
Drugs Targeting the A3 Adenosine Receptor: Human Clinical Study Data

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. A3AR Agonists: Piclidenoson and Namodenoson
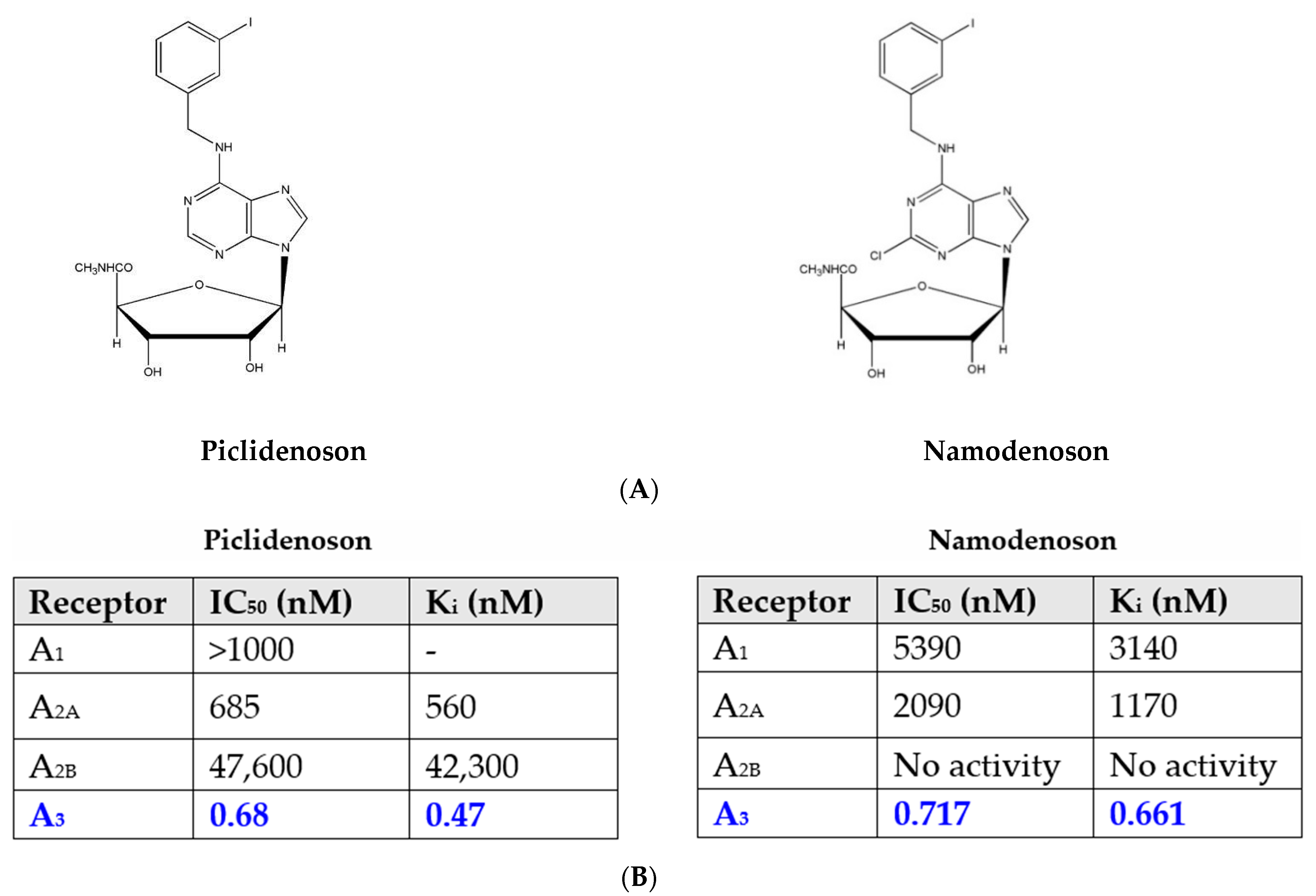
2.1. Piclidenoson and Namodenoson: Pharmacological Activity
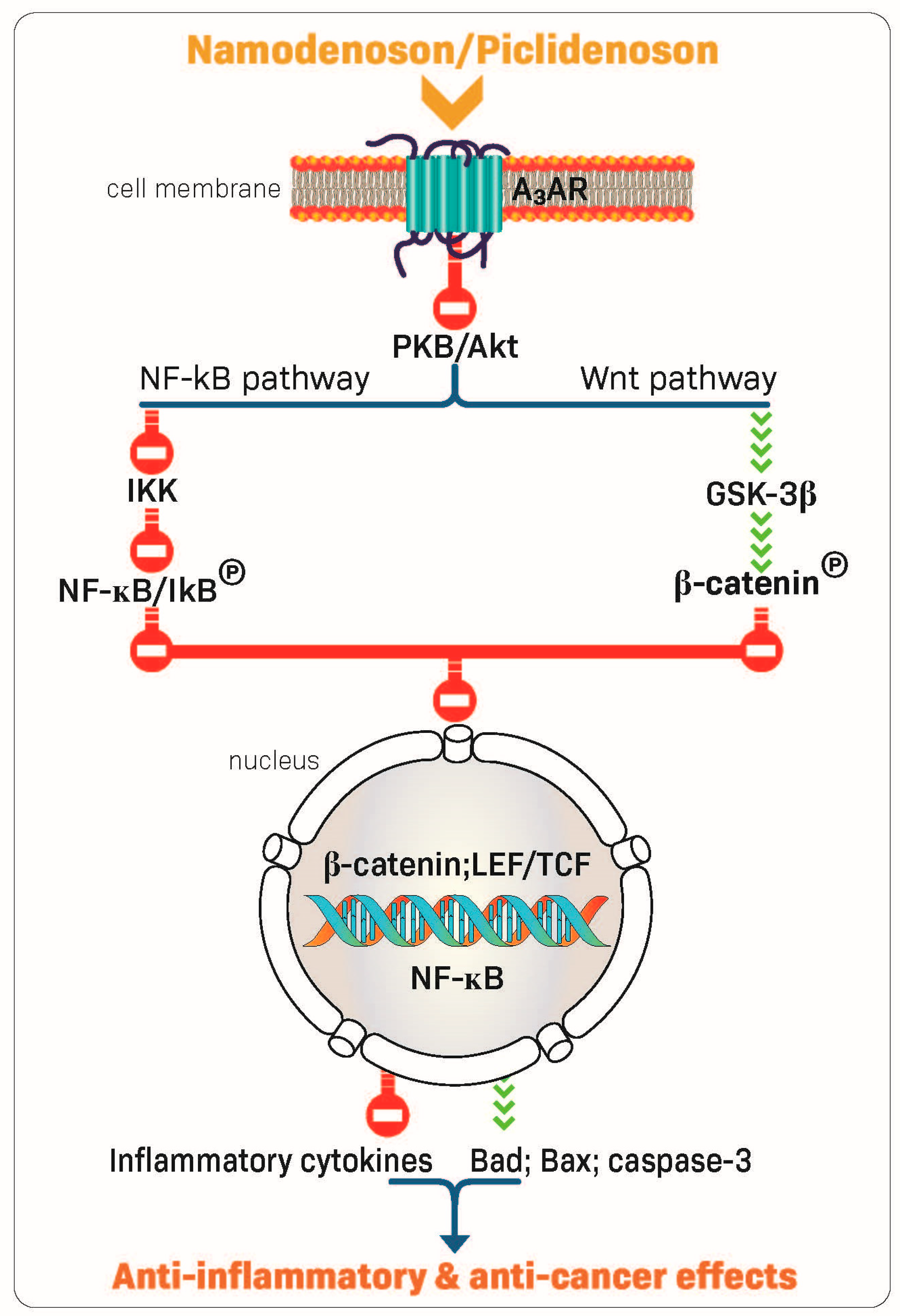
2.2. Piclidenoson and Namodenoson: Mechanism of Action
3. Piclidenoson: Clinical Development
3.1. Piclidenoson in Rheumatoid Arthritis (RA)
3.1.1. Piclidenoson in RA: Phase IIa Study
3.1.2. Piclidenoson in RA: Phase IIb Studies
3.1.3. Piclidenoson in RA: Phase III Study
3.2. Piclidenoson in Psoriasis
3.2.1. Piclidenoson in Psoriasis: Phase II Study
3.2.2. Piclidenoson in Psoriasis: Phase II/III Study
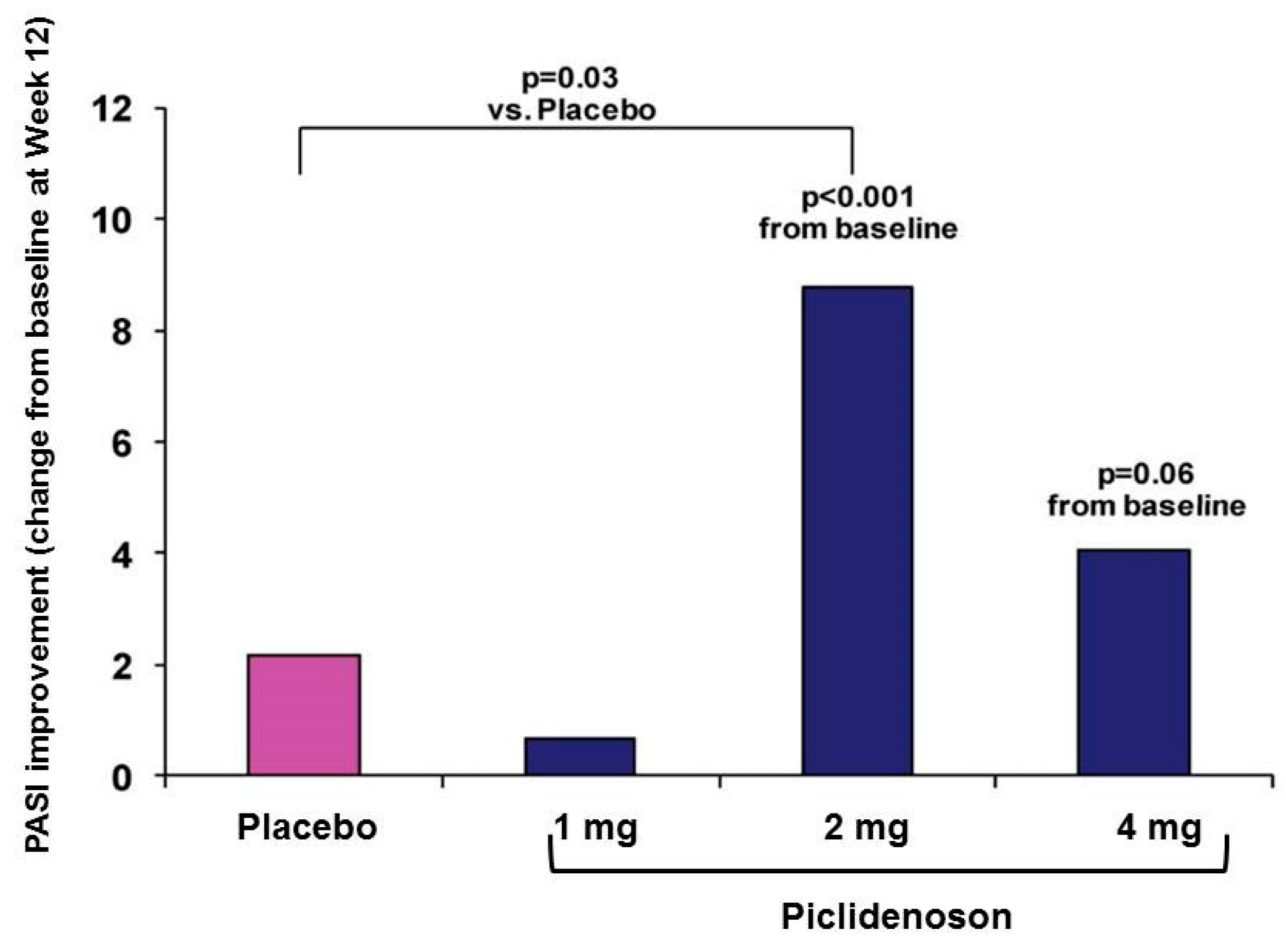
3.2.3. Piclidenoson in Psoriasis: Phase III Study
4. Namodenoson: Clinical Development
4.1. Namodenson in Hepatocellular Carcinoma (HCC)
4.1.1. Namodenoson in HCC: Phase I/II Study
4.1.2. Namodenoson in HCC: Phase II Study
4.2. Namodenson in NASH
Namodenoson in NAFLD/NASH: Phase IIa Study
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Borea, P.A.; Varani, K.; Vincenzi, F.; Baraldi, P.G.; Tabrizi, M.A.; Merighi, S.; Gessi, S. The A3 adenosine receptor: History and perspectives. Pharmacol. Rev. 2015, 67, 74–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazziotta, C.; Rotondo, J.C.; Lanzillotti, C.; Campione, G.; Martini, F.; Tognon, M. Cancer biology and molecular genetics of A3 adenosine receptor. Oncogene 2022, 41, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Cohen, S. The A3 adenosine receptor (A3AR): Therapeutic target and predictive biological marker in rheumatoid arthritis. Clin. Rheumatol. 2016, 35, 2359–2362. [Google Scholar] [CrossRef] [PubMed]
- Madi, L.; Ochaion, A.; Rath-Wolfson, L.; Bar-Yehuda, S.; Erlanger, A.; Ohana, G.; Harish, A.; Merimski, O.; Barer, F.; Fishman, P. The A3 adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clin. Cancer Res. 2004, 10, 4472–4479. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Aghazadeh Tabrizi, M.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K.; et al. A3 adenosine receptors as modulators of inflammation: From medicinal chemistry to therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef]
