
Continued Structural Exploration of Sulfocoumarin as Selective Inhibitor of Tumor-Associated Human Carbonic Anhydrases IX and XII





Abstract
:1. Introduction
2. Results
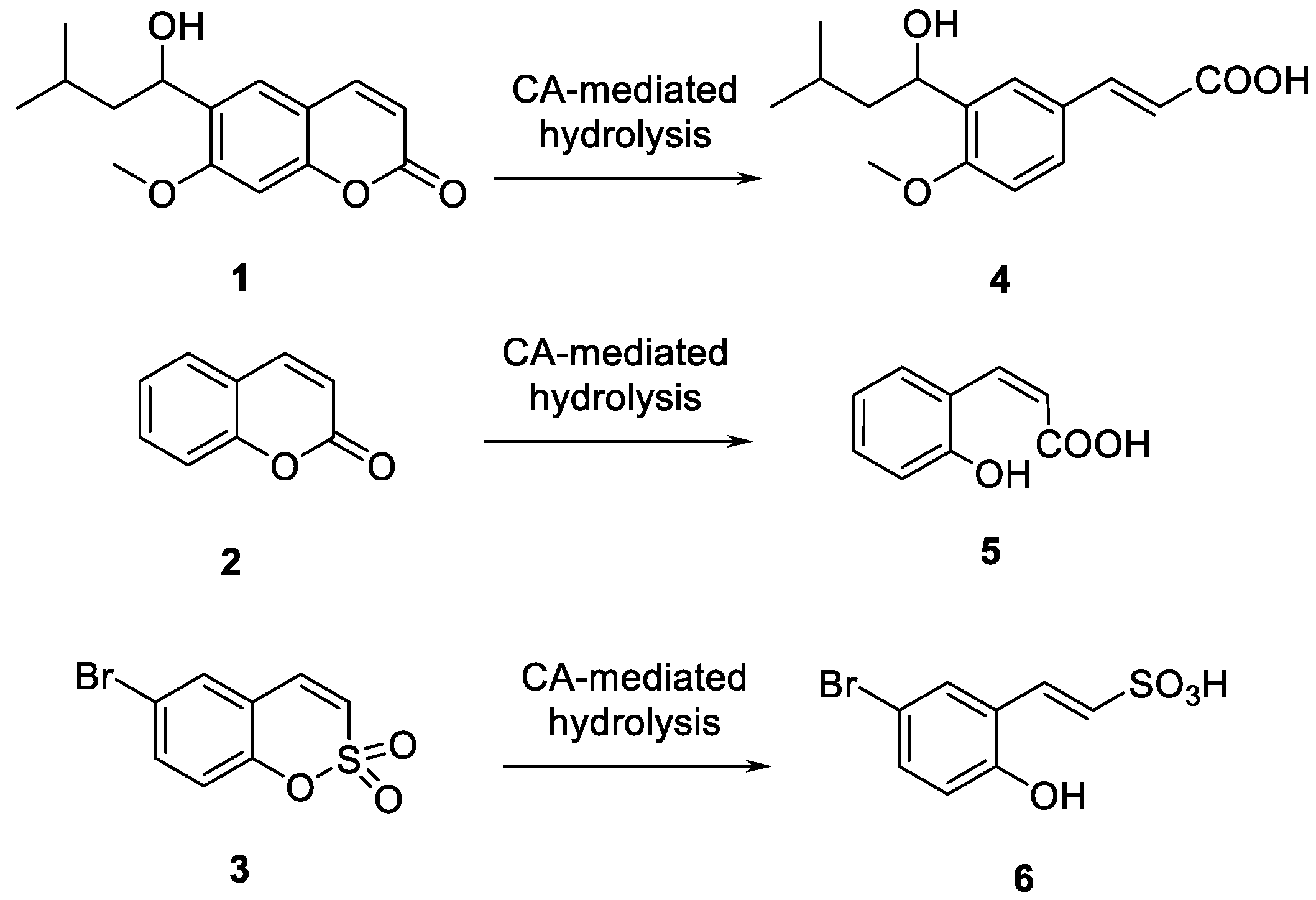
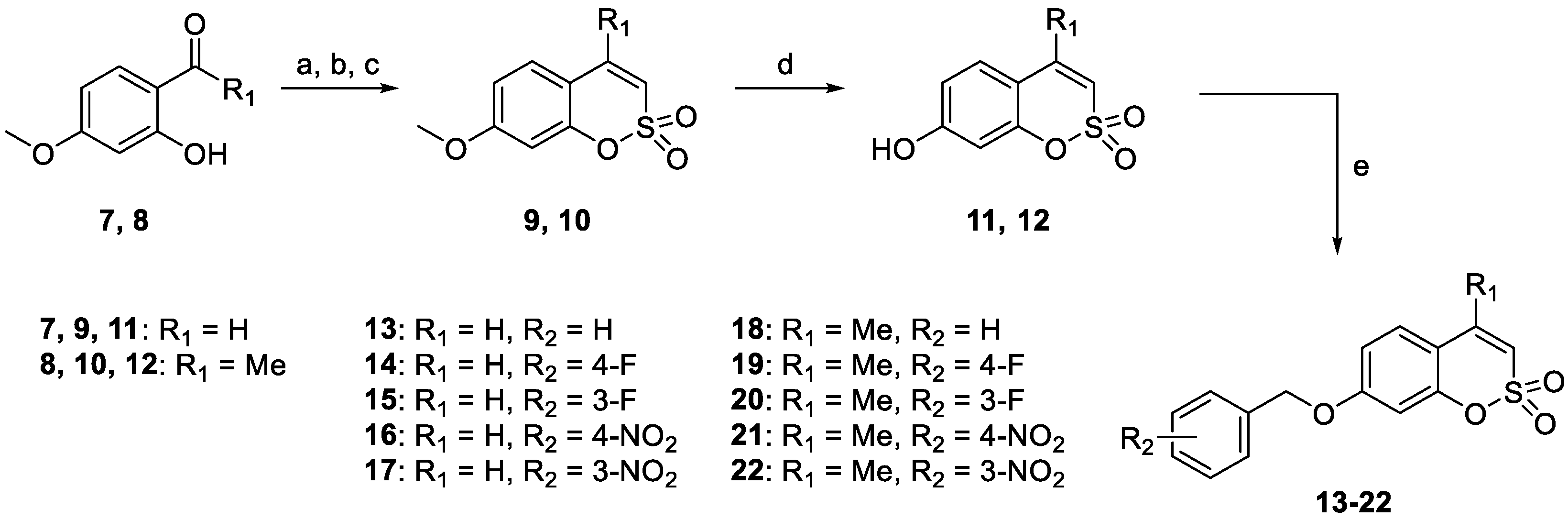
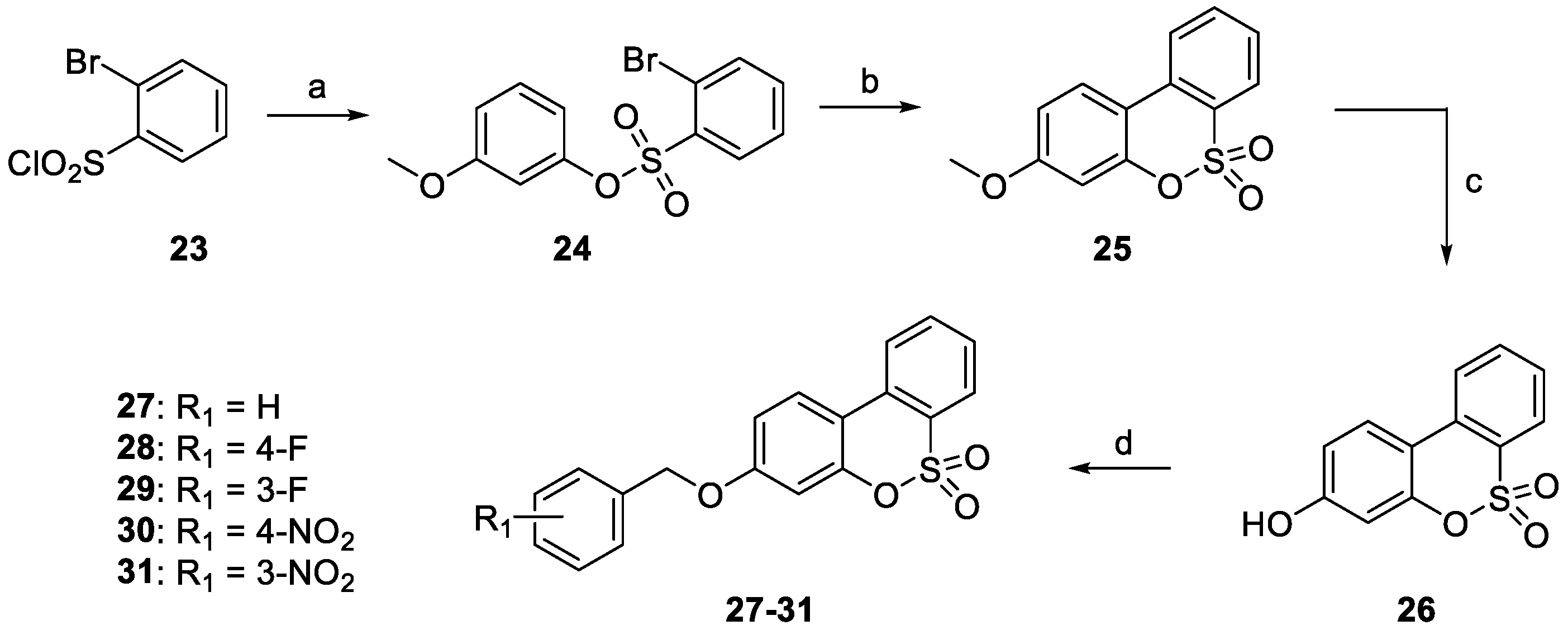
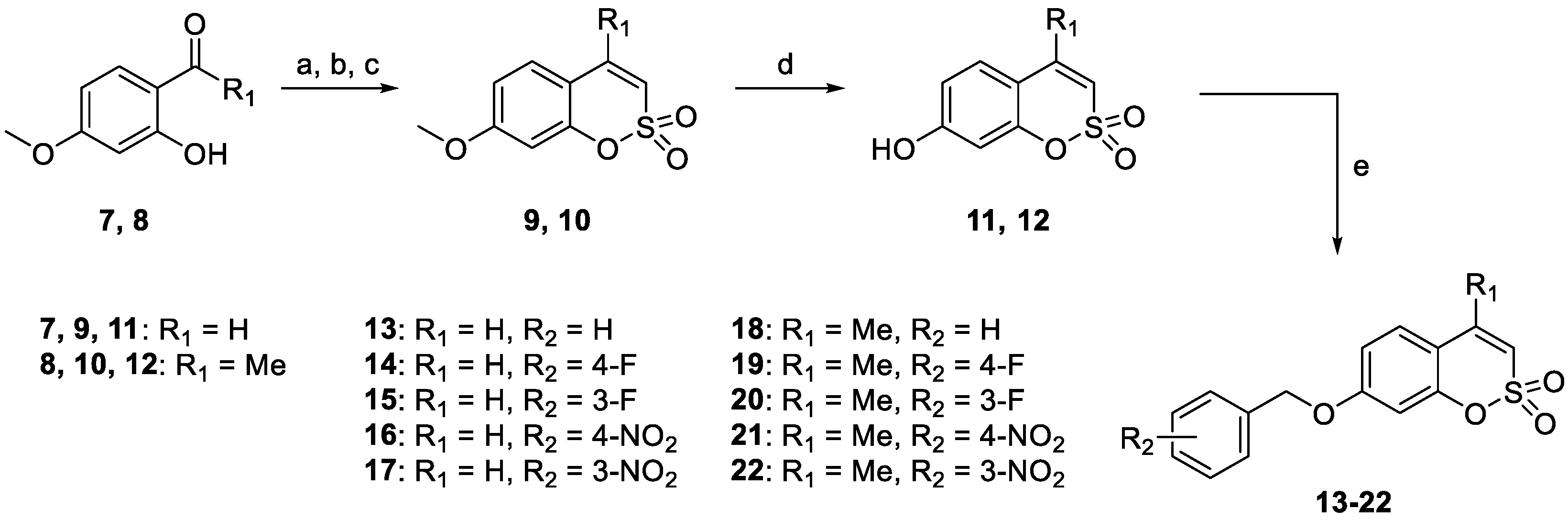
