
Design, Synthesis and Cytotoxic Activity Evaluation of Newly Synthesized Amides-Based TMP Moiety as Potential Anticancer Agents over HepG2 Cells

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
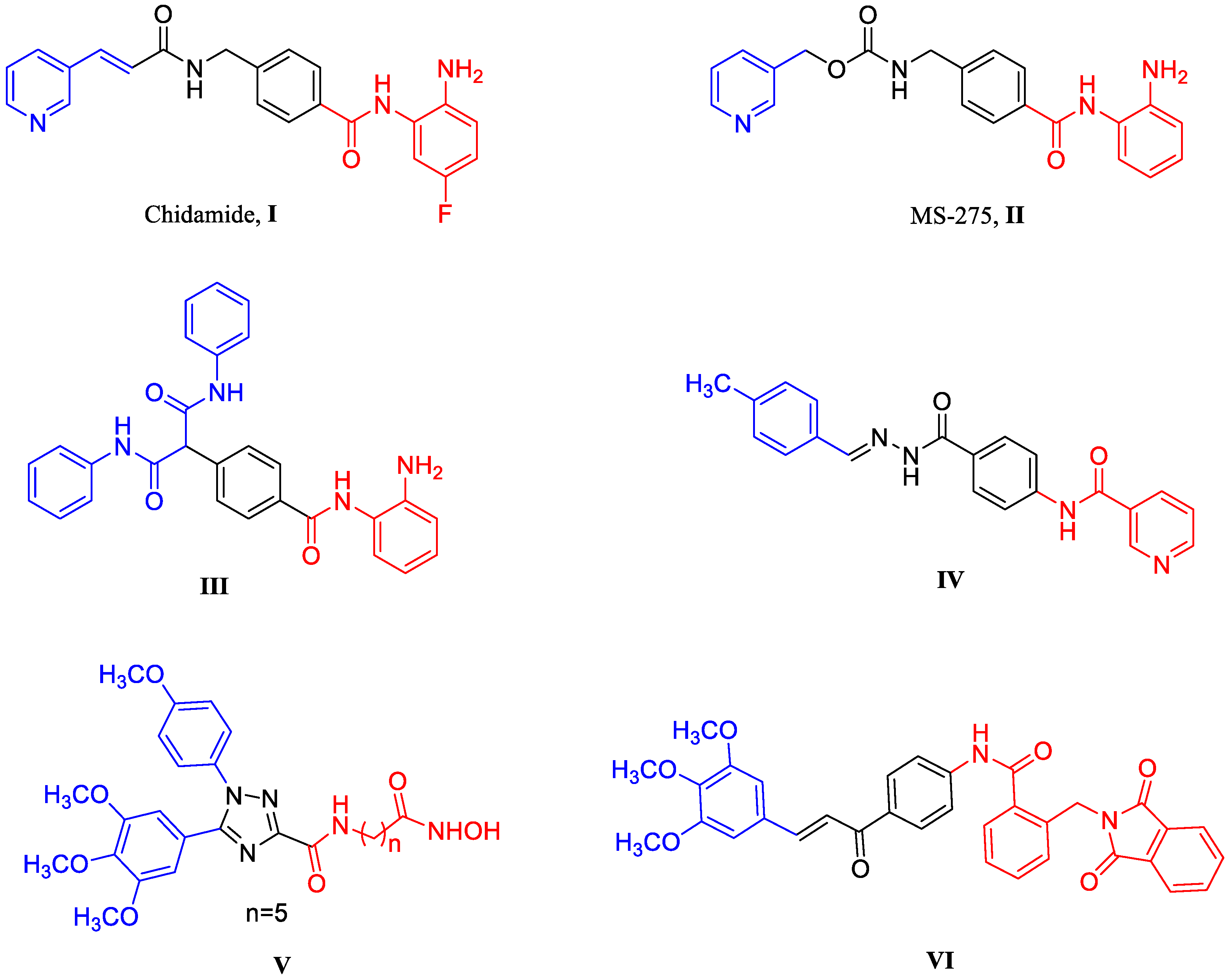
2. Results and Discussion
2.1. Chemistry
2.2. Biology
2.2.1. In Vitro Cytotoxic Activity against HepG2 Cell Line
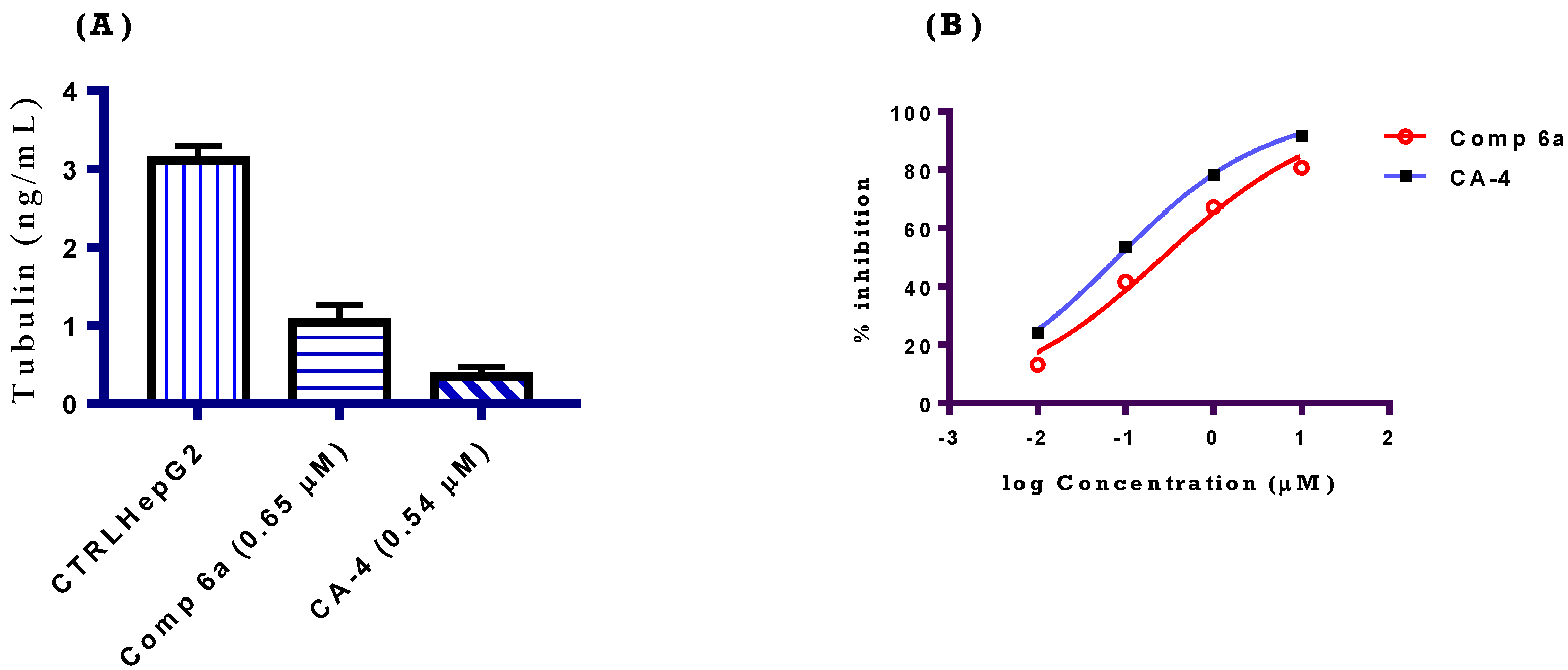
2.2.2. HDAC Inhibitory Activity
2.2.3. Tubulin Polymerization Inhibition Assay
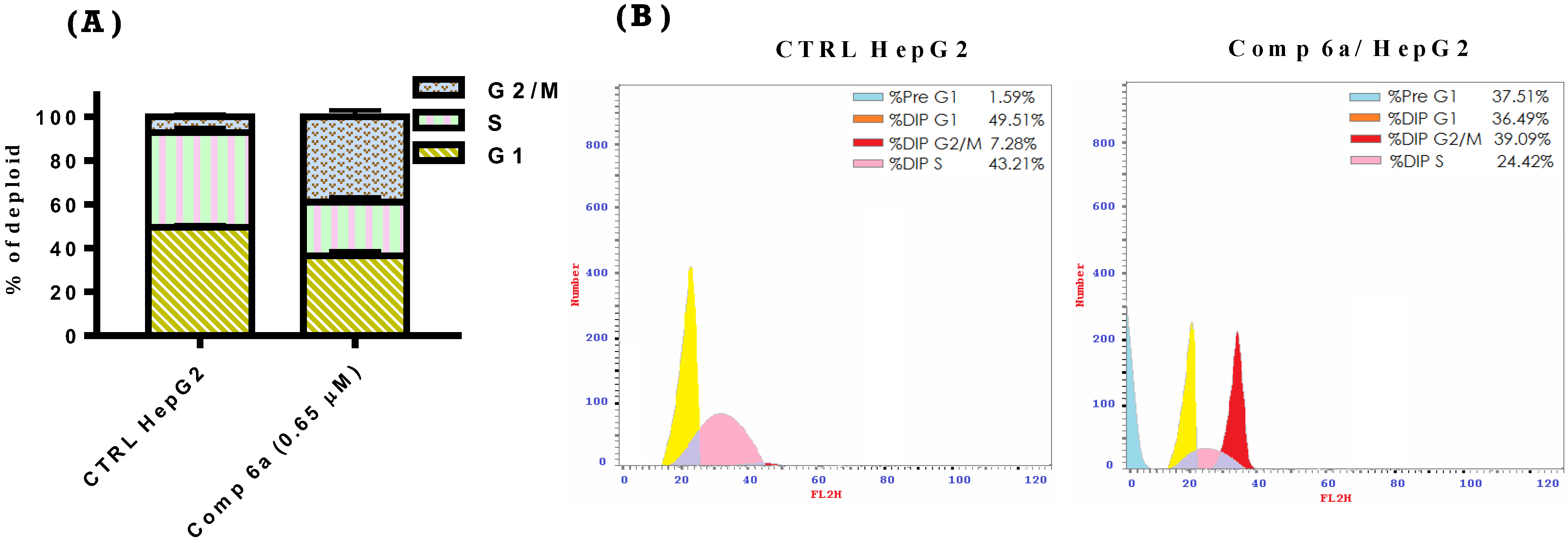
2.2.4. Cell Cycle Analysis
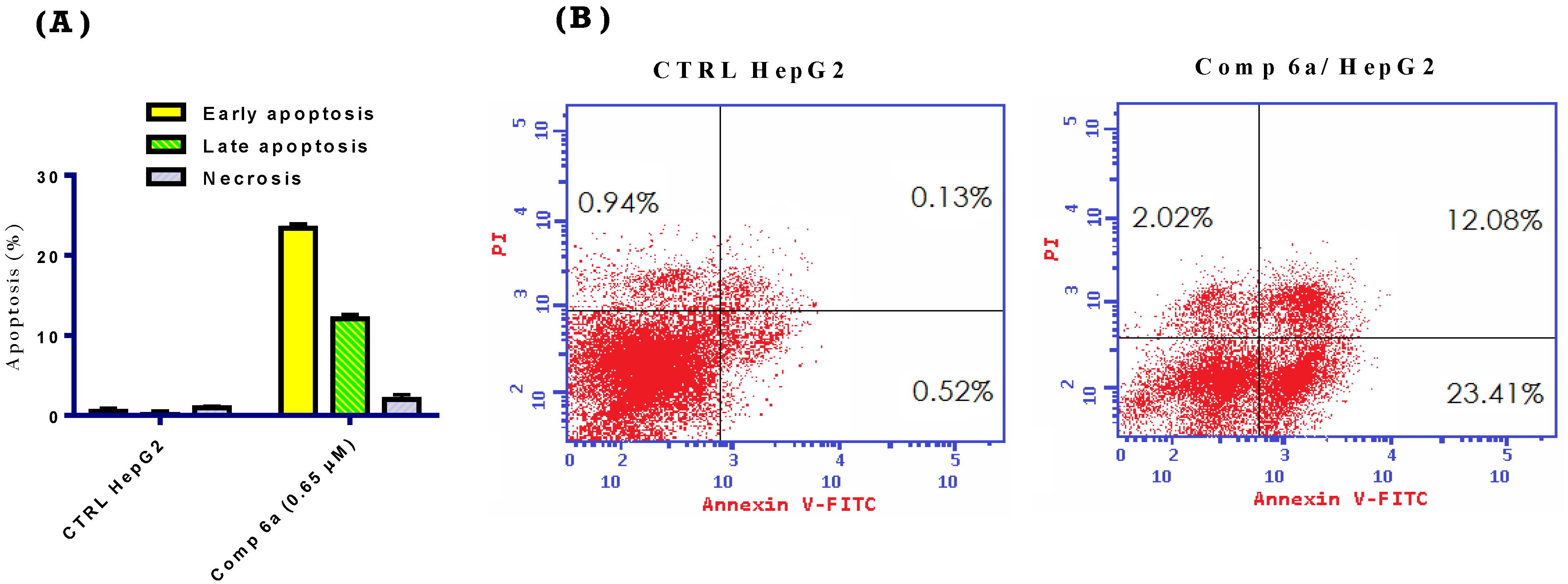
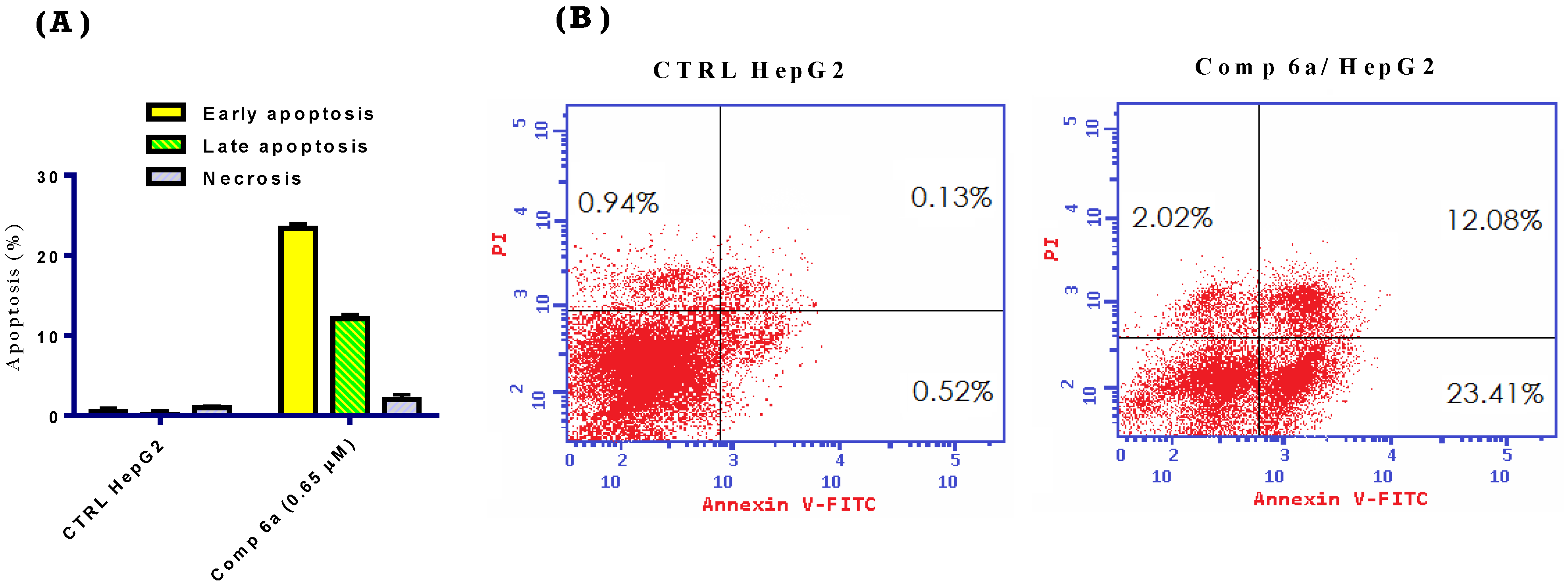
2.2.5. Apoptosis Assay
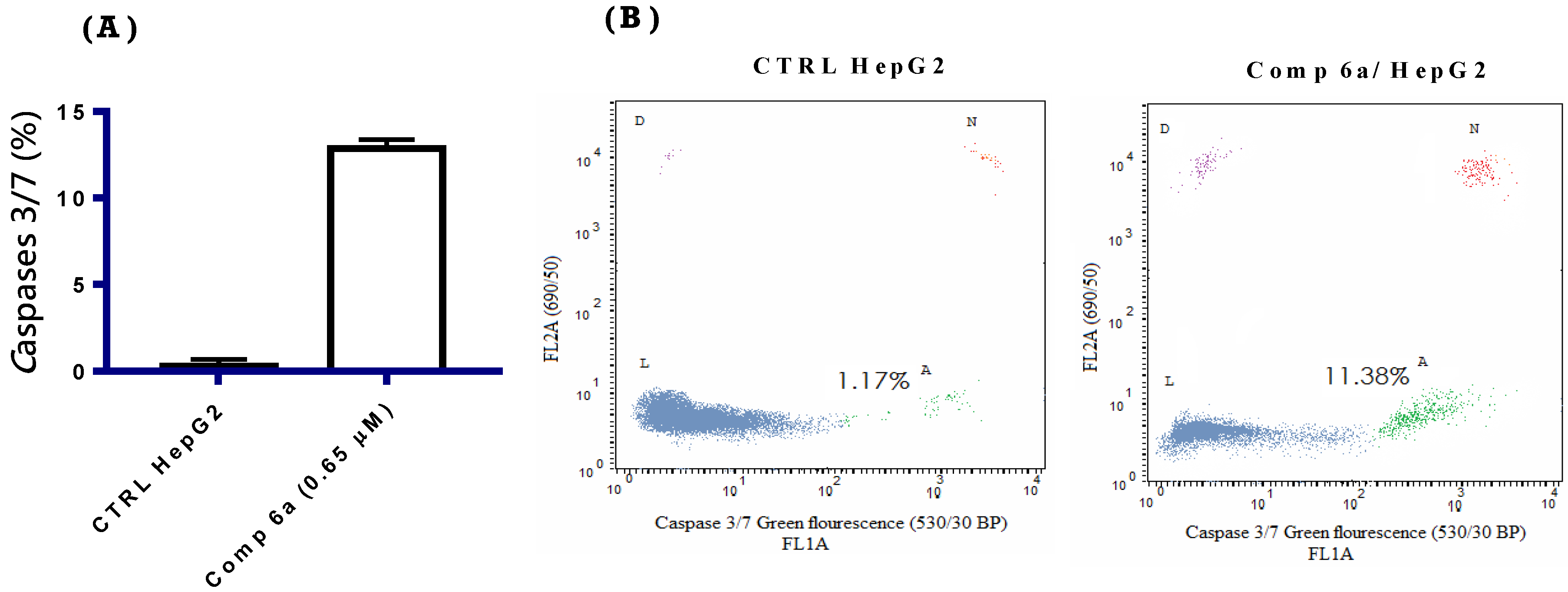
2.2.6. Caspase 3/7 Assay
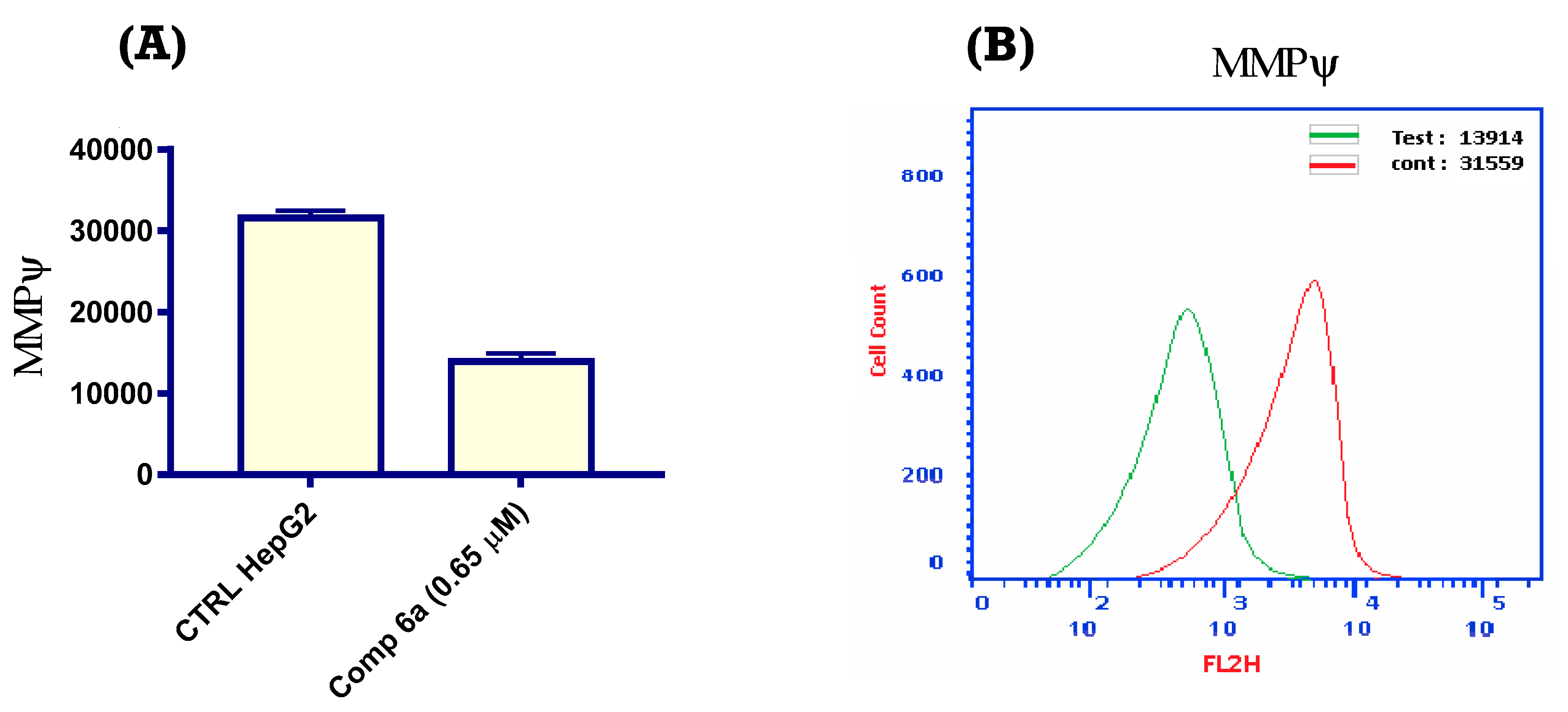
2.2.7. Mitochondrial Membrane Potential (MMP)
3. Conclusions
4. Experimental
4.1. General
4.2. Chemistry
4.2.1. General Procedure for the Preparation of N-(3-(arylamino)-1-(furan-2-yl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamides 2a–c
N-(3-(3-chlorophenylamino)-1-(furan-2-yl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamide (2a)
N-(3-(4-chlorophenylamino)-1-(furan-2-yl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamide (2b)
N-(1-(furan-2-yl)-3-oxo-3-(p-tolylamino)prop-1-en-2-yl)-3,4,5-trimethoxybenzamide (2c)
4.2.2. N-(1-(furan-2-yl)-3-(naphthalen-2-ylamino)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamide (3)
4.2.3. General Procedure for the Preparation of N-(3-(2-(4-Aroyl)hydrazinyl)-1-(furan-2-yl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamides (4a,b)
N-(1-(furan-2-yl)-3-(2-(3-hydroxybenzoyl)hydrazinyl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamide (4a)