- Gessi, S.; Varani, K.; Merighi, S.; Cattabriga, E.; Avitabile, A.; Gavioli, R.; Fortini, C.; Leung, E.; Mac Lennan, S.; Borea, P.A. Expression of A3 adenosine receptors in human lymphocytes: Up-regulation in T cell activation. Mol. Pharmacol. 2004, 65, 711–719. [Google Scholar] [CrossRef]
- Madi, L.; Cohen, S.; Ochayin, A.; Bar-Yehuda, S.; Barer, F.; Fishman, P. Overexpression of A3 adenosine receptor in peripheral blood mononuclear cells in rheumatoid arthritis: Involvement of nuclear factor-kappab in mediating receptor level. J. Rheumatol. 2007, 34, 20–26. [Google Scholar]
- Ochaion, A.; Bar-Yehuda, S.; Cohen, S.; Barer, F.; Patoka, R.; Amital, H.; Reitblat, T.; Reitblat, A.; Ophir, J.; Konfino, I.; et al. The anti-inflammatory target A3 adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn’s disease. Cell. Immunol. 2009, 258, 115–122. [Google Scholar] [CrossRef]
- Silverman, M.H.; Strand, V.; Markovits, D.; Nahir, M.; Reitblat, T.; Molad, Y.; Rosner, I.; Rozenbaum, M.; Mader, R.; Adawi, M.; et al. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: Data from a phase II clinical trial. J. Rheumatol. 2008, 35, 41–48. [Google Scholar]
- Gessi, S.; Cattabriga, E.; Avitabile, A.; Gafa, R.; Lanza, G.; Cavazzini, L.; Bianchi, N.; Gambari, R.; Feo, C.; Liboni, A.; et al. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clin. Cancer Res. 2004, 10, 5895–5901. [Google Scholar] [CrossRef] [Green Version]
- Fishman, P.; Bar-Yehuda, S.; Ardon, E.; Rath-Wolfson, L.; Barrer, F.; Ochaion, A.; Madi, L. Targeting the A3 adenosine receptor for cancer therapy: Inhibition of prostate carcinoma cell growth by A3AR agonist. Anticancer Res. 2003, 23, 2077–2083. [Google Scholar] [PubMed]
- Madi, L.; Bar-Yehuda, S.; Barer, F.; Ardon, E.; Ochaion, A.; Fishman, P. A3 adenosine receptor activation in melanoma cells: Association between receptor fate and tumor growth inhibition. J. Biol. Chem. 2003, 278, 42121–42130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merimsky, O.; Bar-Yehuda, S.; Madi, L.; Fishman, P. Modulation of the A3 adenosine receptor by low agonist concentration induces antitumor and myelostimulatory effects. Drug Dev. Res. 2003, 58, 386–389. [Google Scholar] [CrossRef]