2.1. Chemistry
2.2. Carbonic Anhydrase Inhibition
- -
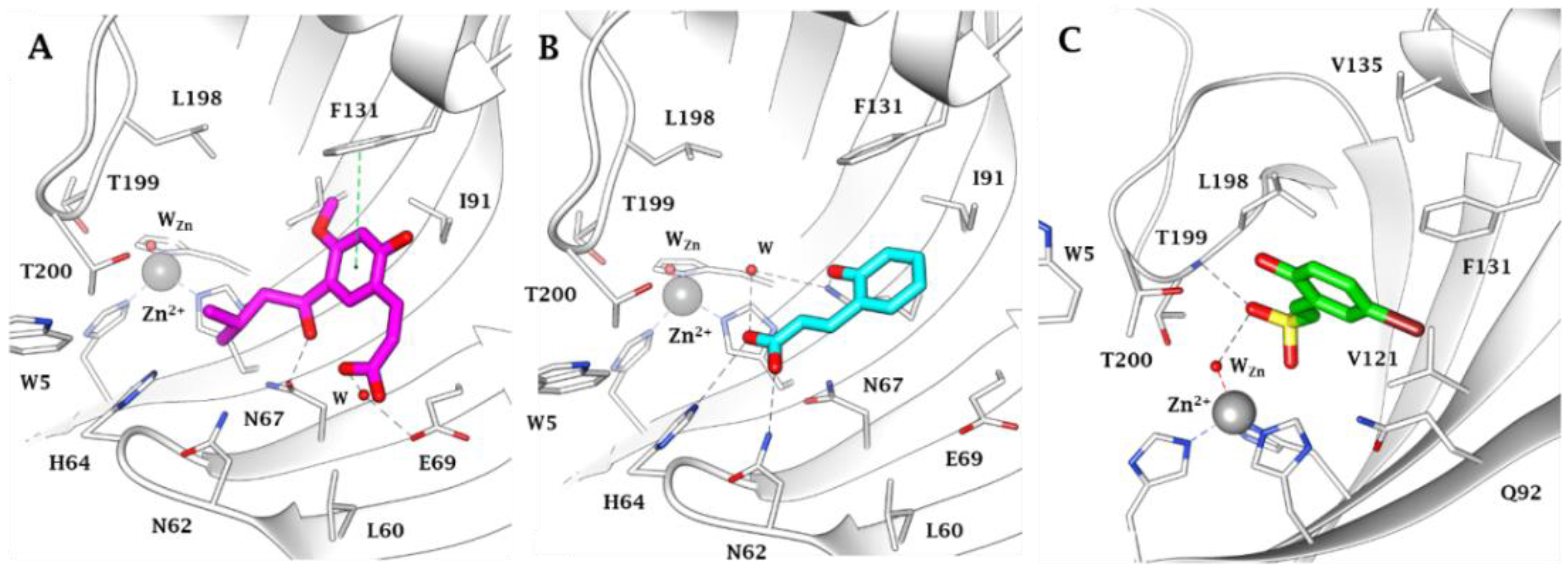
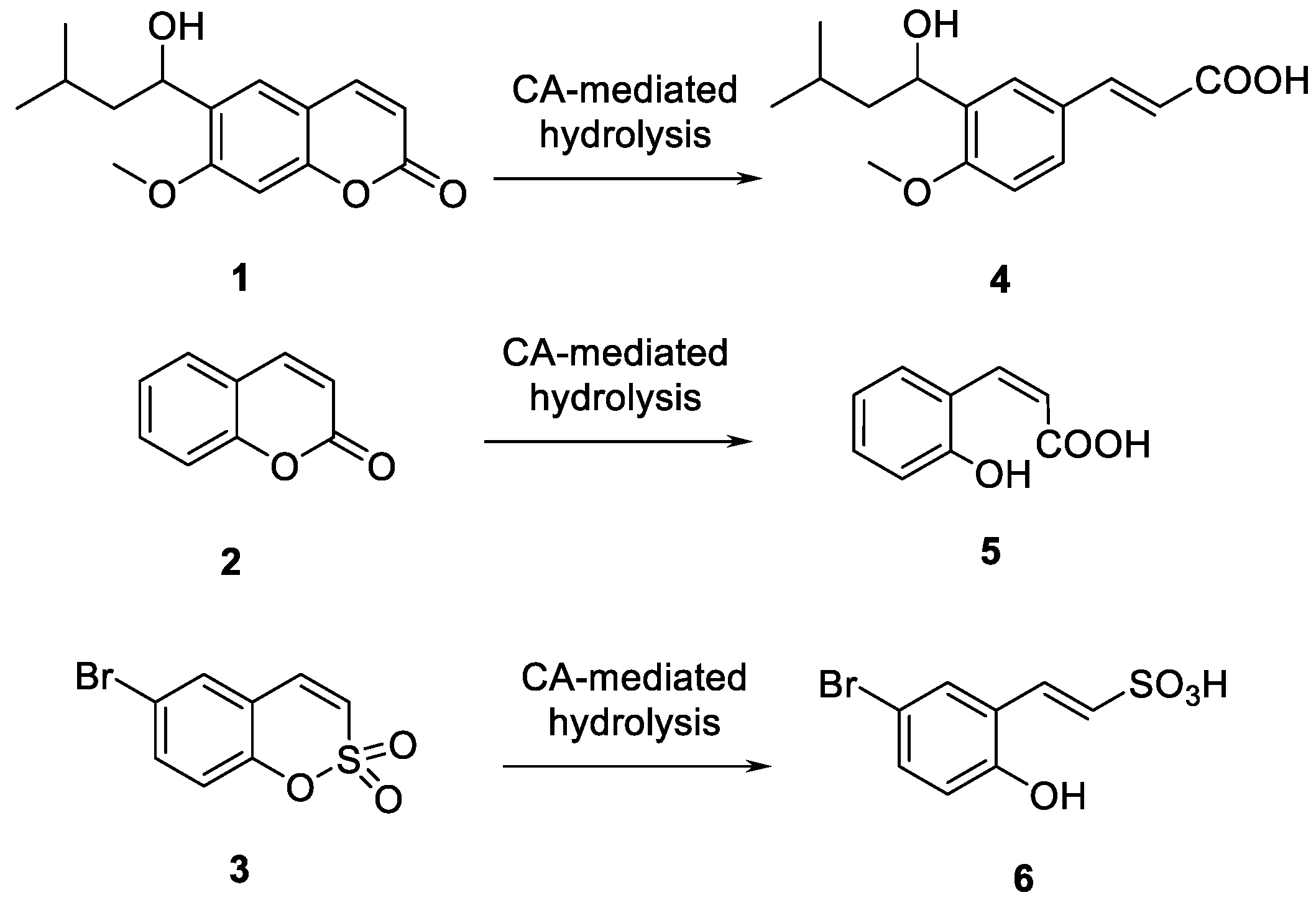
- According to the previous reports [31,32,33,35], isoform hCA I was not inhibited by a large number of substituted sulfocoumarins; however, on the other side, the simplest sulfocoumarin as 11, 12, 26, 36 and 37 showed a weak inhibitory potency. It is likely that compounds 36 (KI = 4625 nM) and 37 (KI = 7139 nM) have a low micromolar inhibition due to the presence of the carboxylic acid moiety, which may work by binding to the H2O molecule coordinated with Zn2+ in the active site [45,46].
