
The Potential of a Protein Model Synthesized Absent of Methionine



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methionine’s Role in Protein Structure
3. Antioxidant Function of Methionine
3.1. Discovering Methionine’s Antioxidant Function
3.2. Reversible Oxidative Ability of Methionine
4. Methionine Modulating Cellular Activity
4.1. Methionine and Calmodulin
4.2. Methionine and Actin
4.3. Methionine and Ion Channels
5. Cysteine Sharing Methionine Function
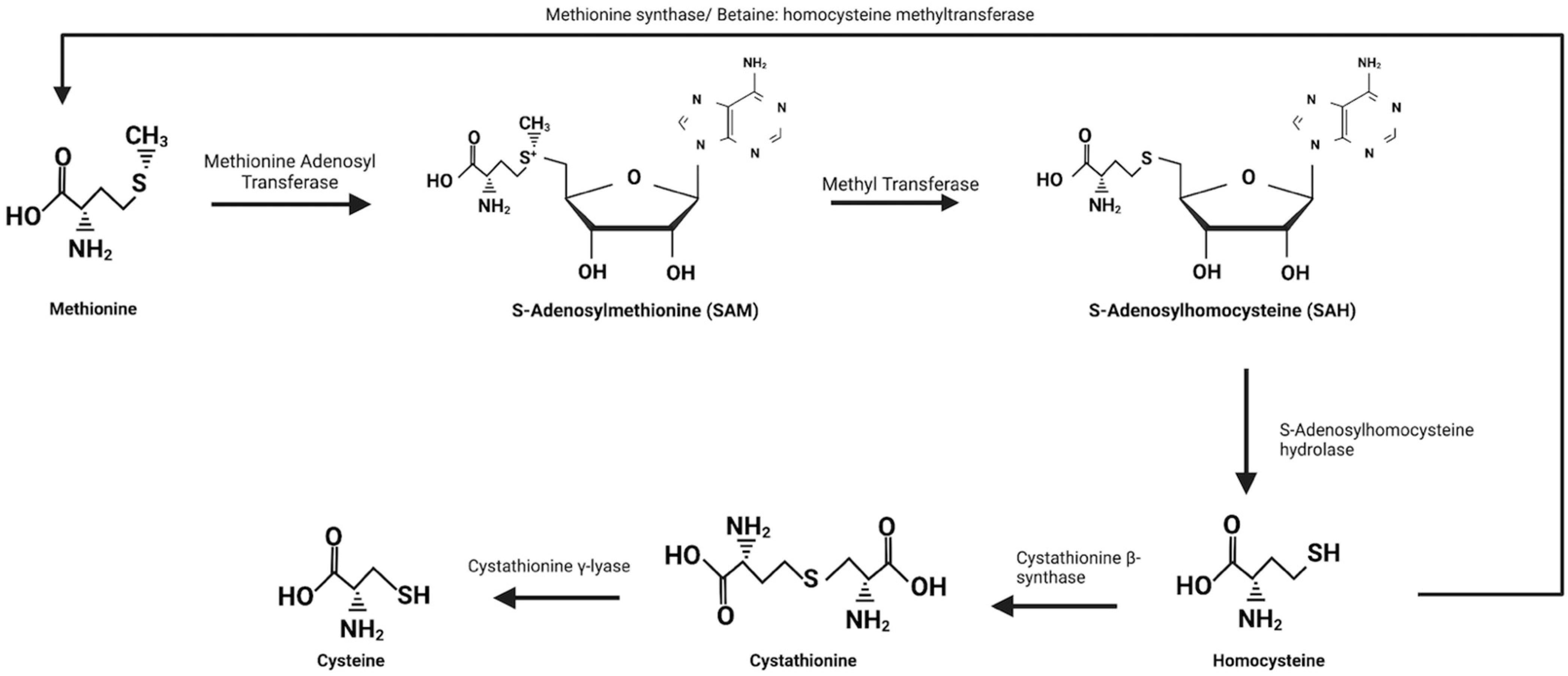
5.1. Transmethylation and Transsulfuration
5.2. Cysteine and Methionine in Protein and Cell Structure
5.3. Cysteine’s Antioxidant Function
5.4. Cysteine Modulating Cell and Protein Activity
6. Alternative Mechanisms for Protein Synthesis Initiation
6.1. Initiating Protein Sythesis with Alternative Amino Acids
6.2. Ribosome Reinitiation
6.3. Internal Ribosome Entry Sites
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whillier, S.; Raftos, J.E.; Chapman, B.; Kuchel, P.W. Role of N-acetylcysteine and cystine in glutathione synthesis in human erythrocytes. Redox Rep. 2009, 14, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Piste, P. Cysteine–master antioxidant. Int. J. Pharm. Chem. Biol. Sci. 2013, 3, 143–149. [Google Scholar]
- Mokhtari, V.; Afsharian, P.; Shahhoseini, M.; Kalantar, S.M.; Moini, A. A review on various uses of N-acetyl cysteine. Cell J. 2017, 19, 11–17. [Google Scholar] [PubMed]
- Neal, R.; Matthews, R.; Lutz, P.; Ercal, N. Antioxidant role of N-acetyl cysteine isomers following high dose irradiation. Free Radic. Biol. Med. 2003, 34, 689–695. [Google Scholar] [CrossRef]
- García-Santamarina, S.; Boronat, S.; Hidalgo, E. Reversible Cysteine Oxidation in Hydrogen Peroxide Sensing and Signal Transduction. Biochemistry 2004, 53, 2560–2580. [Google Scholar] [CrossRef]
- Poole, L.B.; Nelson, K.J. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr. Opin. Chem. Biol. 2008, 12, 18–24. [Google Scholar] [CrossRef]
- Miki, H.; Funato, Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J. Biochem. 2012, 151, 255–261. [Google Scholar] [CrossRef]
- Cross, J.; Templeton, D. Regulation of Signal Transduction through Protein Cysteine Oxidation. Antioxid. Redox Signal. 2006, 8, 1819–1827. [Google Scholar] [CrossRef]
- Isom, D.; Vardy, E.; Oas, T.; Hellinga, H. Picomole-scale characterization of protein stability and function by quantitative cysteine reactivity. Proc. Natl. Acad. Sci. USA 2010, 107, 4908–4913. [Google Scholar] [CrossRef]
- Skryhan, K. The Role of Cysteine Residues in Redox Regulation and Protein Stability of Arabidopsis thaliana Starch Synthase 1. PLoS ONE 2015, 10, e0136997. [Google Scholar] [CrossRef]
- Lim, J.M.; Kim, G.; Levine, R.L. Methionine in Proteins: It’s not Just for Protein Initiation Anymore. Neurochem. Res. 2019, 44, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Zauhar, R.J.; Colbert, C.L.; Morgan, R.S.; Welsh, W.J. Evidence for a strong sulfur-aromatic interaction derived from crystallographic data. Biopolym. Orig. Res. Biomol. 2000, 53, 233–248. [Google Scholar] [CrossRef]