N-(1-(furan-2-yl)-3-(2-isonicotinoylhydrazinyl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamide (4b)
4.2.4. General Procedure for the Synthesis of N-(4-(2-(3-(furan-2-yl)-2-(3,4,5 trimethoxybenzamido)acryloyl)hydrazinecarbonyl)phenyl)nicotinamide (5)
4.2.5. General Procedure for the Synthesis of N-((1Z)-3-(2-(3-(aryl)-2-(3,4,5-trimethoxybenzamido)acryloyl)hydrazinyl)-1-(furan-2-yl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamides 6a–e
N-((1Z)-3-(2-(3-(4-chlorophenyl)-2-(3,4,5-trimethoxybenzamido)acryloyl)hydrazinyl)-1-(furan-2-yl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamide (6a)
N-((1Z)-3-(2-(3-(4-cyanophenyl)-2-(3,4,5-trimethoxybenzamido)acryloyl)hydrazinyl)-1-(furan-2-yl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamide (6b)
N-((1Z)-3-(2-(3-(4-(dimethylamino)phenyl)-2-(3,4,5-trimethoxybenzamido)acryloyl)hydrazinyl)-1-(furan-2-yl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamide (6c)
N-(3-(2-((Z)-3-(furan-2-yl)-2-(3,4,5-trimethoxybenzamido)acryloyl)hydrazinyl)-1-(4-methoxyphenyl)-3-oxoprop-1-en-2-yl)-3,4,5-trimethoxybenzamide (6d)
N-(3-(2-((Z)-3-(furan-2-yl)-2-(3,4,5-trimethoxybenzamido)acryloyl)hydrazinyl)-3-oxo-1-(3,4,5-trimethoxyphenyl)prop-1-en-2-yl)-3,4,5-trimethoxybenzamide (6e)
4.3. Biological Studies
4.3.1. Cytotoxic Activity Evaluation
4.3.2. In Vitro HDAC Inhibition Assay
4.3.3. In Vitro Tubulin Inhibition Assay
4.3.4. Cell Cycle Analysis
4.3.5. Apoptosis Assay
4.3.6. Caspase 3/7 Assay