- Ohana, G.; Bar-Yehuda, S.; Arich, A.; Madi, L.; Dreznick, Z.; Rath-Wolfson, L.; Silberman, D.; Slosman, G.; Fishman, P. Inhibition of primary colon carcinoma growth and liver metastasis by the A3 adenosine receptor agonist CF101. Br. J. Cancer 2003, 89, 1552–1558. [Google Scholar] [CrossRef]
- Fishman, P.; Bar-Yehuda, S.; Ohana, G.; Barer, F.; Ochaion, A.; Erlanger, A.; Madi, L. An agonist to the A3 adenosine receptor inhibits colon carcinoma growth in mice via modulation of GSK-3 beta and NF-kappa b. Oncogene 2004, 23, 2465–2471. [Google Scholar] [CrossRef] [Green Version]
- Bar-Yehuda, S.; Rath-Wolfson, L.; Del Valle, L.; Ochaion, A.; Cohen, S.; Patoka, R.; Zozulya, G.; Barer, F.; Atar, E.; Pina-Oviedo, S.; et al. Induction of an antiinflammatory effect and prevention of cartilage damage in rat knee osteoarthritis by CF101 treatment. Arthritis Rheum. 2009, 60, 3061–3071. [Google Scholar] [CrossRef]
- Ren, T.; Tian, T.; Feng, X.; Ye, S.; Wang, H.; Wu, W.; Qiu, Y.; Yu, C.; He, Y.; Zeng, J.; et al. An adenosine A3 receptor agonist inhibits DSS-induced colitis in mice through modulation of the NF-kappab signaling pathway. Sci. Rep. 2015, 5, 9047. [Google Scholar] [CrossRef] [Green Version]
- Baharav, E.; Bar-Yehuda, S.; Madi, L.; Silberman, D.; Rath-Wolfson, L.; Halpren, M.; Ochaion, A.; Weinberger, A.; Fishman, P. Antiinflammatory effect of a3 adenosine receptor agonists in murine autoimmune arthritis models. J. Rheumatol. 2005, 32, 469–476. [Google Scholar]
- Rath-Wolfson, L.; Bar-Yehuda, S.; Madi, L.; Ochaion, A.; Cohen, S.; Zabutti, A.; Fishman, P. IB-MECA, an A3 adenosine receptor agonist prevents bone resorption in rats with adjuvant induced arthritis. Clin. Exp. Rheumatol. 2006, 24, 400–406. [Google Scholar]
- Bar-Yehuda, S.; Luger, D.; Ochaion, A.; Cohen, S.; Patokaa, R.; Zozulya, G.; Silver, P.B.; de Morales, J.M.; Caspi, R.R.; Fishman, P. Inhibition of experimental auto-immune uveitis by the A3 adenosine receptor agonist CF101. Int. J. Mol. Med. 2011, 28, 727–731. [Google Scholar]
- Fishman, P.; Cohen, S.; Salhab, A.; Amer, J.; Itzhak, I.; Barer, F.; Safadi, R. Namodenoson anti-NAFLD/NASH activity is mediated via de-regulation of the Wnt/β-catenin pathway. Inflamm. Intest. Dis. 2019, 44, 2256–2264. [Google Scholar]