- Valley, C.C. The Methionine-aromatic Motif Plays a Unique Role in Stabilizing Protein Structure. J. Biochem. 2012, 287, 34979–34991. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Mosoni, L.; Berlett, B.S.; Stadtman, E.R. Methionine residues as endogenous antioxidants in proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 15036–15040. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, I.; Mary, J.; Perichon, M.; Friguet, B. Rat peptide methionine sulphoxide reductase: Cloning of the cDNA, and down-regulation of gene expression and enzyme activity during aging. Biochem. J. 2001, 355, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Gabbita, S.P.; Aksenov, M.Y.; Lovell, M.A.; Markesbery, W.R. Decrease in Peptide Methionine Sulfoxide Reductase in Alzheimer’s Disease Brain. J. Neurochem. 2002, 73, 1660–1666. [Google Scholar] [CrossRef]
- Vogt, W. Oxidation of methionyl residues in proteins: Tools, targets, and reversal. Free Radic. Biol. Med. 1995, 18, 93–105. [Google Scholar] [CrossRef]
- Iyer, B.R.; Mahalakshmi, R. Hydrophobic Characteristic Is Energetically Preferred for Cysteine in a Model Membrane Protein. Biophys. J. 2019, 117, 25–35. [Google Scholar] [CrossRef]
- Womack, M.; Kemmerer, K.S.; Rose, W.C. The Relation of Cystine and Methionine to Growth. J. Biol. Chem. 1937, 121, 403–410. [Google Scholar] [CrossRef]
- Womack, M.; Rose, W.C. The Partial Replacement of Dietary Methionine by Cystine for Purposes of Growth. J. Biol. Chem. 1941, 141, 375–379. [Google Scholar] [CrossRef]
- Wingfield, P.T. N-Terminal Methionine Processing. Curr. Prot. Protein Sci. 2017, 88, 6.14.1–6.14.3. [Google Scholar] [CrossRef] [PubMed]
- Boix, E. Role of the N Terminus in RNase A Homologues: Differences in Catalytic Activity, Ribonuclease Inhibitor Interaction and Cytotoxicity. J. Mol. Biol. 1996, 257, 992–1007. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Yamamoto, Y.; Sugawara, T.; Nishimura, O.; Fujino, M. The Additional Methionine Residue at the N-Terminus of Bacterially Expressed Human Interleukin-2 Affects the Interaction between the N- and C-Termini. Biochemistry 2001, 40, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-D. The structural integrity exerted by N-terminal pyroglutamate is crucial for the cytotoxicity of frog ribonuclease from Rana pipiens. Nucleic Acids Res. 2003, 31, 5247–5255. [Google Scholar] [CrossRef][Green Version]
- Reid, K.S.C.; Lindley, P.F.; Thornton, J.M. Sulphur-aromatic interactions in proteins. FEBS Lett. 1985, 190, 209–213. [Google Scholar] [CrossRef]
- Orabi, E.A.; English, A.M. Modeling Protein S–Aromatic Motifs Reveals Their Structural and Redox Flexibility. J. Phys. Chem. B 2018, 122, 3760–3770. [Google Scholar] [CrossRef]
- Lewis, A.K. Oxidation increases the strength of the methionine-aromatic interaction. Nat. Chem. Biol. 2016, 12, 860–866. [Google Scholar] [CrossRef]
- Chao, C.-C.; Ma, Y.-S.; Stadtman, E.R. Modification of protein surface hydrophobicity and methionine oxidation by oxidative systems. Proc. Nat. Acad. Sci. USA 1997, 94, 2969–2974. [Google Scholar] [CrossRef]
- Brot, N.; Weissbach, H. Biochemistry and physiological role of methionine sulfoxide residues in proteins. Arch. Biochem. Biophys. 1983, 223, 271–281. [Google Scholar] [CrossRef]
- Levine, R.L.; Berlett, B.S.; Moskovitz, J.; Mosoni, L.; Stadtman, E.R. Methionine residues may protect proteins from critical oxidative damage. Mech. Aging Dev. 1999, 107, 323–332. [Google Scholar] [CrossRef]
- Luo, S.; Levine, R. Testing the Hypothesis That “Methionine Residues in Proteins Are Antioxidants”. FASEB J. 2008, 22, 758.1. [Google Scholar] [CrossRef]