4.3.7. Mitochondrial Membrane Potential (MMP) Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Liu, Q.; Zhang, B.; Wang, Y.; Wang, X.; Gou, S. Discovery of phthalazino[1,2-b]-quinazolinone derivatives as multi-target HDAC inhibitors for the treatment of hepatocellular carcinoma via activating the p53 signal pathway. Eur. J. Med. Chem. 2021, 229, 114058. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, X.; Zhao, H.; Liu, M.; Du, J.; Zhang, J.; Yang, X.; Hou, X.; Fang, H. Discovery of DNA-Targeting HDAC Inhibitors with Potent Antitumor Efficacy In Vivo That Trigger Antitumor Immunity. J. Med. Chem. 2022, 65, 3667–3683. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-S.; Chou, C.-H.; Wu, Y.-H.; Yang, M.-H.; Chu, S.-H.; Chao, Y.-S.; Chen, C.-N. CC-01 (chidamide plus celecoxib) modifies the tumor immune microenvironment and reduces tumor progression combined with immune checkpoint inhibitor. Sci. Rep. 2022, 12, 1–18. [Google Scholar] [CrossRef]
- Dhiman, A.; Sharma, R.; Singh, R.K. Target-based anticancer indole derivatives and insight into structure—Activity relationship: A mechanistic review update (2018–2021). Acta Pharm. Sin. B, 2022; In Press. [Google Scholar]
- Wang, Y.-Q.; Wang, P.-Y.; Wang, Y.-T.; Yang, G.-F.; Zhang, A.; Miao, Z.-H. An Update on Poly(ADP-ribose)polymerase-1 (PARP-1) Inhibitors: Opportunities and Challenges in Cancer Therapy. J. Med. Chem. 2016, 59, 9575–9598. [Google Scholar] [CrossRef]
- Tian, Y.; Xie, Z.; Liao, C. Design, synthesis and anticancer activities of novel dual poly(ADP-ribose) polymerase-1/histone deacetylase-1 inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127036. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Xu, Y.; Jin, X.; Zhang, Q.; Ouyang, F.; Han, L.; Zhan, M.; Li, X.; Liang, B.; Huang, X. Structure modification and biological evaluation of indole-chalcone derivatives as anti-tumor agents through dual targeting tubulin and TrxR. Eur. J. Med. Chem. 2021, 227, 113897. [Google Scholar] [CrossRef]