- -
- Similar to hCA I, for hCA II we did not observe any inhibition by all the substituted sulfocoumarines, except for 9–12, 26, 36 and 37. Compounds 11 and 12 showed a low micromolar inhibition, respectively KI = 2896 nM and KI = 3857 nM, whilst analogs 9 and 10 have the KI in the medium micromolar range, which can likely be associated to the presence of a free phenol moiety on products 11 and 12, which is protected in 9 and 10.
- -
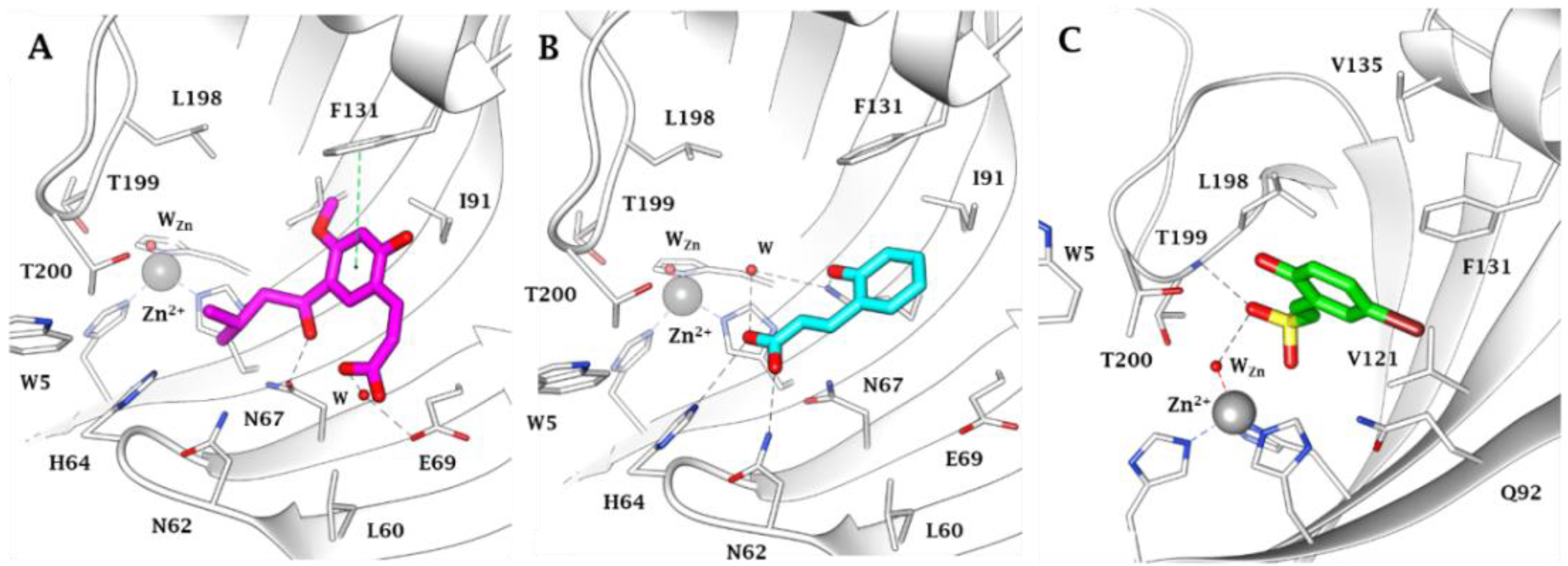
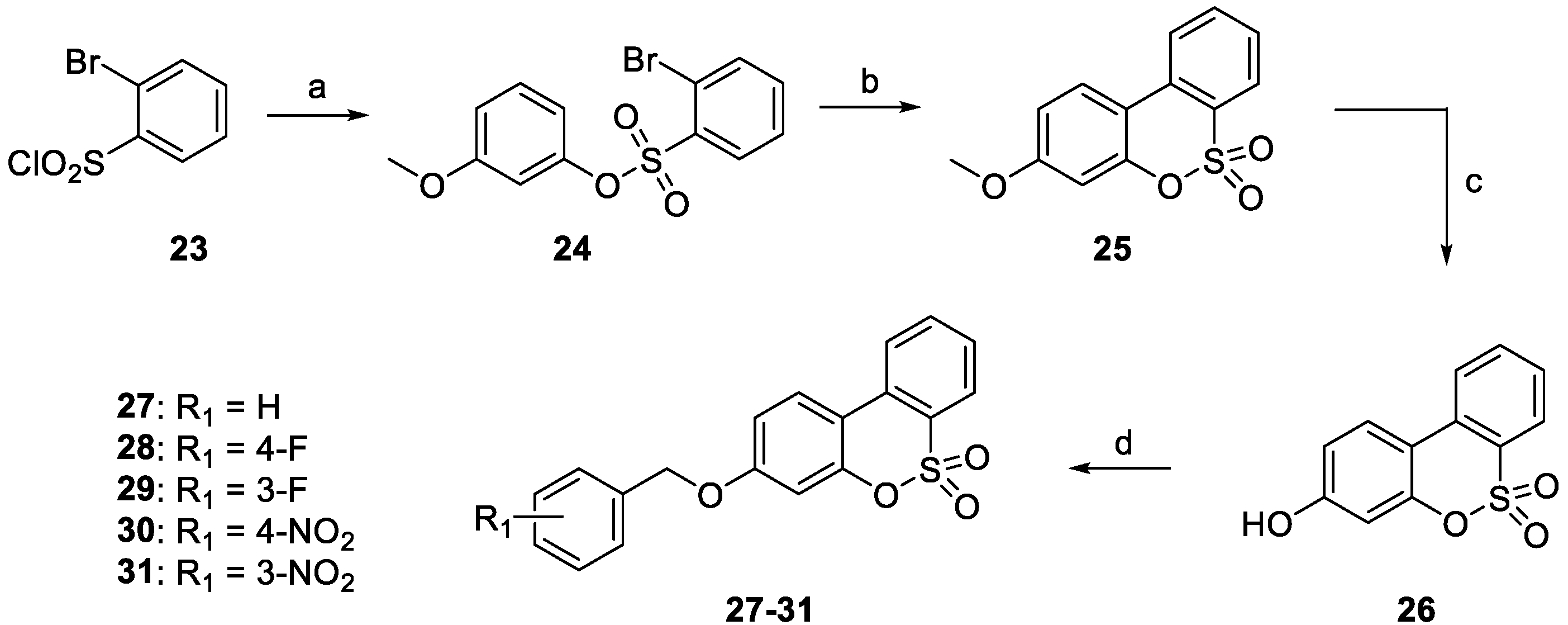
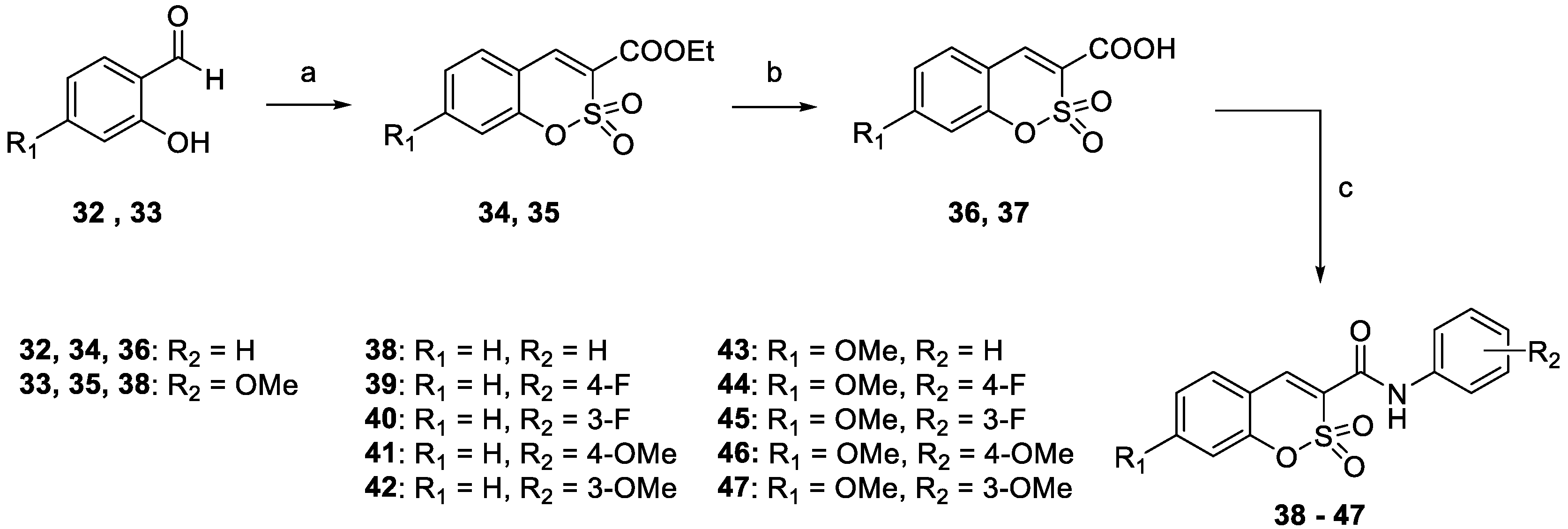
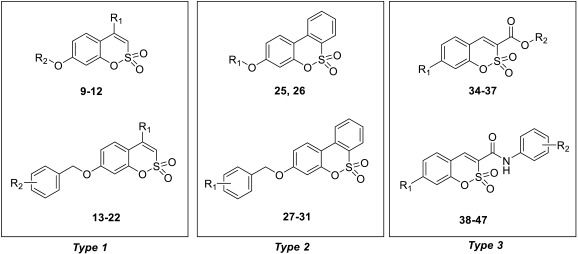
- The target hCA IX resulted in being the most inhibited isoforms by sulfocoumarins reported here. Considering the three different types of products, type 1 shows the most efficient inhibition against this isozyme, with KI values in the low-medium nanomolar range, between 14.3 nM and 97.2 nM. In detail, the first-in-class compound in term of inhibition potency is the number 19, which shows a fluorine atom in para position on the benzyloxy moiety and a methyl group as R1 substituent (KI = 14.3 nM), whilst the elimination of the benzyloxy moiety (10) gives us the less effective compound (KI = 97.2 nM). Interestingly, the general behaviour of these compounds owned on type 1 was that, indeed, the insertion of the methyl group as an R1 substituent produced a worsening of the KI values; in fact, it was demonstrated by products 16 (KI = 68.9 nM) and 17 (KI = 40.2 nM) compared to the corresponding 21 (KI = 89.7 nM) and 22 (KI = 61.4 nM). This rule seems to be broken only by derivatives 18 (KI = 49.8 nM) and 19 (KI = 14.3 nM), which resulted in better inhibitors with respect to analogs 13 (KI = 55.6 nM) and 14 (KI = 22.9 nM). Sulfocoumarines belonging to type 2, even with a benzyloxy moiety, suffered from the addition of a further aromatic ring on the scaffold that improves the steric hindrance on the sulfonate ring, which induced a worsening of the KI values. Indeed, the inhibition constants are in the high nanomolar range, between 615.1 nM and 1286 nM. The best compound result to be 29 (KI = 615.1 nM) with a fluorine atom in meta position on the benzyloxy moiety. The movement of the R1 group from meta to para position produces a worsening of the KI values. Examples are 29 (KI = 615.1 nM) and 31 (KI = 898.1 nM), respectively, with 28 (KI = 754.9 nM) and 30 (KI = 964.7 nM). Interestingly, it is a significant decline of the inhibition data of the sulfocoumarines belonging to type 3, with an amide as linker on position 3 of the sulfonate ring. It is likely that the presence of bulky groups with the CAI portion results in a worsening of the KI values, in a micromolar range between 499.7 nM and 64380 nM. The most potent compounds of this class are 43 (KI = 2429 nM) and the simplest sulfocoumarines 34 (KI = 1631 