- Netzer, N. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 2009, 462, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Toennies, G.; Kolb, J.J. Methionine Studies. J. Biol. Chem. 1939, 128, 399–405. [Google Scholar] [CrossRef]
- Lim, J.C.; You, Z.; Kim, G.; Levine, R.L. Methionine sulfoxide reductase A is a stereospecific methionine oxidase. Proc. Natl. Acad. Sci. USA 2011, 10472–10477. [Google Scholar] [CrossRef] [PubMed]
- Boschi-Muller, S.; Gand, A.; Branlant, G. The methionine sulfoxide reductases: Catalysis and substrate specificities. Arch. Biochem. Biophys. 2008, 474, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Nagano, N.; Ota, M.; Nishikawa, K. Strong hydrophobic nature of cysteine residues in proteins. FEBS Lett. 1999, 458, 69–71. [Google Scholar] [CrossRef]
- Manta, B.; Gladyshev, V.N. Regulated methionine oxidation by monooxygenases. Free Radic. Biol. Med. 2017, 109, 141–155. [Google Scholar] [CrossRef]
- Lin, Z. Free methionine-(R)-sulfoxide reductase from Escherichia coli reveals a new GAF domain function. Proc. Natl. Acad. Sci. USA 2007, 104, 9597–9602. [Google Scholar] [CrossRef]
- Lee, B.C.; Le, D.T.; Gladyshev, V.N. Mammals Reduce Methionine-S-sulfoxide with MsrA and Are Unable to Reduce Methionine-R-sulfoxide, and This Function Can Be Restored with a Yeast Reductase. J. Biol. Chem. 2008, 283, 28361–28369. [Google Scholar] [CrossRef]
- Le, D.T. Functional Analysis of Free Methionine-R-sulfoxide Reductase from Saccharomyces cerevisiae. J. Biol. Chem. 2009, 284, 4354–4364. [Google Scholar] [CrossRef]
- Dobrovolska, O.; Rychkov, G.; Shumilina, E.; Nerinovski, K.; Schmidt, A.; Shabalin, K.; Yakimov, A.; Dikiy, A. Structural Insights into Interaction between Mammalian Methionine Sulfoxide Reductase B1 and Thioredoxin. J. Biomed. Biotechnol. 2012, 2012, 586539. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J.; Bar-Noy, S.; Williams, W.; Requena, J.; Berlett, B.; Stadtman, E. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc. Natl. Acad. Sci. USA 2001, 93, 12920–12925. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.; Daniel, D.S.; Parida, B.K.; Jagannath, C.; Dhandayuthapani, S. Methionine Sulfoxide Reductase A (MsrA) Deficiency Affects the Survival of Mycobacterium smegmatis within Macrophages. J. Bacteriol. 2004, 186, 3590–3598. [Google Scholar] [CrossRef] [PubMed]
- John, G.S. Peptide methionine sulfoxide reductase from Escherichia coli and Mycobacterium tuberculosis protects bacteria against oxidative damage from reactive nitrogen intermediates. Proc. Natl. Acad. Sci. USA 2001, 98, 9901–9906. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J.; Flescher, E.; Berlett, B.S.; Azare, J.; Poston, J.M.; Stadtman, E.R. Overexpression of peptide-methionine sulfoxide reductase in Saccharomyces cerevisiae and human T cells provides them with high resistance to oxidative stress. Proc. Natl. Acad. Sci. USA 1998, 95, 14071–14075. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proc. Natl. Acad. Sci. USA 2002, 99, 2748–2753. [Google Scholar] [CrossRef] [PubMed]
- Picot, C.R.; Perichon, M.; Cintrat, J.-C.; Friguet, B.; Petropoulos, I. The peptide methionine sulfoxide reductases, MsrA and MsrB (hCBS-1), are downregulated during replicative senescence of human WI-38 fibroblasts. FEBS Lett. 2004, 558, 74–78. [Google Scholar] [CrossRef]