- Dushanan, R.; Weerasinghe, S.; Dissanayake, D.P.; Senthilinithy, R. Cracking a cancer code histone deacetylation in epigenetic: The implication from molecular dynamics simulations on efficacy assessment of histone deacetylase inhibitors. J. Biomol. Struct. Dyn. 2020, 40, 2352–2368. [Google Scholar] [CrossRef]
- Doke, M.; Pendyala, G.; Samikkannu, T. Psychostimulants and opioids differentially influence the epigenetic modification of histone acetyltransferase and histone deacetylase in astrocytes. PLoS ONE 2021, 16, e0252895. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Zhang, D.; Wei, W.; Bæk, M.; Liu, W.; Gao, J.; Danková, D.; Nielsen, A.L.; Bolding, J.E.; Yang, L.; et al. Class I histone deacetylases (HDAC1–3) are histone lysine delactylases. Sci. Adv. 2022, 8, eabi6696. [Google Scholar] [CrossRef]
- Wang, K.; Tang, R.; Wang, S.; Xiong, Y.; Wang, W.; Chen, G.; Zhang, K.; Li, P.; Tang, Y.-D. Isoform-Selective HDAC Inhibitor Mocetinostat (MGCD0103) Alleviates Myocardial Ischemia/Reperfusion Injury Via Mitochondrial Protection Through the HDACs/CREB/PGC-1α Signaling Pathway. J. Cardiovasc. Pharmacol. 2021, 79, 217–228. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mohammad, G. Epigenetic modifications in diabetes. Metabolism 2022, 126, 154920. [Google Scholar] [CrossRef] [PubMed]