- Cohen, S.; Fishman, P. Targeting the A3 adenosine receptor to treat cytokine release syndrome in cancer immunotherapy. Drug Des. Devel. Ther. 2019, 13, 491–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Yehuda, S.; Stemmer, S.M.; Madi, L.; Castel, D.; Ochaion, A.; Cohen, S.; Barer, F.; Zabutti, A.; Perez-Liz, G.; Del Valle, L.; et al. The A3 adenosine receptor agonist CF102 induces apoptosis of hepatocellular carcinoma via de-regulation of the Wnt and NF-kappab signal transduction pathways. Int. J. Oncol. 2008, 33, 287–295. [Google Scholar] [PubMed]
- Ledderose, C.; Hefti, M.M.; Chen, Y.; Bao, Y.; Seier, T.; Li, L.; Woehrle, T.; Zhang, J.; Junger, W.G. Adenosine arrests breast cancer cell motility by A3 receptor stimulation. Purinergic Signal. 2016, 12, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishman, P.; Bar-Yehuda, S.; Barer, F.; Madi, L.; Multani, A.F.; Pathak, S. The A3 adenosine receptor as a new target for cancer therapy and chemoprotection. Exp. Cell Res. 2001, 269, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Yehuda, S.; Madi, L.; Barak, D.; Mittelman, M.; Ardon, E.; Ochaion, A.; Cohn, S.; Fishman, P. Agonists to the A3 adenosine receptor induce G-CSF production via NF-κB activation: A new class of myeloprotective agents. Exp. Hematol. 2002, 30, 1390–1398. [Google Scholar] [CrossRef]
- Cohen, S.; Stemmer, S.M.; Zozulya, G.; Ochaion, A.; Patoka, R.; Barer, F.; Bar-Yehuda, S.; Rath-Wolfson, L.; Jacobson, K.A.; Fishman, P. CF102 an A3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J. Cell. Physiol. 2011, 226, 2438–2447. [Google Scholar] [CrossRef] [Green Version]
- van Troostenburg, A.R.; Clark, E.V.; Carey, W.D.; Warrington, S.J.; Kerns, W.D.; Cohn, I.; Silverman, M.H.; Bar-Yehuda, S.; Fong, K.L.; Fishman, P. Tolerability, pharmacokinetics and concentration-dependent hemodynamic effects of oral CF101, an A3 adenosine receptor agonist, in healthy young men. Int. J. Clin. Pharmacol. Ther. 2004, 42, 534–542. [Google Scholar] [CrossRef]