- Novoselov, S.V. Regulation of Selenoproteins and Methionine Sulfoxide Reductases A and B1 by Age, Calorie Restriction, and Dietary Selenium in Mice. Antioxid. Redox Signal. 2010, 12, 829–838. [Google Scholar] [CrossRef]
- Dragicevic, N.; Smith, A.; Lin, X.; Yuan, F.; Copes, N.; Delic, V.; Tan, J.; Cao, C.; Shytle, R.D.; Bradshaw, P.C. Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J. Alzheimers Dis. 2011, 26, 507–521. [Google Scholar] [CrossRef]
- Zhao, G.; Yao-Yue, C.; Qin, G.-W.; Guo, L.-H. Luteolin from Purple Perilla mitigates ROS insult particularly in primary neurons. Neurobiol. Aging 2012, 33, 176–186. [Google Scholar] [CrossRef]
- Sanei, M.; Saberi-Demneh, A. Effect of curcumin on memory impairment: A systematic review. Phytomedicine 2019, 52, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Catanesi, M. L-Methionine Protects against Oxidative Stress and Mitochondrial Dysfunction in an In Vitro Model of Parkinson’s Disease. Antioxidants 2021, 10, 1467. [Google Scholar] [CrossRef] [PubMed]
- Squier, T.; Bigelow, D. Protein oxidation and age-dependent alterations in calcium homeostasis. Front. Biosci.-Landmark 2000, 5, d504. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.C.; Kim, G.; Levine, R.L. Stereospecific oxidation of calmodulin by methionine sulfoxide reductase A. Free Radic. Biol. Med. 2013, 61, 257–264. [Google Scholar] [CrossRef]
- Marimoutou, M.; Springer, D.; Liu, C.; Kim, G.; Levine, R. Oxidation of Methionine 77 in Calmodulin Alters Mouse Growth and Behavior. Antioxidants 2018, 7, 140. [Google Scholar] [CrossRef]
- Dalle-Donne, I. The actin cytoskeleton response to oxidants: From small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic. Biol. Med. 2001, 31, 1624–1632. [Google Scholar] [CrossRef]
- Milzani, A. The oxidation produced by hydrogen peroxide on Ca-ATP-G-actin. Protein Sci. 2000, 9, 1774–1782. [Google Scholar] [CrossRef]
- Giridharan, S.S.P.; Caplan, S. MICAL-Family Proteins: Complex Regulators of the Actin Cytoskeleton. Antioxid. Redox Signal. 2014, 20, 2059–2073. [Google Scholar] [CrossRef]
- Hung, R.-J.; Pak, C.W.; Terman, J.R. Direct Redox Regulation of F-Actin Assembly and Disassembly by Mical. Science 2011, 334, 1710–1713. [Google Scholar] [CrossRef]
- Lee, B.; Péterfi, Z.; Hoffmann, F.; Fomenko, D.; Hoffmann, P.; Gladyshev, V. MsrB1 and MICALs Regulate Actin Assembly and Macrophage Function via Reversible Stereoselective Methionine Oxidation. Mol. Cell 2013, 51, 397–404. [Google Scholar] [CrossRef]
- Hung, R.-J.; Spaeth, C.S.; Yesilyurt, H.G.; Terman, J.R. SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat. Cell Biol. 2013, 15, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Dichiara, T.J.; Reinhart, P.H. Redox Modulation of Ca2+-Activated K+ Channels. J. Neurosci. 1997, 17, 4942–4955. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-W.; Nara, M.; Wang, Y.-X.; Kotlikoff, M.I. Redox Regulation of Large Conductance Ca2+-activated K+ Channels in Smooth Muscle Cells. J. Gen. Physiol. 1997, 110, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Findlay, I. Contrasting effects of intracellular redox couples on the regulation of maxi-K channels in isolated myocytes from rabbit pulmonary artery. J. Gen. Physiol. 1997, 500, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.D.; Daggett, H.; Hanner, M.; Garcia, M.; McManus, O.; Brot, N.; Weissbach, H.; Heinemann, S.; Hoshi, T. Oxidative Regulation of Large Conductance Calcium-Activated Potassium Channels. J. Gen. Physiol. 2001, 117, 253–274. [Google Scholar] [CrossRef]