nM), 35 (KI = 1085 nM), 36 (KI = 499.7 nM) and 37 (KI = 853.7 nM) showed an ester or carboxylic acid moieties in position 3. Sulfocoumarines with a bromine atom in a meta position as the R2 group (40, 45) resulted in the worst values of inhibition, respectively KI = 40,840 nM and 64,380 nM. The hCA XII resulted in becoming the second most efficiently inhibited isoform by sulfocoumarines, with it being particularly possible to observe a similar trend of the KI values as already shown for the hCA IX. Considering products belonging to type 1, they show the most potent inhibition for this isoform, between 8.6 nM and 136.0 nM. The presence of a methyl group in R1 position produced an improvement of the KI values for derivatives 18 (KI = 8.6 nM), without substituent on the benzyloxy moiety, and 20 (KI = 25.7 nM), with fluorine in the meta position. For all the other products, a worsening of Ki values, such as for 14 (KI = 19.2 nM) and 19 (KI = 41.7 nM), or for 11 (KI = 42.5 nM) and 12 (KI = 55.0 nM), was observed. On the other side, the movement of the fluorine atom from meta to para position (19) resulted in a KI value of 41.7 nM. Regarding the nitro group, compound 17 (KI = 77.5 nM) with R2 group in a meta position show an improvement in term of inhibition than its analogue 16 (KI = 94.7 nM) with R2 group in a para position. For sulfocoumarines 18–22, the fluorine atom resulted in being better than nitro group as a substituent on the benzyloxy moiety; indeed, 19 (KI = 41.7 nM) and 20 (KI = 25.7 nM) are strongest inhibitors in term of potency than 21 (KI = 100.8 nM) and 22 (KI = 136.0 nM). For compounds belonging to type 2, the addition of an aromatic ring on the sulfocoumarin scaffold determines a worsening of the KI values in the medium–high nanomolar range, between 414.2 nM and 1369 nM. The presence of a bulky group such as the nitro group leads to a worsening in terms of inhibition potency, particularly products 30 and 31 have KI of 1056 nM and 1369 nM. The simplest sulfocoumarin with a free phenol moiety, 26, showed the best inhibition for type 2 series, 414.2 nM. Sulfocoumarins belonging to type 3 that present a further enhancement of the steric hindrance on the sulfonate ring, have KI values in the range of micromolar (KI = 408.2–78,320 nM). Compound 40 and 45, with a bromine atom in the meta position, show the worst inhibition potencies on hCA XII, respectively 78,320 nM and 76,540 nM; on the other side, sulfocoumarines with the less bulky group in R2 position result in being a good inhibitor on this isoform, while, indeed, 38 and 43 with a hydrogen atom show KI values in the range of low micromolar—respectively, 2301 nM and 1594 nM. The best inhibitors belonging to the type 3 series are compounds 36 (KI = 563.7 nM) and 37 (KI = 408.2 nM), likely thanks to the free carbocylic acid moiety.