- Palamaris, K.; Moutafi, M.; Gakiopoulou, H.; Theocharis, S. Histone Deacetylase (HDAC) Inhibitors: A Promising Weapon to Tackle Therapy Resistance in Melanoma. Int. J. Mol. Sci. 2022, 23, 3660. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef] [PubMed]
- Pojani, E.; Barlocco, D. Romidepsin (FK228), A histone deacetylase Inhibitor and its analogues in cancer chemotherapy. Curr. Med. Chem. 2021, 28, 1290–1303. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, M.; Song, W.; Cai, Q.; Zhang, L.; Sun, X.; Zou, L.; Zhang, H.; Wang, L.; Xue, H. Chidamide plus prednisone, etoposide, and thalidomide for untreated angioimmunoblastic T-cell lymphoma in a Chinese population: A multicenter phase II trial. Am. J. Hematol. 2022, 97, 623–629. [Google Scholar] [CrossRef]
- Hauschild, A.; Trefzer, U.; Garbe, C.; Kaehler, K.; Ugurel, S.; Kiecker, F.; Eigentler, T.; Krissel, H.; Schadendorf, D. A phase II multicenter study on the histone deacetylase (HDAC) inhibitor MS-275, comparing two dosage schedules in metastatic melanoma. J. Clin. Oncol. 2006, 24, 8044. [Google Scholar] [CrossRef]
- Siliphaivanh, P.; Harrington, P.; Witter, D.J.; Otte, K.; Tempest, P.; Kattar, S.; Kral, A.M.; Fleming, J.C.; Deshmukh, S.V.; Harsch, A.; et al. Design of novel histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4619–4624. [Google Scholar] [CrossRef]
- Hamoud, M.M.; Pulya, S.; Osman, N.A.; Bobde, Y.; Hassan, A.E.; Abdel-Fattah, H.A.; Ghosh, B.; Ghanim, A.M. Design, synthesis, and biological evaluation of novel nicotinamide derivatives as potential histone deacetylase-3 inhibitors. N. J. Chem. 2020, 44, 9671–9683. [Google Scholar] [CrossRef]