- Kassmann, M.; Hansel, A.; Leipold, E.; Birkenbell, J.; Lu, S.Q.; Hoshi, T.; Heinemann, S. Oxidation of multiple methionine residues impairs rapid sodium channel inactivation. Pflügers Arch.-Eur. J. Physiol. 2008, 456, 1085–1095. [Google Scholar] [CrossRef][Green Version]
- Ma, J.-H.; Luo, A.-T.; Zhang, P.-H. Effect of hydrogen peroxide on persistent sodium current in guinea pig ventricular myocytes1. Acta Pharm. Sin. 2005, 26, 828–834. [Google Scholar] [CrossRef]
- Song, Y.; Shryock, J.C.; Wagner, S.; Maier, L.S.; Belardinelli, L. Blocking Late Sodium Current Reduces Hydrogen Peroxide-Induced Arrhythmogenic Activity and Contractile Dysfunction. J. Pharm. Exp. Thera 2006, 318, 214–222. [Google Scholar] [CrossRef]
- Hoshi, T.; Heinemann, S.H. Regulation of cell function by methionine oxidation and reduction. J. Physiol. 2001, 531, 1–11. [Google Scholar] [CrossRef]
- Du Vigneaud, V.; Kilmer, G.W.; Rachele, J.R.; Cohn, M. On the Mechanism of the conversion in vivo of methionine to cystine. J. Biol. Chem. 1944, 155, 645–651. [Google Scholar] [CrossRef]
- Cantoni, G.L. S-Adenosylmethionine: A New Intermediate Formed Enzymatically from L-Methionine and Adenosinetriphosphate. J. Biol. Chem. 1953, 204, 403–416. [Google Scholar] [CrossRef]
- Shoveller, A.K.; Brunton, J.A.; House, J.D.; Pencharz, P.B.; Ball, R.O. Dietary Cysteine Reduces the Methionine Requirement by an Equal Proportion in Both Parenterally and Enterally Fed Piglets. J. Nutr. 2003, 133, 4215–4224. [Google Scholar] [CrossRef] [PubMed]
- Di Buono, M.; Wykes, L.J.; Ball, R.O.; Pencharz, P.B. Dietary cysteine reduces the methionine requirement in men. Am. J. Clin. Nutr. 2001, 74, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.; Olszowy, P.; Ciborowski, P. 2—Biomolecules. In Proteomic Profiling and Analytical Chemistry, 2nd ed.; Ciborowski, P., Silberring, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 7–24. [Google Scholar] [CrossRef]
- Ziegler, D.M.; Poulsen, L.L. Protein disulfide bond synthesis: A possible intracellular mechanism. Trends Biochem. Sci. 1977, 2, 79–81. [Google Scholar] [CrossRef]
- Pal, D.; Chakrabarti, P. Different Types of Interactions Involving Cysteine Sulfhydryl Group in Proteins. J. Biomol. Struct. Dyn. 1998, 15, 1059–1072. [Google Scholar] [CrossRef]
- Duan, G.; Smith, V.H.; Weaver, D.F. Characterization of aromatic-thiol π-type hydrogen bonding and phenylalanine-cysteine side chain interactions through ab initio calculations and protein database analyses. Mol. Phys. 2001, 99, 1689–1699. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Rhee, S. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Meister, A. Glutathione Metabolism. Methods Enzym. 1995, 251, 3–7. [Google Scholar]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Et Biophys. Acta 2008, 1780, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Poland, B.; Honzatko, R. Protein Sulfhydryls and Their Role in Antioxidant Function of Protein S-Thiolation. Arch. Biochem. Biophys. 1995, 319, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.E.; Roth, D.; Winther, J.R. Quantifying the global cellular thiol–disulfide status. Proc. Natl. Acad. Sci. USA 2009, 106, 422–427. [Google Scholar] [CrossRef]