3. Materials and Methods
3.1. Chemistry
3.2. Carbonic Anhydrase Inhibition
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Capasso, C. An overview of the bacterial carbonic anhydrases. Metabolites 2017, 7, 56. [Google Scholar]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C.T. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med. Chem. 2011, 3, 1165–1180. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin. Investig. Drugs 2018, 27, 963–970. [Google Scholar] [CrossRef]
- Supuran, C.T. Experimental Carbonic Anhydrase Inhibitors for the Treatment of Hypoxic Tumors. J. Exp. Pharmacol. 2020, 12, 603–617. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discov. 2017, 12, 61–88. [Google Scholar] [CrossRef]
- Fabrizi, F.; Mincione, F.; Somma, T.; Scozzafava, G.; Galassi, F.; Masini, E. A new approach to antiglaucoma drugs: Carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J. Enzym. Inhib. Med. Chem. 2012, 27, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Carbonic anhydrase inhibitors: An update on experimental agents for the treatment and imaging of hypoxic tumors. Expert Opin Investig Drugs. 2021, 30, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Nerella, S.G.; Singh, P.; Arifuddin, M.; Supuran, C.T. Anticancer carbonic anhydrase inhibitors: A patent and literature update 2018-2022. Expert Opin. Ther. Pat. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Pochet, L.; Masereel, B.; Scozzafava, A.; Supuran, C.T. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J. Med. Chem. 2010, 53, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.K.; Ostlund, C.; Ahmadi, A.; Kyle, A.; Auf dem Keller, U.; Leung, S.; Huntsman, D.G.; et al. Targeting Tumor Hypoxia: Suppression of Breast Tumor Growth and Metastasis by Novel Carbonic Anhydrase IX Inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [Green Version]
- Touisni, N.; Maresca, A.; McDonald, P.C.; Lou, Y.; Scozzafava, A.; Dedhar, S.; Winum, J.Y.; Supuran, C.T. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J. Med. Chem. 2011, 54, 8271–8277. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.A.; Vullo, D.; Maresca, A.; Supuran, C.T.; Poulsen, S.A. Natural product coumarins that inhibit human carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1539–1543. [Google Scholar] [CrossRef] [Green Version]
- Balboni, G.; Congiu, C.; Onnis, V.; Maresca, A.; Scozzafava, A.; Winum, J.Y.; Supuran, C.T. Flavones and structurally related 4-chromenones inhibit carbonic anhydrases by a different mechanism of action compared to coumarins. Bioorg. Med. Chem. Lett. 2012, 22, 3063–3066. [Google Scholar] [CrossRef]
- Kumar, A.; Siwach, K.; Supuran, C.T.; Sharma, P.K. A decade of tail-approach based design of selective as well as potent tumor associated carbonic anhydrase inhibitors. Bioorg Chem. 2022, 126, 105920. [Google Scholar] [CrossRef]
- Nocentini, A.; Angeli, A.; Carta, F.; Winum, J.Y.; Zalubovskis, R.; Carradori, S.; Capasso, C.; Donald, W.A.; Supuran, C.T. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J. Enzym. Inhib. Med. Chem. 2021, 36, 561–580. [Google Scholar] [CrossRef]
- Bonardi, A.; Falsini, M.; Catarzi, D.; Varano, F.; Di Cesare Mannelli, L.; Tenci, B. Structural investigations on coumarins leading to chromeno[4,3-c]pyrazol-4-ones and pyrano[4,3-c]pyrazol-4-ones: New scaffolds for the design of the tumor-associated carbonic anhydrase isoforms IX and XII. Eur. J. Med. Chem. 2018, 146, 47–59. [Google Scholar] [CrossRef]
- Angapelly, S.; Sri Ramya, P.V.; Angeli, A.; Supuran, C.T.; Arifuddin, M. Sulfocoumarin-, coumarin-, 4-sulfamoylphenyl-bearing indazole-3-carboxamide hybrids: Synthesis and selective inhibition of tumor-associated carbonic anhydrase isozymes IX and XII. ChemMedChem 2017, 12, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Giovannuzzi, S.; D’Ambrosio, M.; Luceri, C.; Osman, S.M.; Pallecchi, M.; Bartolucci, G.; Nocentini, A.; Supuran, C.T. Aromatic Sulfonamides including a Sulfonic Acid Tail: New Membrane Impermeant Carbonic Anhydrase Inhibitors for Targeting Selectively the Cancer-Associated Isoforms. Int. J. Mol. Sci. 2021, 23, 461. [Google Scholar] [CrossRef] [PubMed]
- Bozdag, M.; Alafeefy, A.M.; Altamimi, A.M.; Vullo, D.; Carta, F.; Supuran, C.T. Coumarins and other fused bicyclic heterocycles with selective tumor-associated carbonic anhydrase isoforms inhibitory activity. Bioorg. Med. Chem. 2017, 25, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Tanc, M.; Carta, F.; Scozzafava, A.; Supuran, C.T. a-Carbonic anhydrases possess thioesterase activity. ACS Med. Chem. Lett. 2015, 6, 292–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Tiwari, M.; Supuran, C.T. Novel coumarins and benzocoumarins acting as isoform-selective inhibitors against the tumor-associated carbonic anhydrase IX. J. Enzym. Inhib. Med. Chem. 2014, 2, 292–296. [Google Scholar] [CrossRef]