- Hao, S.-Y.; Qi, Z.-Y.; Wang, S.; Wang, X.-R.; Chen, S.-W. Synthesis and bioevaluation of N-(3,4,5-trimethoxyphenyl)-1H-pyrazolo[3,4-b]pyridin-3-amines as tubulin polymerization inhibitors with anti-angiogenic effects. Bioorganic Med. Chem. 2020, 31, 115985. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Sun, Z.; Zhao, F.; Shan, G.; Meng, Q. Recent advances in research of colchicine binding site inhibitors and their interaction modes with tubulin. Futur. Med. Chem. 2021, 13, 839–858. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; He, M.; Li, Y.; Peng, Z.; Wang, G. A review on synthetic chalcone derivatives as tubulin polymerisation inhibitors. J. Enzym. Inhib. Med. Chem. 2021, 37, 9–38. [Google Scholar] [CrossRef]
- Ebenezer, O.; Shapi, M.; Tuszynski, J.A. A Review of the Recent Developments of Molecular Hybrids Targeting Tubulin Polymerization. Int. J. Mol. Sci. 2022, 23, 4001. [Google Scholar] [CrossRef] [PubMed]
- Aboeldahab, A.M.A.; Beshr, E.A.M.; Shoman, M.E.; Rabea, S.M.; Aly, O.M. Spirohydantoins and 1,2,4-triazole-3-carboxamide derivatives as inhibitors of histone deacetylase: Design, synthesis, and biological evaluation. Eur. J. Med. Chem. 2018, 146, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Mourad, A.A.E.; Mourad, M.A.E.; Jones, P.G. Novel HDAC/Tubulin Dual Inhibitor: Design, Synthesis and Docking Studies of α-Phthalimido-Chalcone Hybrids as Potential Anticancer Agents with Apoptosis-Inducing Activity. Drug Des. Dev. Ther. 2020, 14, 3111–3130. [Google Scholar] [CrossRef]
- Bukhari, S.N.; Ejaz, H.; Elsherif, M.A.; Junaid, K.; Zaki, I.; Masoud, R.E. Design and Synthesis of Some New Furan-Based Derivatives and Evaluation of In Vitro Cytotoxic Activity. Molecules 2022, 27, 2606. [Google Scholar] [CrossRef]