- Stemmer, S.M.; Benjaminov, O.; Medalia, G.; Ciuraru, N.B.; Silverman, M.H.; Bar-Yehuda, S.; Fishman, S.; Harpaz, Z.; Farbstein, M.; Cohen, S.; et al. CF102 for the treatment of hepatocellular carcinoma: A phase I/II, open-label, dose-escalation study. Oncologist 2013, 18, 25–26. [Google Scholar] [CrossRef]
- Lee, J.Y.; Jhun, B.S.; Oh, Y.T.; Lee, J.H.; Choe, W.; Baik, H.H.; Ha, J.; Yoon, K.S.; Kim, S.S.; Kang, I. Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of PI 3-kinase/AKT and NF-kappab activation in murine BV2 microglial cells. Neurosci. Lett. 2006, 396, 1–6. [Google Scholar] [CrossRef]
- Hasko, G.; Nemeth, Z.H.; Vizi, E.S.; Salzman, A.L.; Szabo, C. An agonist of adenosine A3 receptors decreases interleukin-12 and interferon-gamma production and prevents lethality in endotoxemic mice. Eur. J. Pharmacol. 1998, 358, 261–268. [Google Scholar] [CrossRef]
- Cohen, S.; Barer, F.; Itzhak, I.; Silverman, M.H.; Fishman, P. Inhibition of IL-17 and IL -23 in human keratinocytes by the A3 adenosine receptor agonist piclidenoson. J. Immunol. Res. 2018, 2018, 2310970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishman, P.; Bar-Yehuda, S.; Madi, L.; Rath-Wolfson, L.; Ochaion, A.; Cohen, S.; Baharav, E. The PI3K-NF-kappab signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Res. Ther. 2006, 8, R33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochaion, A.; Bar-Yehuda, S.; Cohen, S.; Amital, H.; Jacobson, K.A.; Joshi, B.V.; Gao, Z.G.; Barer, F.; Patoka, R.; Del Valle, L.; et al. The A3 adenosine receptor agonist CF502 inhibits the PI3K, PKB/AKT and NF-kappab signaling pathway in synoviocytes from rheumatoid arthritis patients and in adjuvant-induced arthritis rats. Biochem. Pharmacol. 2008, 76, 482–494. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Barer, F.; Bar-Yehuda, S.; AP, I.J.; Jacobson, K.A.; Fishman, P. A(3) adenosine receptor allosteric modulator induces an anti-inflammatory effect: In vivo studies and molecular mechanism of action. Mediators Inflamm. 2014, 2014, 708746. [Google Scholar] [CrossRef] [Green Version]
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 2012, 17, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Radu, A.F.; Bungau, S.G. Management of rheumatoid arthritis: An overview. Cells 2021, 10, 2857. [Google Scholar] [CrossRef]