- Requejo, R.; Hurd, T.R.; Costa, N.J.; Murphy, M.P. Cysteine residues exposed on protein surfaces are the dominant intramitochondrial thiol and may protect against oxidative damage. FEBS J. 2010, 277, 1465–1480. [Google Scholar] [CrossRef]
- Ruppersberg, J.P. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature 1991, 352, 711–714. [Google Scholar] [CrossRef]
- Heinemann, S.; Rettig, J.; Wunder, F.; Pongs, O. Molecular and functional characterization of a rat brain Kv β 3 potassium channel subunit. FEBS Lett. 1995, 377, 383–389. [Google Scholar]
- Ciorba, M.; Heinemann, S.; Weissbach, H.; Hoshi, T. Modulation of potassium channel function by methionine oxidation and reduction. Proc. Natl. Acad. Sci. USA 1997, 94, 9932–9937. [Google Scholar] [CrossRef]
- Yellen, G. The voltage-gated potassium channels and their relatives. Nature 2002, 419, 35–42. [Google Scholar] [CrossRef]
- Daloso, D.M.; Müller, K.; Obata, T.; Florian, A.; Tohge, T.; Bottcher, A.; Riondet, C.; Bariat, L.; Carrari, F.; Nunes-Nesi, A.; et al. Thioredoxin, a master regulator of the tricarboxylic acid cycle in plant mitochondria. Proc. Nat. Acad. Sci. USA 2015, 11, 1392–1400. [Google Scholar] [CrossRef]
- Nietzel, T.; Mostertz, J.; Hochgräfe, F.; Schwarzländer, M. Redox regulation of mitochondrial proteins and proteomes by cysteine thiol switches. Mitochondrion 2017, 33, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Zito, E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 2015, 83, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Sevier, C.S.; Qu, H.; Heldman, N.; Gross, E.; Fass, D.; Kaiser, C. Modulation of Cellular Disulfide-Bond Formation and the ER Redox Environment by Feedback Regulation of Ero1. Cell 2007, 129, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Riemer, J.; Christensen, B.; Sørensen, E.S.; Ellgaard, L. A novel disulphide switch mechanism in Ero1α balances ER oxidation in human cells. EMBO J. 2008, 27, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.M.; Chakravarthi, S.; Langton, K.P.; Sheppard, A.M.; Lu, H.; Bulleid, N.J. Low reduction potential of Ero1α regulatory disulphides ensures tight control of substrate oxidation. EMBO J. 2008, 27, 2988–2997. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Stortchevoi, A.; Köhrer, C.; Varshney, U.; RajBhandary, U.L. Initiator tRNA and its role in initiation of protein synthesis. Cold Spring Harb. Symp Quant. Biol. 2001, 66, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Chattapadhyay, R.; Pelka, H.; Schulman, L.H. Initiation of in vivo protein synthesis with non-methionine amino acids. Biochemistry 1990, 29, 4263–4268. [Google Scholar] [CrossRef]
- Pallanck, L.; Schulman, L.H. Anticodon-dependent aminoacylation of a noncognate tRNA with isoleucine, valine, and phenylalanine in vivo. Proc. Natl. Acad. Sci. USA 1991, 88, 3872–3876. [Google Scholar] [CrossRef]
- Pak, M.; Pallanck, L.; Schulman, L.H. Conversion of a methionine initiator tRNA into a tryptophan-inserting elongator tRNA in vivo. Biochemistry 1992, 31, 3303–3309. [Google Scholar] [CrossRef]