- Zaki, I.; Abu El-ata, S.A.; Fayad, E.; Abu Ali, O.A.; Abu Almaaty, A.H.; Saad, A.S. Evaluation of Synthetic 2,4-Disubstituted-benzo[g]quinoxaline Derivatives as Potential Anticancer Agents. Pharmaceuticals 2021, 14, 853. [Google Scholar] [CrossRef]
- Zaki, I.; Abou-Elkhair, R.A.I.; Abu Almaaty, A.H.A.; Abu Ali, O.; Fayad, E.; Ahmed Gaafar, A.G.; Zakaria, M.Y. Design and Synthesis of Newly Synthesized Acrylamide Derivatives as Potential Chemotherapeutic Agents against MCF-7 Breast Cancer Cell Line Lodged on PEGylated Bilosomal Nano-Vesicles for Improving Cytotoxic Activity. Pharmaceuticals 2021, 14, 1021. [Google Scholar] [CrossRef]
- Liu, Y.; Meng, Y.; Bian, J.; Liu, B.; Li, X.; Guan, Q.; Li, Z.; Zhang, W.; Wu, Y.; Zuo, D. 2-Methoxy-5((3,4,5-trimethosyphenyl) seleninyl) phenol causes G2/M cell cycle arrest and apoptosis in NSCLC cells through mitochondrial apoptotic pathway and MDM2 inhibition. J. Biochem. Mol. Toxicol. 2022, 3, e23066. [Google Scholar] [CrossRef]
- Nuth, M.; Benakanakere, M.R.; Ricciardi, R.P. Discovery of a potent cytotoxic agent that promotes G 2/M phase cell cycle arrest and apoptosis in a malignant human pharyngeal squamous carcinoma cell line. Int. J. Oncol. 2022, 60, 1–10. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp No | IC50 Value (μM) | |
|---|---|---|
| HepG2 | HL-7702 | |
| 2a | 68.90 ± 3.32 | NT |
| 2b | 23.28 ± 1.03 | NT |
| 2c | 22.03 ± 0.96 | NT |
| 3 | 16.24 ± 0.79 | NT |
| 4a | 13.37 ± 0.56 | NT |
| 4b | 8.36 ± 0.51 | NT |
| 5 | 3.25 ± 0.25 | NT |
| 6a | 0.65 ± 0.03 | 9.62 ± 0.23 |
| 6b | 0.92 ± 0.10 | 11.09 ± 0.18 |
| 6c | 1.12 ± 0.12 | 9.88 ± 0.14 |
| 6d | 3.81 ± 0.18 | NT |