- Felson, D.T.; Lavalley, M.P. The ACR20 and defining a threshold for response in rheumatic diseases: Too much of a good thing. Arthritis Res. Ther. 2014, 16, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Curtis, J.R.; Trivedi, M.; Haraoui, B.; Emery, P.; Park, G.S.; Collier, D.H.; Aras, G.A.; Chung, J. Defining and characterizing sustained remission in patients with rheumatoid arthritis. Clin. Rheumatol. 2018, 37, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Stoilov, R.M.; Licheva, R.N.; Mihaylova, M.K.; Reitblat, T.; Dimitrov, E.A.; Shimbova, K.M.; Bhatia, G.; Pispati, A.; Gurman-Balbir, A.; Bagaria, B.R.; et al. Therapeutic effect of oral CF101 in patients with rheumatoid arthritis: A randomized, double-blind, placebo-controlled phase II study. Immunome Res. 2014, 11, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Reitblat, T.; Gurman- Balbir, A.; Harpaz, Z.; Farbstein, M.; Silverman, M.; Kerns, W.; Fishman, P. The efficacy and safety of piclidenoson vs methotrexate in early rheumatoid arthritis: Phase 3 randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2021, 73. [Google Scholar]
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the pathogenesis of psoriasis: From keratinocyte perspective. Cell Death Dis. 2022, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Feldman, S.R.; Krueger, G.G. Psoriasis assessment tools in clinical trials. Ann. Rheum. Dis. 2005, 64 (Suppl. SII), ii65–ii68. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Akerman, L.; Ziv, M.; Kadurina, M.; Gospodinov, D.; Pavlotsky, F.; Yankova, R.; Kouzeva, V.; Ramon, M.; Silverman, M.H.; et al. Treatment of plaque-type psoriasis with oral CF101: Data from an exploratory randomized phase 2 clinical trial. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 361–367. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Gospodinov, D.K.; Gheorghe, N.; Mateev, G.S.; Rusinova, M.V.; Hristakieva, E.; Solovastru, L.G.; Patel, R.V.; Giurcaneanu, C.; Hitova, M.C.; et al. Treatment of plaque-type psoriasis with oral CF101: Data from a phase II/III multicenter, randomized, controlled trial. J. Drugs Dermatol. 2016, 15, 931–938. [Google Scholar]
