej Kobylarczyk, Dorota Glosz, Emilia Kuzniak-Glanowska, Dominika Tabor, Monika Srebro-Hooper, Jakub J. Zakrzewski, Katarzyna Dziedzic-Kocurek, Tadeusz M. Muzioł, and Robert Podgajny. 2022. "Supramolecular cis-“Bis(Chelation)” of [M(CN)6]3− (M = CrIII, FeIII, CoIII) by Phloroglucinol (H3PG)" Molecules 27, no. 13: 4111. https://doi.org/10.3390/molecules27134111
APA StyleJędrzejowska, K., Kobylarczyk, J., Glosz, D., Kuzniak-Glanowska, E., Tabor, D., Srebro-Hooper, M., Zakrzewski, J. J., Dziedzic-Kocurek, K., Muzioł, T. M., & Podgajny, R. (2022). Supramolecular cis-“Bis(Chelation)” of [M(CN)6]3− (M = CrIII, FeIII, CoIII) by Phloroglucinol (H3PG). Molecules, 27(13), 4111. https://doi.org/10.3390/molecules27134111