| 6e | 3.98 ± 0.09 | NT |
| SAHA | 2.91 ± 0.15 | NT |
| CA-4 | 0.54 ± 0.04 | 8.86 ± 0.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Warhi, T.; Aldhahrani, A.; Althobaiti, F.; Fayad, E.; Abu Ali, O.A.; Albogami, S.; Abu Almaaty, A.H.; Khedr, A.I.M.; Bukhari, S.N.A.; Zaki, I. Design, Synthesis and Cytotoxic Activity Evaluation of Newly Synthesized Amides-Based TMP Moiety as Potential Anticancer Agents over HepG2 Cells. Molecules 2022, 27, 3960. https://doi.org/10.3390/molecules27123960
Al-Warhi T, Aldhahrani A, Althobaiti F, Fayad E, Abu Ali OA, Albogami S, Abu Almaaty AH, Khedr AIM, Bukhari SNA, Zaki I. Design, Synthesis and Cytotoxic Activity Evaluation of Newly Synthesized Amides-Based TMP Moiety as Potential Anticancer Agents over HepG2 Cells. Molecules. 2022; 27(12):3960. https://doi.org/10.3390/molecules27123960
Chicago/Turabian StyleAl-Warhi, Tarfah, Adil Aldhahrani, Fayez Althobaiti, Eman Fayad, Ola A. Abu Ali, Sarah Albogami, Ali H. Abu Almaaty, Amgad I. M. Khedr, Syed Nasir Abbas Bukhari, and Islam Zaki. 2022. "Design, Synthesis and Cytotoxic Activity Evaluation of Newly Synthesized Amides-Based TMP Moiety as Potential Anticancer Agents over HepG2 Cells" Molecules 27, no. 12: 3960. https://doi.org/10.3390/molecules27123960
APA StyleAl-Warhi, T., Aldhahrani, A., Althobaiti, F., Fayad, E., Abu Ali, O. A., Albogami, S., Abu Almaaty, A. H., Khedr, A. I. M., Bukhari, S. N. A., & Zaki, I. (2022). Design, Synthesis and Cytotoxic Activity Evaluation of Newly Synthesized Amides-Based TMP Moiety as Potential Anticancer Agents over HepG2 Cells. Molecules, 27(12), 3960. https://doi.org/10.3390/molecules27123960