- Papp, K.; Reich, K.; Leonardi, C.L.; Kircik, L.; Chimenti, S.; Langley, R.G.; Hu, C.; Stevens, R.M.; Day, R.M.; Gordon, K.B.; et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: Results of a phase III, randomized, controlled trial (efficacy and safety trial evaluating the effects of apremilast in psoriasis [esteem] 1). J. Am. Acad. Dermatol. 2015, 73, 37–49. [Google Scholar] [CrossRef]
- Apremilast [Package Insert]; Amgen: Thousand Oaks, CA, USA, 2021.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Subramaniam, S.; Kelley, R.B.; Venook, A.P. A review of hepatocellular carcinoma (HCC) staging systems. Chin. Clin. Oncol. 2013, 2, 33–45. [Google Scholar]
- Rimassa, L.; Worns, M.A. Navigating the new landscape of second-line treatment in advanced hepatocellular carcinoma. Liver Int. 2020, 40, 1800–1811. [Google Scholar] [CrossRef]
- Stemmer, S.M.; Manojlovic, N.S.; Marinca, M.V.; Petrov, P.; Cherciu, N.; Ganea, D.; Ciuleanu, T.E.; Pusca, I.A.; Beg, M.S.; Purcell, W.T.; et al. Namodenoson in advanced hepatocellular carcinoma and Child-Pugh B cirrhosis: Randomized placebo-controlled clinical trial. Cancers 2021, 13, 187. [Google Scholar] [CrossRef]
- Vuppalanchi, R.; Noureddin, M.; Alkhouri, N.; Sanyal, A.J. Therapeutic pipeline in nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Safadi, R.; Braun, M.; Francis, A.; Milgrom, Y.; Massarwa, M.; Hakimian, D.; Hazou, W.; Issachar, A.; Harpaz, Z.; Farbstein, M.; et al. Randomised clinical trial: A phase 2 double-blind study of namodenoson in non-alcoholic fatty liver disease and steatohepatitis. Aliment. Pharmacol. Ther. 2021, 54, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fishman, P. Drugs Targeting the A3 Adenosine Receptor: Human Clinical Study Data. Molecules 2022, 27, 3680. https://doi.org/10.3390/molecules27123680
Fishman P. Drugs Targeting the A3 Adenosine Receptor: Human Clinical Study Data. Molecules. 2022; 27(12):3680. https://doi.org/10.3390/molecules27123680
Chicago/Turabian StyleFishman, Pnina. 2022. "Drugs Targeting the A3 Adenosine Receptor: Human Clinical Study Data" Molecules 27, no. 12: 3680. https://doi.org/10.3390/molecules27123680
APA StyleFishman, P. (2022). Drugs Targeting the A3 Adenosine Receptor: Human Clinical Study Data. Molecules, 27(12), 3680. https://doi.org/10.3390/molecules27123680