- Carta, F.; Maresca, A.; Scozzafava, A.; Supuran, C.T. 5- and 6-membered (thio)lactones are prodrug type carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 267–270. [Google Scholar] [CrossRef]
- Isik, S.; Vullo, D.; Bozdag, M.; Carta, F.; Supuran, C.T. 7-Amino-3,4-dihydro-1H-quinoline-2-one, a compound similar to the substituted coumarins, inhibits a-carbonic anhydrases without hydrolysis of the lactam ring. J. Enzym. Inhib. Med. Chem. 2015, 30, 773–777. [Google Scholar]
- Bozdag, M.; Bua, S.; Osman, S.M.; AlOthman, Z.; Supuran, C.T. Carbonic anhydrase I, II, IV and IX inhibition with a series of 7-amino-3,4-dihydroquinolin-2(1H)-one derivatives. J. Enzym. Inhib. Med. Chem. 2017, 32, 885–892. [Google Scholar] [CrossRef] [Green Version]
- Tars, K.; Vullo, D.; Kazaks, A.; Leitans, J.; Lends, A.; Grandane, A.; Zalubovskis, R.; Scozzafava, A.; Supuran, C.T. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): A class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J. Med. Chem. 2013, 56, 293–300. [Google Scholar] [CrossRef]
- Ferraroni, M.; Carta, F.; Scozzafava, A.; Supuran, C.T. Thioxocoumarins show an alternative carbonic anhydrase inhibition mechanism compared to coumarins. J. Med. Chem. 2016, 59, 462–473. [Google Scholar] [CrossRef]
- Grandane, A.; Tanc, M.; Zalubovskis, R.; Supuran, C.T. Synthesis of 6-tetrazolyl-substituted sulfocoumarins acting as highly potent and selective inhibitors of the tumor-associated carbonic anhydrase isoforms IX and XII. Bioorg. Med. Chem. 2014, 22, 1522–1528. [Google Scholar] [CrossRef] [PubMed]
- Grandane, A.; Tanc, M.; Zalubovskis, R.; Supuran, C.T. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg. Med. Chem. Lett. 2014, 24, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Grandane, A.; Tanc, M.; Zalubovskis, R.; Supuran, C.T. Synthesis of 6-aryl-substituted sulfocoumarins and investigation of their carbonic anhydrase inhibitory action. Bioorg. Med. Chem. 2015, 23, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Tanc, M.; Carta, F.; Scozzafava, A.; Supuran, C.T. 6-Substituted 1,2-benzoxathiine-2,2-dioxides are isoformselective inhibitors of human carbonic anhydrases IX, XII and VA. Org. Biomol. Chem. 2015, 13, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Grandane, A.; Tanc, M.; Di Cesare Mannelli, L.; Carta, F.; Ghelardini, C.; Zalubovskis, R. 6-Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J. Med. Chem. 2015, 58, 3975–3983. [Google Scholar] [CrossRef]
- Nocentini, A.; Ceruso, M.; Carta, F.; Supuran, C.T. 7-Aryl-triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrase IX and XII. J. Enzym. Inhib. Med. Chem. 2016, 31, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Nocentini, A.; Carta, F.; Tanc, M.; Selleri, S.; Supuran, C.T.; Bazzicalupi, C. Deciphering the mechanism of human carbonic anhydrases inhibition with sulfocoumarins: Computational and experimental studies. Chemistry 2018, 24, 7840–7844. [Google Scholar] [CrossRef]
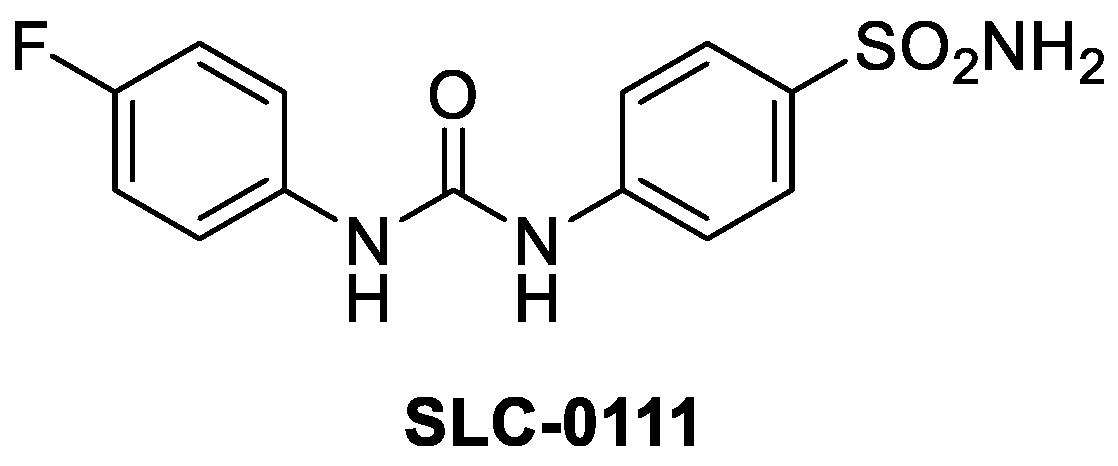
- McDonald, P.C.; Chia, S.; Bedard, P.L.; Chu, Q.; Lyle, M.; Tang, L.; Singh, M.; Zhang, Z.; Supuran, C.T.; Renouf, D.J.; et al. A Phase 1 Study of SLC-0111, a Novel Inhibitor of Carbonic Anhydrase IX, in Patients with Advanced Solid Tumors, Am. J. Clin. Oncol. 2020, 43, 484–490. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Z.; Shi, J.; Chen, H.; Mi, B.; Li, P.; Gong, P. Synthesis and cytotoxicity of novel 10-substituted dihydroartemisinin derivatives containing N-arylphenyl-ethenesulfonamide groups. Molecules 2013, 18, 2864–2877. [Google Scholar] [CrossRef] [Green Version]
- Pacchiano, F.; Carta, F.; McDonald, P.C. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [Green Version]
- Pustenko, A.; Stepanovs, D.; Žalubovskis, R.; Vullo, D.; Kazaks, A.; Leitans, J.; Tars, K.; Supuran, C.T. 3H-1,2-benzoxathiepine 2,2-dioxides: A new class of isoform-selective carbonic anhydrase inhibitors. J. Enzyme. Inhib. Med. Chem. 2017, 32, 767–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, L.; Peeters, S.; Lieuwes, N.G. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother. Oncol. 2011, 99, 424–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pustenko, A.; Nocentini, A.; Balašova, A.; Krasavin, M.; Zalubovskis, R.; Supuran, C.T. 7-Acylamino-3H-1,2-benzoxathiepine 2,2-dioxides as new isoform-selective carbonic anhydrase IX and XII inhibitors. J. Enzyme. Inhib. Med. Chem. 2020, 35, 650–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Nocentini, A.; Elsayed, Z.M.; Al-Warhi, T.; Aljaeed, N.; Alotaibi, O.J.; Al-Sanea, M.M.; Abdel-Aziz, H.A.; Supuran, C.T. Benzofuran-Based Carboxylic Acids as Carbonic Anhydrase Inhibitors and Antiproliferative Agents against Breast Cancer. CS Med. Chem. Lett. 2020, 11, 1022–1027. [Google Scholar] [CrossRef]