- Varshney, U.; Rajbhandary, U.L. Initiation of protein synthesis from a termination codon. Proc. Natl. Acad. Sci. USA 1990, 87, 1586–1590. [Google Scholar] [CrossRef]
- Tharp, J.M.; Krahn, N.; Varshney, U.; Söll, D. Hijacking Translation Initiation for Synthetic Biology. ChemBioChem 2020, 21, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Tharp, J.M. Initiation of Protein Synthesis with Non-Canonical Amino Acids In Vivo. Angew. Chem. 2020, 132, 3146–3150. [Google Scholar] [CrossRef]
- Tharp, J.M.; Walker, J.A.; Söll, D.; Schepartz, A. Initiating protein synthesis with noncanonical monomers in vitro and in vivo. Met. Enzym. 2021, 656, 495–519. [Google Scholar]
- Drabkin, H.J.; Rajbhandary, U.L. Initiation of Protein Synthesis in Mammalian Cells with Codons Other than AUG and Amino Acids Other than Methionine. Mol. Cell Biol. 1998, 18, 5140–5147. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.; Hellen, C.U.T.; Pestova, T.V. Termination and Post-termination Events in Eukaryotic Translation. Adv. Protein Chem. Struct. Biol. 2012, 86, 45–93. [Google Scholar]
- Lee, S. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2012, 109, E2424–E2432. [Google Scholar] [CrossRef]
- Johnstone, T.G.; Bazzini, A.A.; Giraldez, A.J. Upstream ORF s are prevalent translational repressors in vertebrates. EMBO J. 2016, 35, 706–723. [Google Scholar] [CrossRef]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational Regulation of Gene Expression during Conditions of Cell Stress. Mol. Cell 2010, 40, 228–237. [Google Scholar] [CrossRef]
- Mohammad, M.P.; Pondělíčková, V.M.; Zeman, J.; Gunišová, S.; Valášek, L.S. In vivo evidence that eIF3 stays bound to ribosomes elongating and terminating on short upstream ORFs to promote reinitiation. Nucleic Acids Res. 2017, 45, 2658–2674. [Google Scholar] [CrossRef]
- Kwan, T.; Thompson, S.R. Noncanonical Translation Initiation in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2019, 11, a032672. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, I.I. Non-Canonical Translation Initiation Mechanisms Employed by Eukaryotic Viral mRNAs. Biochemistry 2021, 86, 1060–1094. [Google Scholar] [CrossRef] [PubMed]
- Walters, B.; Thompson, S.R. Cap-independent translational control of carcinogenesis. Front. Oncol. 2016, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Weingarten-Gabbay, S.; Elias-Kirma, S.; Nir, R.; Gritsenko, A.A.; Stern-Ginossar, N.; Yakhini, Z.; Weinberger, A.; Segal, E. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science 2016, 351, 6270. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savino, R.J.; Kempisty, B.; Mozdziak, P. The Potential of a Protein Model Synthesized Absent of Methionine. Molecules 2022, 27, 3679. https://doi.org/10.3390/molecules27123679
Savino RJ, Kempisty B, Mozdziak P. The Potential of a Protein Model Synthesized Absent of Methionine. Molecules. 2022; 27(12):3679. https://doi.org/10.3390/molecules27123679
Chicago/Turabian StyleSavino, Ronald J., Bartosz Kempisty, and Paul Mozdziak. 2022. "The Potential of a Protein Model Synthesized Absent of Methionine" Molecules 27, no. 12: 3679. https://doi.org/10.3390/molecules27123679
APA StyleSavino, R. J., Kempisty, B., & Mozdziak, P. (2022). The Potential of a Protein Model Synthesized Absent of Methionine. Molecules, 27(12), 3679. https://doi.org/10.3390/molecules27123679