- Sechi, M.; Innocenti, A.; Pala, N.; Rogolino, D.; Carcelli, M.; Scozzafava, A.; Supuran, C.T. Inhibition of a-class cytosolic human carbonic anhydrases I, II, IX and XII, and b-class fungal enzymes by carboxylic acids and their derivatives: New isoform-I selective nanomolar inhibitors. Med. Chem. Lett. 2012, 22, 5801–5806. [Google Scholar] [CrossRef] [Green Version]
- Bonardi, A.; Nocentini, A.; Bua, S.; Combs, J.; Lomelino, C.; Andring, J.; Lucarini, L.; Sgambellone, S.; Masini, E.; McKenna, R.; et al. Sulfonamide Inhibitors of Human Carbonic Anhydrases Designed through a Three-Tails Approach: Improving Ligand/Isoform Matching and Selectivity of Action. J. Med. Chem. 2020, 63, 7422–7444. [Google Scholar] [CrossRef]
- Petreni, A.; Bonardi, A.; Lomelino, C.; Osman, S.M.; ALOthman, Z.A.; Eldehna, W.M.; El-Haggar, R.; McKenna, R.; Nocentini, A.; Supuran, C.T. Inclusion of a 5-fluorouracil moiety in nitrogenous bases derivatives as human carbonic anhydrase IX and XII inhibitors produced a targeted action against MDA-MB-231 and T47D breast cancer cells. Eur. J. Med. Chem. 2020, 190, 112112. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | KI (nM) a,b | |||||
| R1 | R2 | CA I | CA II | CA IX | CA XII | |
| 9 | H | OCH3 | >100,000 | 15,360 | 74.9 | 61.4 |
| 10 | CH3 | OCH3 | >100,000 | 8814 | 97.2 | 73.9 |
| 11 | H | H | 16,538 | 2896 | 55.8 | 42.5 |
| 12 | CH3 | H | 9961 | 3857 | 81.7 | 55.0 |
| 13 | H | H | >100,000 | >100,000 | 55.6 | 28.3 |
| 14 | H | 4-F | >100,000 | >100,000 | 22.9 | 19.2 |
| 15 | H | 3-F | >100,000 | >100,000 | 28.4 | 46.0 |
| 16 | H | 4-NO2 | >100,000 | >100,000 | 69.8 | 94.7 |
| 17 | H | 3-NO2 | >100,000 | >100,000 | 40.2 | 77.5 |
| 18 | CH3 | H | >100,000 | >100,000 | 49.8 | 8.6 |
| 19 | CH3 | 4-F | >100,000 | >100,000 | 14.3 | 41.7 |
| 20 | CH3 | 3-F | >100,000 | >100,000 | 33.2 | 25.7 |
| 21 | CH3 | 4-NO2 | >100,000 | >100,000 | 89.7 | 100.8 |
| 22 | CH3 | 3-NO2 | >100,000 | >100,000 | 61.4 | 136.0 |
| 25 | OCH3 | - | >100,000 | >100,000 | 1286 | 965.8 |
| 26 | H | - | 42650 | 8997 | 558.9 | 414.2 |
| 27 | H | - | >100,000 | >100,000 | 680.5 | 910.7 |
| 28 | 4-F | - | >100,000 | >100,000 | 754.9 | 653.8 |
| 29 | 3-F | - | >100,000 | >100,000 | 615.1 | 566.4 |
| 30 | 4-NO2 | - | >100,000 | >100,000 | 964.7 | 1056 |
| 31 | 3-NO2 | - | >100,000 | >100,000 | 898.1 | 1369 |
| 34 | H | CH2CH3 | >100,000 | >100,000 | 1631 | 2594 |
| 35 | OCH3 | CH2CH3 | >100,000 | >100,000 | 1085 | 1528 |
| 36 | H | H | 4625 | 996.5 | 499.7 | 563.7 |
| 37 | OCH3 | H | 7139 | 728.1 | 853.7 | 408.2 |
| 38 | H | H | >100,000 | >100,000 | 3288 | 2301 |
| 39 | H | 4-Br | >100,000 | >100,000 | 34,350 | 58,980 |
| 40 | H | 3-Br | >100,000 | >100,000 | 40,840 | 78,320 |
| 41 | H | 4-OCH3 | >100,000 | >100,000 | 8231 | 8550 |
| 42 | H | 3-OCH3 | >100,000 | >100,000 | 5424 | 3380 |
| 43 | OCH3 | H | >100,000 | >100,000 | 2429 | 1594 |
| 44 | OCH3 | 4-Br | >100,000 | >100,000 | 51,460 | 42,520 |
| 45 | OCH3 | 3-Br | >100,000 | >100,000 | 64,380 | 76,540 |
| 46 | OCH3 | 4-OCH3 | >100,000 | >100,000 | 4367 | 6547 |
| 47 | CH3 | 3-OCH3 | >100,000 | >100,000 | 6558 | 2469 |
| AAZ | - | - | 250 | 12.0 | 25.0 | 5.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giovannuzzi, S.; Capasso, C.; Nocentini, A.; Supuran, C.T. Continued Structural Exploration of Sulfocoumarin as Selective Inhibitor of Tumor-Associated Human Carbonic Anhydrases IX and XII. Molecules 2022, 27, 4076. https://doi.org/10.3390/molecules27134076
Giovannuzzi S, Capasso C, Nocentini A, Supuran CT. Continued Structural Exploration of Sulfocoumarin as Selective Inhibitor of Tumor-Associated Human Carbonic Anhydrases IX and XII. Molecules. 2022; 27(13):4076. https://doi.org/10.3390/molecules27134076
Chicago/Turabian StyleGiovannuzzi, Simone, Clemente Capasso, Alessio Nocentini, and Claudiu T. Supuran. 2022. "Continued Structural Exploration of Sulfocoumarin as Selective Inhibitor of Tumor-Associated Human Carbonic Anhydrases IX and XII" Molecules 27, no. 13: 4076. https://doi.org/10.3390/molecules27134076
APA StyleGiovannuzzi, S., Capasso, C., Nocentini, A., & Supuran, C. T. (2022). Continued Structural Exploration of Sulfocoumarin as Selective Inhibitor of Tumor-Associated Human Carbonic Anhydrases IX and XII. Molecules, 27(13), 4076. https://doi.org/10.3390/molecules27134076